

Генетика, 2019, T. 55, № 2, стр. 229-233
Мажорная мутация в гене SPAST у пациентов с аутосомно-доминантной спастической параплегией из Республики Башкортостан
И. М. Хидиятова 1, 2, *, А. Ф. Ахметгалеева 1, Е. В. Сайфуллина 3, Р. Ф. Идрисова 4, М. А. Янкина 1, В. В. Шавалиева 2, Р. В. Магжанов 3, Э. К. Хуснутдинова 1, 2
1 Институт биохимии и генетики Уфимского федерального исследовательского центра
Российской академии наук
450054 Уфа, Россия
2 Башкирский государственный университет
450076 Уфа, Россия
3 Башкирский государственный медицинский университет
450000 Уфа, Россия
4 Республиканская клиническая больница им. Г.Г. Куватова
450005 Уфа, Россия
* E-mail: imkhid@mail.ru
Поступила в редакцию 16.02.2018
После доработки 10.04.2018
Принята к публикации 20.03.2018
Аннотация
Наследственные спастические параплегии (НСП) – группа нейродегенеративных болезней с преимущественным поражением пирамидного тракта. На сегодняшний день мутации, ответственные за заболевание, идентифицированы более чем в 70 генных локусах. К наиболее частым причинам развития НСП относятся мутации в гене спастина (SPAST), однако мажорные мутации являются редкостью для данного заболевания. При исследовании пациентов из 63 неродственных семей с НСП, проживающих в Республике Башкортостан (РБ), в гене SPAST была идентифицирована мутация c.283delG (p.Ala95Profs*66), обнаруженная в семьях татарской этнической принадлежности с высокой частотой. В общей выборке неродственных больных из РБ ее частота составила 19%, а в выборке пациентов-татар – 44%. Было установлено, что во всех семьях с данной мутацией НСП наследуется по аутосомно-доминантному типу, а клинические симптомы заболевания в большинстве случаев соответствуют типичному неосложненному фенотипу, характерному для НСП формы SPG4.
Наследственные спастические параплегии (НСП) – генетически и клинически гетерогенная группа дегенеративных заболеваний нервной системы, обусловленных дистальным поражением длинных аксонов кортикоспинального тракта. Клинически НСП проявляются спастичностью мышц нижних конечностей. В зависимости от того, является ли основной симптом единственным или сочетается с другими неврологическими или экстраневральными симптомами, выделяют неосложненные или осложненные формы [1]. Распространенность НСП, как в целом, так и ее отдельных генетических форм, значительно варьирует в различных популяциях, составляя от 0.5 до 12 на 100 000 населения (http://www.hspersunite.org.au); в Республике Башкортостан (РБ) – 3.5 на 100 000 населения [2].
В настоящее время известно более 70 генетических локусов, идентифицировано 59 генов, мутации в которых обусловливают развитие НСП с различными типами наследования [3, 4] (http:// neuromuscular.wustl.edu/spinal/fsp.html). По современной номенклатуре генные локусы и соответствующие формы НСП обозначают аббревиатурой SPG (от англ. Spastik Paraplegia Gene), с порядковыми номерами в хронологической последовательности [5]. Эпидемиологические и молекулярно-генетические исследования НСП в отдельных регионах и этнических группах представляются весьма актуальными, позволяющими разрабатывать наиболее эффективные подходы ДНК-диагностики и медико-генетического консультирования в семьях с данной патологией.
Мутации в гене спастина (SPAST) являются наиболее частой причиной НСП, они ответственны за 45% случаев аутосомно-доминантных НСП (АД НСП), 12–18% спорадических случаев заболевания [6]. Ранее нами были представлены результаты исследований гена SPAST у пациентов с НСП из РБ, проведенных частично методами прямого секвенирования (в отдельных экзонах гена), частично – методами SSCP-анализа с последующим секвенированием образцов с измененной электрофоретической подвижностью одноцепочечной ДНК. В ходе этих исследований были идентифицированы новые, ранее неописанные мутации с.322del29 (p.Val108SerfsX18), с.885del10 (p.Thr295ThrfsX16), c.1114A>G (p.Arg372Gly) [7, 8].
В настоящее время общую обследуемую выборку пациентов с НСП из РБ представляют члены 63 неродственных семей (из них 27 – татарских; пять – башкирских; 14 – русских; по одной семье – чувашской, украинской, марийской; восемь – метисных семей, а также шесть семей с неустановленной этнической принадлежностью). С целью продолжения поиска генетических причин развития НСП у жителей исследуемого региона в трех неродственных семьях с аутосомно-доминантной формой заболевания у пробандов было проведено таргетное секвенирование экзома, включающее анализ кодирующих последовательностей более 700 генов, ответственных за возникновение ряда неврологических заболеваний, в том числе 54 генов НСП.
