

Генетика, 2019, T. 55, № 5, стр. 516-523
Полиморфизм однонуклеотидных замен в генах hsp65 и MACPPE12 Mycobacterium avium subsp. hominissuis
Д. А. Старкова 1, *, T. Iwamoto 2, А. А. Вязовая 1, В. М. Молчанов 3, В. Ю. Журавлев 4, Б. И. Вишневский 4, О. В. Нарвская 1, 4
1 Санкт-Петербургский научно-исследовательский институт эпидемиологии и микробиологии
им. Пастера
197101 Санкт-Петербург, Россия
2 Институт здравоохранения, кафедра инфекций
6500046 Кобе, Япония
3 Санкт-Петербургский химико-фармацевтический университет
197376 Санкт-Петербург, Россия
4 Санкт-Петербургский научно-исследовательский институт фтизиопульмонологии
193063 Санкт-Петербург, Россия
* E-mail: dariastarkova13@gmail.com
Поступила в редакцию 17.07.2018
После доработки 15.08.2018
Принята к публикации 24.08.2018
Аннотация
Mycobacterium avium subsp. hominissuis (MAH) – обитатели окружающей среды, которые являются оппортунистическими патогенами животных и человека. Целью исследования являлся анализ профилей однонуклеотидных замен (SNPs) в генах hsp65 и MACPPE12 для характеристики российской популяции МАН в контексте изучения филогенетических связей и эволюции географически удаленных популяций M. avium subsp. hominissuis. Нами проведено секвенирование продуктов амплификации hsp65 и MACPPE12 40 штаммов MAH, выделенных от больных микобактериозом. Нуклеотидные последовательности выравнивали на референсный геном M. avium subsp. hominissuis 104 (accession no. NC_008595.1). Сравнивали мутационные профили российских и зарубежных штаммов MAH. Профили однонуклеотидных замен в гене hsp65 соответствовали секвоварам трех типов: code 1, code 2 и code 3. Большинство штаммов MAH (72.5%) принадлежали к секвовару code 1, характерному для референс-штамма. В гене MACPPE12 выявлены SNPs в 20 позициях. Профили SNPs MACPPE12 были сгруппированы в девять “нуклеотидных” секвоваров: NA01, NA02, NA03, NA06, NA10, NA13, NA14, NA19 и NA_Rus01. Из 20 SNPs восемь являлись несинонимичными, что обусловило формирование семи “аминокислотных” секвоваров: AA01, AA02, AA04, AA07, AA08, AA13 и AA_Rus01. При этом в состав секвовара AA02 входили три варианта профиля синонимичных нуклеотидных замен: NA02, NA03 и NA06. Половина штаммов MAH принадлежали к секвовару AA02 (тип NA02). Таким образом, нами установлена относительная консервативность нуклеотидной последовательности гена hsp65 и полиморфизм гена MACPPE12, а также выявлен доминирующий кластер АА02 (тип NA02)/code 1 и уникальный вариант АA_Rus01 (NA_Rus01) среди штаммов российской популяции МАН. Сравнительный анализ профилей SNP sгенов hsp65 и MACPPE12 позволил выявить различия и сходство между штаммами географически удаленных популяций МАН, что вносит вклад в характеристику глобальной популяции вида M. avium.
Бактерии рода Mycobacterium семейства Mycobacteriaceae – неподвижные, аэробные, грамположительные палочки, которые характеризуются кислото- и щелочеустойчивостью при окраске карболовым фуксином, высоким содержанием липидов в клеточной стенке. Содержание Г + Ц в молекуле ДНК составляет 62–70%. В настоящее время род включает более 130 видов, подвидов и комплексов [1–3].
К представителям рода Mycobacterium относят возбудителей туберкулеза человека и животных (M. tuberculosis, M. bovis, M. africanum и др.), а также сапрофитные и условно-патогенные виды нетуберкулезных микобактерий, в частности Mycobacterium avium [1–3].
M. avium – медленнорастущие кислотоустойчивые микобактерии, типичные обитатели окружающей среды, которые, однако, являются оппортунистическими патогенами диких и домашних животных, птиц и человека [1, 4].
Согласно современным представлениям, вид M. avium включает несколько подвидов, ассоциированных с определенным кругом хозяев, экологическими и географическими характеристиками штаммов, которые имеют специфические геномные паттерны. Так, Mycobacterium avium subsp. hominissuis (MAH) может вызывать заболевания у людей (в том числе у ВИЧ-инфицированных) и животных (крупного рогатого скота, свиней); M. avium subspp. avium, silvaticum и paratuberculosis поражают птиц и животных [1, 5, 6].