Секвенирование проведено на приборе MiSeq, Illumina. Обработка данных секвенирования была проведена с использованием автоматизированного алгоритма, включающего выравнивание прочтений на референсную последовательность генома человека (hg19), постпроцессинг выравнивания, выявление вариантов и фильтрацию вариантов по качеству, а также аннотацию выявленных вариантов по всем известным транскриптам каждого гена из базы RefSeq с применением ряда методов предсказания патогенности замен (SIFT, PolyPhen2-HDIV, PolyPhen2-HVAR, MutationTaster, LRT), а также методов расчета эволюционной консервативности позиций (PhyloP, PhastCons). Для оценки популяционных частот выявленных вариантов использованы выборки проектов “1000 геномов”, ESP6500 и Exome Aggregation Consortium.
Предположительно патогенный вариант, обнаруженный в гене SPAST, был подтвержден секвенированием по Сэнгеру. Далее был проведен скрининг на его наличие/отсутствие у других членов семей обследованных пациентов, затем – в 63 неродственных семьях с НСП из РБ, а также в контрольной выборке здоровых индивидов (60 чел.). У всех обследуемых лиц кровь была получена с их информированного согласия. Исследования одобрены биоэтическим комитетом ИБГ УФИЦ РАН.
В результате проведенного исследования у всех трех неродственных пациентов идентифицирована однонуклеотидная гетерозиготная делеция в первом экзоне гена SPAST: c.283delG (p.Ala95Profs*66). Делеция подтверждена секвенированием по Сэнгеру (рис. 1). Было установлено, что данная делеция может быть выявлена с помощью рестриктазы BsaJI, для которой при наличии мутации сайт рестрикции теряется. Также ее можно идентифицировать методом SSCP-анализа или электрофореза двухцепочечной ДНК в полиакриламидном геле, получив первоначально достаточно короткие для данных методов фрагменты (ранее, в предшествующем исследовании, мы не выявили изменений картины SSCP-анализа образцов с данной делецией при длине исследуемого фрагмента 320 пн). Так, при использовании эндонуклеазы MspI исходный амплификат разрезается на фрагменты размерами 128, 124, 35, 22, 6, 6 пн, при этом участок ДНК с делецией с.283delG попадает во фрагмент 35 пн. При наличии делеции в гетерозиготном состоянии образуются соответствующие фрагменты 35 и 34 пн, разделяющиеся электрофоретически в 8%-ном ПААГ. Этим методом был проведен дальнейший скрининг на наличие данной делеции в гене SPAST в семьях пациентов и в контрольной выборке. В семьях трех обследуемых пациентов делеция c.283delG была обнаружена только у больных родственников. В общей обследуемой выборке пациентов с НСП (63 неродственных пациента и члены их семей) делеция была выявлена у больных из 12 неродственных семей. Примечательным оказалось то, что все эти семьи принадлежат к этнической группе татар. В результате последующего скрининга на наличие/отсутствие делеции с.283delG в контрольной группе здоровых неродственных индивидов татарской этнической принадлежности (60 чел.) данная мутация не была обнаружена.
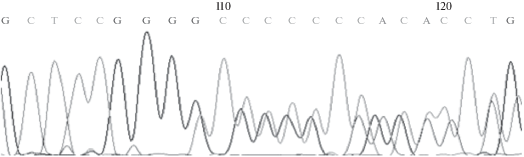
Делеция c.283delG (p.Ala95Profs*66) в гене SPAST не зарегистрирована в контрольных выборках “1000 геномов”, ESP6500 и ExAC, но, описанная в одной британской семье с НСП [9], представлена в базе мутаций HGMD (www.hgmd.org).
Клиническая картина всех обследованных пациентов с мутацией p.Ala95Profs*66 соответствовала неосложненной форме НСП, что характерно для большинства случаев формы SPG4, описанных в ряде работ [6, 9–11].
Белок спастин является представителем семейства ААА – белков, АТФаз особого класса с множественными видами клеточной активности [12]. Главная функция спастина – обеспечение динамики микротрубочек цитоскелета. Нарушение этого процесса приводит к развитию заболевания формы SPG4. В результате делеции с.283delG в первом экзоне гена SPAST, приводящей к сдвигу рамки считывания, синтезируется укороченный белок, в котором отсутствуют домены, выполняющие основные функции белка: MIT-домен (116–194 АА), от которого зависит способность спастина участвовать в цитокинезе и эндосомальном транспорте [13, 14]; MTBD-домен (270–328 АА), играющий важную роль в морфогенезе ЭПР [15–18]; АТФазный домен (342–599 АА), ответственный за разборку спастином микротрубочек – одну из главных функций белка [19].