Генетическое разнообразие MAH обусловлено полиморфизмом кодирующих (rpoB, gyrB, hsp65, область ITS гена 16S-23S rRNA) и некодирующих (MATR-VNTR, IS901, IS900, IS1245, IS1311) последовательностей генома. Показано, что геном штаммов MAH характеризуется наибольшим числом единичных однонуклеотидных замен (Single Nucleotide Polymorphisms, SNPs) по сравнению с M. avium других подвидов [7].
Анализ полиморфизма участка конститутивного гена hsp65, кодирующего белок теплового шока (heat shock protein) с молекулярной массой 65 кДa, используют для геноидентификации MAH [8–10].
Известно, что микобактерии имеют два родственных семейства генов – PE и PPE. Наименования этих генов отражают присутствие ProGlu (PE) и ProProGlu (PPE) мотивов в консервативных доменах N-терминальной области соответствующих глицин-богатых белковых антигенов. Показано, что белки РЕ и РРЕ, экспрессируемые на поверхности бактериальных клеток, ассоциированы с вирулентностью микобактерий и формированием клеточного и гуморального иммунного ответа [11]. Среди ортологов РРЕ особый интерес представляет ген МАСРРЕ12, поскольку он является уникальным для подвида hominissuis [12, 13].
Для дифференциации штаммов (генотипирования) МАН используют присутствие однонуклеотидных замен в генах hsp65 и МАСРРЕ12 [4, 7, 8].
Ранее на основе 13-локусного MATR-VNTR-типирования и IS1245-RFLP-типирования нами выявлена генетическая неоднородность 90 штаммов MAH, выделенных от больных микобактериозом из числа иммунокомпетентных (микобактериоз легких) и ВИЧ-инфицированных (диссеминированная форма микобактериоза) [14, 15].
Целью настоящего исследования является анализ профилей однонуклеотидных замен в генах hsp65 и MACPPE12 для характеристики российской популяции МАН в контексте изучения филогенетических связей и эволюции географически удаленных популяций M. avium subsp. hominissuis.
МАТЕРИАЛЫ И МЕТОДЫ
Изучены 90 штаммов M. avium, полученных от двух групп пациентов (72 штамма от иммунокомпетентных больных микобактериозом легких и 19 штаммов от ВИЧ-инфицированных с диссеминированной формой инфекции) в 2008–2011 гг. в Санкт-Петербургском научно-исследовательском институте фтизиопульмонологии (СПбНИИФ). Культивирование микобактерий осуществляли в лаборатории СПбНИИФ общепринятым методом на среде Левенштейна–Йенсена при 37°С. Видимый рост культур медленнорастущих микобактерий наблюдали в течение 21 дня.
Процедуру выделения хромосомной ДНК из чистых культур M. avium проводили с использованием стандартного протокола [16].
Все штаммы M. avium были отнесены к подвиду hominissuis с помощью полимеразной цепной реакции (ПЦР) для детекции инсерционных элементов IS901, IS900 [17]. Аллельный полиморфизм 90 штаммов MAH оценивали методом MATR-VNTR-типирования по 13 вариабельным локусам: TR292, TRX3, TR25, TR47, MATR-1, MATR-4, MATR-5, MATR-6, MATR-8, MATR-11, MATR14, MATR-15, MATR-16 [15, 18, 19].
Для дальнейшей оценки генетической вариабельности штаммов MAH методом секвенирования продуктов амплификации генов hsp65 и MACPPE12 из 90 штаммов были отобраны 40 (в том числе 19 – от ВИЧ-позитивных пациентов), из которых 24 имели индивидуальный MATR-VNTR-профиль IP (Individual Profile), остальные 16 входили в состав кластеров Mа1–Mа8 [15].
Секвенирование продукта амплификации 3'‑концевого участка гена hsp65 1059 пн (начиная с позиции 574) проводили с использованием прямого MAChspF_574 (CGGTTCGACAAGGGTTACAT) и обратного MAChsp65R (ACGGACTCAGAAGTCCATGC) праймеров [4, 20].
Секвенирование продукта амплификации размером 1337 пн гена MACPPE12 (расположен в локусе Mav_2006 референс-штамма M. avium subsp. hominissuis 104 (accession no. in GenBank, NC_008595.1)) проводили с использованием прямого MAV_2006F (TGCGTGGTAACAAAAGCAAC) и обратного MAV_2006R (CTTGCTGCGTAATGCGATAA) праймеров [13].