Исследования роли спастина в динамике микротрубочек показали, что при мутациях, приводящих к преждевременному окончанию синтеза белка, НСП развивается из-за недостатка его количества [20], о чем свидетельствует и отсутствие детектируемых уровней количества укороченных спастинов в клеточных линиях [21–23]. Следовательно, гаплонедостаточность – самый ожидаемый механизм для объяснения развития спастической параплегии в случае с синтезом укороченного белка [6, 24]. Кроме того, появились и новые данные, указывающие на возможность и другого отрицательного типа воздействия укороченных белков спастина на нейроны: усеченная изоформа M1 спастина, необходимая для взаимодействия трубочек ЭПР и ЭПР с микротрубочками, является более стабильной, чем усеченная M87 изоформа, и может оказывать токсический эффект на нейроны путем нарушения аксонального ретроградного транспорта и из-за ограниченной способности нейронов устранять поврежденные органеллы и белки [25].
Таким образом, учитывая все описанные выше сведения – локализацию делеции в первом экзоне гена, приводящую к сдвигу рамки считывания и преждевременной терминации синтеза белка, наличие ее только у пациентов с НСП и отсутствие у здоровых членов семей, а также в контрольной группе здоровых индивидов, можно с достаточной уверенностью считать ее патогенной мутацией. В гене спастина на сегодняшний день описано более 680 различных мутаций (www.hgmd.org), в том числе приводящих к появлению преждевременного стоп-кодона и нарушению синтеза полноразмерного белка: это нонсенс-мутации [26], различные делеции со сдвигом рамки считывания [6, 11, 27, 28], дупликации и инсерции [26, 29]. Однако в основном все описанные мутации были идентифицированы в отдельных семьях с НСП, и в целом мажорные мутации являются редкостью для данного заболевания. Обнаруженная нами делеция c.283delG (p.Ala95Profs*66) была идентифицирована в 12 семьях татарской этнической принадлежности, ее частота составила среди татарских семей 44%, а среди всех обследованных неродственных семей с НСП из Республики Башкортостан – 19%. Распространение мутации в семьях татарской этнической принадлежности, проживающих в РБ, предположительно можно связать с эффектом основателя, что характерно для многих известных мутаций, частых среди больных с наследственной патологией, проживающих в этом регионе. В целом делеция c.283delG (p.Ala95Profs*66) в гене SPAST расширила спектр идентифицированных мутаций у пациентов с НСП из РБ и, являясь самой частой (мажорной), вносит существенный вклад в создание алгоритма ДНК-диагностики НСП в данном регионе.
Работа выполнена на оборудовании ЦКП “Биомика” (Отделение биохимических методов исследований и нанобиотехнологии РЦКП “Агидель”) и УНУ “КОДИНК”. Исследование поддержано грантом РФФИ р_а № 17-44-020951; образцы ДНК для исследования взяты из “Коллекции биологических материалов человека” ИБГ УФИЦ РАН, поддержанной Программой биоресурсных коллекций ФАНО России (соглашение № 007-030164/2).
Список литературы
Harding A.E. Classification of the hereditary ataxias and paraplegias // The Lancet. 1983. V. 321. № 8334. P. 1151–1155.
Магжанов Р.В., Сайфуллина Е.В., Идрисова Р.Ф. и др. Эпидемиологическая характеристика наследственных спастических параплегий в Республике Башкортостан // Мед. генетика. 2013. № 7. С. 12–16.
Novarino G., Fenstermaker A.G., Zaki M.S. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders // Science. 2014. V. 343. № 6170. P. 506–511. doi 10.1126/science.1247363
Klebe S., Stevanin G., Depienne C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: from SPG1 to SPG72 and still counting // Rev. Neurol. 2015. V. 171. № 6. P. 505–530. doi 10.1016/j.neurol.2015.02.017
Fink J.K. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms // Acta Neuropathologica. 2013. V. 126. № 3. P. 307–328. doi 10.1007/s00401-013-1115-8
Fonknechten N., Mavel D., Byrne B. et al. Spectrum of SPG4 mutations in autosomal dominant spastic paraplegia // Hum. Mol. Genet. 2000. V. 9. № 4. P. 637–644.
Ахметгалеева А.Ф., Хидиятова И.М., Сайфуллина Е.В. и др. Две новые мутации в гене SPG4 у пациентов с аутосомно-доминантной спастической параплегией // Генетика. 2016. Т. 52. № 6. С. 691–696.
Ахметгалеева А.Ф., Хидиятова И.М., Сайфуллина Е.В. и др. Клинический случай спорадической спастической параплегии при новой мутации в гене SPAST // Мед. генетика. 2016. Т. 15. № 7. С. 11–13.
Lindsey J.C., Lusher M.E., McDermott C.J. et al. Mutation analysis of the spastin gene (SPG4) in patients with hereditary spastic paraparesis // J. Med. Genet. 2000. V. 37. № 10. P. 759–765.