Обработка хроматограмм секвенирования и выравнивание сиквенсов на референсный геном M. avium subsp. hominissuis 104, трансляция генов и идентификация нуклеотидных и аминокислотных замен были выполнены с использованием пакета программ SnapGene® (версия 4.1), Unipro UGENE 1.12 (Россия) и Highest stringency. Обработанные последовательности были выровнены на референсный геном методом локального парного выравнивания (алгоритм Смита–Ватермана) с автоматической идентификацией нуклеотидных замен. Поиск аминокислотных замен осуществлялся путем сравнения аминокислотных последовательностей продуктов трансляции референсного генома и анализируемых сиквенсов.
Степень родства между штаммами MAH оценивали с использованием алгоритма Neighbor Joining (NJ) и графически отображали в виде дендрограммы, построенной на сайте URL: http:// www.miru-vntrplus.org.
РЕЗУЛЬТАТЫ
Видимый рост нефотохромогенных микобактерий на среде Левенштейна–Йенсена, положительные результаты биохимических тестов (каталазная активность, способность восстанавливать теллурит калия, наличие никотинамидазы и пиразинамидазы) позволили судить о принадлежности 40 чистых культур медленнорастущих нетуберкулезных микобактерий к группе Mycobacterium avium complex. Все штаммы были идентифицированы до вида M. avium с использованием тест-системы GenoType®Mycobacterium CM/AS (Hain Lifescience, Германия) и отнесены к подвиду hominissuis, поскольку не содержали мобильных элементов IS901 и IS900 [14, 15].
Результаты секвенирования ПЦР-продукта 3'‑участка гена hsp65 (1090 пн), с одной стороны, подтвердили принадлежность исследуемых штаммов M. avium к подвиду hominissuis, с другой – продемонстрировали их относительный геномный полиморфизм. Так, профили однонуклеотидных замен в гене hsp65 (табл. 1) соответствовали секвоварам трех типов: code 1, code 2 и code 3 [4, 20]. Большинство штаммов MAH (n = 29; 72.5%) были отнесены к секвовару code 1, характерному для референс-штамма M. avium subsp. hominissuis 104. Секвовары code 2 и code 3 включали 3 (7.5%) и 8 (20%) штаммов соответственно. Следует отметить, что к секвовару code 1 принадлежали 15 из 19 штаммов MAH, выделенных от ВИЧ-позитивных больных микобактериозом с диссеминированной формой инфекции; к секвовару code 3 – остальные штаммы.
Таблица 1.
Профили SNPs в гене hsp65 клинических изолятов МАН по сравнению со штаммом M. avium subsp. hominissuis 104
| hsp65 секвовары | Позиции нуклеотидных замен в штамме M. avium subsp. hominissuis 104 | Число щтаммов МАН | ||||
|---|---|---|---|---|---|---|
| 1128 | 1218 | 1269 | 1272 | 1536 | ||
| Code 1 | C | A | G | C | A | 29 |
| Code 2 | · | G | · | · | G | 3 |
| Code 3 | G | G | C | G | G | 8 |
Секвенирование нуклеотидной последовательности гена MACPPE12 40 штаммов MAH выявило однонуклеотидные замены в 20 позициях (табл. 2) по сравнению с референс-штаммом M. avium subsp. hominissuis 104. Как видно из табл. 2, профили SNPs были сгруппированы в девять “нуклеотидных” секвоваров: NA01, NA02, NA03, NA06, NA10, NA13, NA14, NA19 и NA_Rus01 (Nucleic Acid, нуклеиновая кислота). Из 20 SNPs восемь являлись несинонимичными (nsSNPs), что обусловило наличие семи “аминокислотных” секвоваров: AA01, AA02, AA04, АА07, AA08, AA13 и AA_Rus01 (Amino Acid, аминокислота). При этом в состав секвовара AA02 входили три варианта профиля синонимичных нуклеотидных замен (sSNPs): NA02, NA03 и NA06. Следует отметить, что обозначения выявленных нами секвоваров по аминокислотному и нуклеотидному типам приведены согласно Iwamoto et al. [13]. Однако секвовар, обозначенный АA_Rus01 (NA_Rus01), являлся уникальными для российской выборки штаммов MAH.
Таблица 2.
Профили SNPs в гене MACPPE12 клинических изолятов МАН по сравнению со штаммом M. avium subsp. hominissuis 104
| Аминокислотный (AA) тип профиля SNP | Нуклеотидный (NA) тип профиля SNP | Позиции нуклеотидных замен в штамме M. avium subsp. hominissuis 104 | Число штаммов МАН | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 66 | 272 | 468 | 558 | 605 | 733 | 744 | 765 | 822 | 829 | 831 | 866 | 867 | 868 | 870 | 877 | 924 | 978 | 1150 | 1330 | |||
| S (Gly-Gly) | N (Lys-Arg) | S (Gly-Gly) | S (Val-Val) | N (Ser-Leu) | S (Leu-Leu) | S (Ala-Ala) | S (Gly-Gly) | S (Thr-Thr) | N (Ser-Gly) | S (Ser-Ser) | N (Leu-Gln) | S (Leu-Leu) | N (Glu-Lys) | S (Glu-Glu) | N (Ala-Pro) | S (Gly-Gly) | S (Ala-Ala) | N (Ala-Thr) | N (Arg-Gly) | |||
| AA01 | NA01 | A | A | T | T | С | С | G | G | G | A | С | T | G | G | G | G | С | G | G | A | 4 |
| AA02 | NA02 | · | · | · | · | · | · | · | · | · | · | · | · | · | · | · | · | · | · | · | G | 20 |
| NA03 | · | · | С | · | · | T | · | · | · | · | · | · | · | · | · | · | · | · | · | G | 1 | |
| NA06 | G | · | · | · | · | · | · | · | · | · | · | · | · | · | · | · | · | · | · | G | 1 | |
| AA04 | NA10 | · | · | · | · | T | · | · | · | · | · | · | · | · | · | · | · | · | · | · | G | 1 |
| AA07 | NA13 | G | · | С | С | · | · | · | · | · | G | T | · | · | · | A | · | · | · | · | G | 5 |
| AA08 | NA14 | · | G | С | С | · | · | · | · | · | G | T | · | · | · | · | С | T | A | · | G | 2 |
| AA13 | NA19 | · | · | С | С | · | · | A | A | A | G | T | A | A | A | · | · | · | · | A | . | 5 |
| AA_ Rus01 | NA_ Rus01 | · | · | С | С | · | · | · | · | · | G | T | · | · | · | A | · | · | · | · | G | 1 |
Половина из 40 штаммов MAH были отнесены к доминирующему секвовару AA02 типа NA02. К этому же секвовару принадлежали девять (47.4%) из 19 штаммов, выделенных от ВИЧ-позитивных больных микобактериозом; к типам NA03 и NA06 секвовара AA02 – по одному штамму MAH.
Секвовары AA07 и AA13 включали по пять штаммов соответственно; остальные секвовары – AA01, AA04, AA08, AA_Rus01 содержали от одного до трех штаммов MAH.
Характеристика полиморфизма клинических изолятов МАН по генам МАСPPE12 и hsp65 представлена в табл. 3. Данные табл. 3 демонстрируют, что в российской популяции МАН доминирующим является кластер АА02 (тип NA02)/code 1.
Таблица 3.
Характеристика полиморфизма клинических изолятов МАН по генам МАСPPE12 и hsp65
| Секвовары MACPPE12 | Секвовары hsp65 | |||
|---|---|---|---|---|
| АА-тип | NA-тип | code 1 | code 2 | code 3 |
| AA01 | NA01 | 2 | 1 | 1 |
| AA02 | NA02 | 15 | 2 | 3 |
| NA03 | 1 | – | – | |
| NA06 | 1 | – | – | |
| AA13 | NA19 | 3 | – | 2 |
| AA04 | NA10 | 1 | – | – |
| AA07 | NA13 | 3 | – | 2 |
| AA08 | NA14 | 2 | – | – |
| AA_Rus01 | NA_Rus01 | 1 | – | – |
ОБСУЖДЕНИЕ
Анализ полиморфизма участка конститутивного гена hsp65 успешно используют для геноидентификации М. avium уже более 10 лет. Однако присутствие и распределение однонуклеотидных мутаций в различных участках hsp65 позволило использовать последовательность данного гена не только для внутривидовой идентификации, но и дифференциации штаммов (генотипирования) возбудителя [7–10]. Различные варианты рекомбинационных событий между аллелями в геноме MAH привели к формированию гетерогенности популяции в целом [7].