Hentati A., Deng H.X., Zhai H. et al. Novel mutations in spastin gene and absence of correlation with age at onset of symptoms // Neurology. 2000. V. 55. № 9. P. 1388–1390.
Basri R.1., Yabe I., Soma H. et al. Four mutations of the spastin gene in Japanese families with spastic paraplegia // J. Hum. Genet. 2006. V. 51. № 8. P. 711–715. doi 10.1007/s10038-006-0412-7
Lumb J.H., Connell J.W., Allison R., Reid E. The AAA ATPase spastin links microtubule severing to membrane modelling // Biochim. Biophys. Acta 2012. V. 1823. № 1. P. 192–197. doi 10.1016/j.bbamcr.2011.08.010
Guizetti J., Schermelleh L., Mäntler J. et al. Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments // Science. 2011. V. 331. № 6024. P. 1616–1620. doi 10.1126/science.1201847
Allison R.1., Lumb J.H., Fassier C. et al. An ESCRT-spastin interaction promotes fission of recycling tubules from the endosome // J. Cell Biol. 2013. V. 202. № 3. P. 527–543. doi 10.1083/jcb.201211045
White S.R., Lauring B. AAA+ ATPases: achieving diversity of function with conserved machinery // Traffic. 2007. V. 8. № 12. P. 1657–1667. doi 10.1111/j.1600-0854.2007.00642.x
Park S.H., Zhu P.P., Parker R.L., Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network // J. Clin. Invest. 2010. V. 120. № 4. P. 1097–1110. doi 10.1172/JCI40979
Blackstone C., O’Kane C.J., Reid E. Hereditary spastic paraplegias: membrane traffic and the motor pathway // Nat. Rev. Neurosci. 2011. V. 12. № 1. P. 31–42. doi 10.1038/nrn2946
Blackstone C. Cellular pathways of hereditary spastic paraplegia // Annu. Rev. Neurosci. 2012. V. 35. P. 25–47. doi 10.1146/annurev-neuro-062111-150400
Evans K.J., Gomes E.R., Reisenweber S.M. et al. Linking axonal degeneration to microtubule remodeling by Spastin-mediated microtubule severing // J. Cell. Biol. 2005. V. 168. № 4. P. 599–606.
Errico A., Ballabio A., Rugarli E.I. Spastin, the protein, mutated in autosomal dominant hereditary spastic paraplegia, is involved in microtubule dynamics // Hum. Mol. Genet. 2002. V. 11. № 2. P. 153–163.
Solowska J.M., Baas P.W. Hereditary spastic paraplegia SPG4: what is known and not known about the disease // Brain. 2015. P. 2471–2484.
Rebbapragada I., Lykke-Andersen J. Execution of nonsense-mediated mRNA decay: what defines a substrate? // Curr. Opin. Cell Biol. 2009. V. 21. № 3. P. 394–402. doi 10.1016/j.ceb.2009.02.007
Lykke-Andersen S., Jensen T.H. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes // Nat. Rev. Mol. Cell Biol. 2015. V. 16. № 11. P. 665–677. doi 10.1038/nrm4063
Burger J., Fonknechten N., Hoeltzenbein M. et al. Hereditary spastic paraplegia caused by mutations in the SPG4 gene // Eur. J. Hum. Genet. 2000. V. 8. № 10. P. 771–776. doi 10.1038/sj.ejhg.5200528
Solowska J.M., Rao A.N., Baas P.W. Truncating mutations of SPAST associated with hereditary spastic paraplegia indicate greater accumulation and toxicity of the M1 isoform of spastin // Mol. Biol. Cell. 2017. V. 28. № 13. P. 1728–1737. doi 10.1091/mbc.E17-01-0047
de Bot S.T., van den Elzen R.T., Mensenkamp A.R. Hereditary spastic paraplegia due to SPAST mutations in 151 Dutch patients: new clinical aspects and 27 novel mutations // J. Neurol. Neurosurg. Psychiatry. 2010. V. 81. № 10. P. 1073–1078. doi 10.1136/jnnp.2009.201103
Sauter S., Miterski B., Klimpe S. et al. Mutation analysis of the spastin gene (SPG4) in patients in Germany with autosomal dominant hereditary spastic paraplegia // Hum. Mutat. 2002. V. 20. № 2. P. 127–132. doi 10.1002/humu.10105
Magariello A., Muglia M., Patitucci A. et al. Novel spastin (SPG4) mutations in Italian patients with hereditary spastic paraplegia // Neuromusc. Disorders. 2006. V. 16. № 6. P. 387–390. doi 10.1016/j.nmd.2006.03.009
Crippa F., Panzeri C., Martinuzzi A. et al. Eight novel mutations in SPG4 in a large sample of patients with hereditary spastic paraplegia // Arch. Neurol. 2006. V. 5. P. 750–755. doi 10.1001/archneur.63.5.750
Дополнительные материалы отсутствуют.