Так, сравнительный анализ мутационных профилей hsp65 российских и зарубежных штаммов, выделенных от человека, выявил различия популяций MAH. Установлено, что в Японии и Корее явно доминировали секвовары code 15, code 16 и code 2, тогда как наиболее распространенный секвовар code 1, выявленный более чем у половины российских штаммов (72.5%), встречался лишь у 4.7% штаммов из Японии и отсутствовал в корейской выборке [4, 21]. Вместе с тем секвовары code 1 и code 2 превалировали среди штаммов MAH в США и Канаде [20].
Обращает на себя внимание, что code 1 в Японии доминирует у штаммов MAH, выделенных от свиней (76%) [4]. Секвовары code 1 (23.1%) и code 2 (65.4%) преобладают у штаммов МАН крупного рогатого скота в Швейцарии [22].
Принимая во внимание генетическое родство штаммов M. avium hominissuis – оппортунистических патогенов домашних животных и человека, ряд авторов указывают на общность источников инфекции и возможность передачи возбудителя от животных к человеку [1, 5, 13, 23]. Однако до сих пор остается открытым вопрос о передаче возбудителя от человека к человеку.
Полагают, что вирулентность микобактерий связана с семейством генов, контролирующих экспрессию белков PE (Pro-Glu) и PPE (Pro-Pro-Glu), функции которых у штаммов M. avium изучены недостаточно [12, 13].
В зарубежной литературе имеется ограниченное число публикаций, посвященных генетическому разнообразию M. avium hominissuis, что затрудняет сравнительный анализ полученных данных. Так, Iwamoto et al. [13] приводят результаты секвенирования гена MACPPE12 штаммов MAH, выделенных от разных источников окружающей среды, в Японии и Корее. Сравнение выборок продемонстрировало доминирование секвовара АА02 у штаммов МАН, выделенных от человека в России, Японии и Корее. Примечательно, что секвовар АА03, который является вторым по частоте встречаемости в Японии, не обнаружен в изученной нами выборке, в то время как характерный лишь для корейской выборки секвовар АА13 выявлен у штаммов МАН российской популяции.
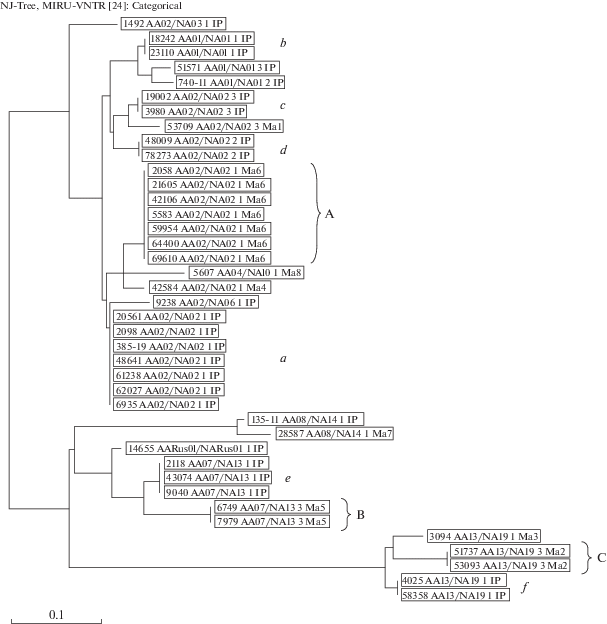
Сопоставление результатов MATR-VNTR типирования штаммов MAH по 13 локусам (TR292, TRX3, TR25, TR47, MATR-1, MATR-4, MATR-5, MATR-6, MATR-8, MATR-11, MATR-14, MATR-15, MATR-16) [15] с результатами секвенирования генов МАСРРЕ12 и hsp65 отражено на дендрограмме (рис. 1). Как видно из дендрограммы, наиболее крупный кластер А включает семь штаммов доминирующего в российской популяции МАН кластера Ма6 (с числовым профилем 2 222 223 145 443') секвоваров АА02(NA02)/code 1. Кластеры В и С включают по два штамма кластера Ма5 (2422223142443') секвоваров АА07(NA13)/code 3 и кластера Ма2 (2422213145443') секвоваров АА13(NA19)/code 3 соответственно. Штаммы кластеров a, b, c, d, e, f группировались по принципу идентичности нуклеотидных последовательностей генов МАСРРЕ12 (АА- и NА-типы) и hsp65 (code 1–3), но имели при этом индивидуальные числовые профили (IP), различавшиеся по числу копий более чем в двух локусах MATR-VNTR. Учитывая различную скорость эволюции кодирующих и некодирующих областей генома, данное обстоятельство свидетельствует о гетерогенности группы штаммов, входящих в состав кластеров а–f, но не исключает филогенетического родства между ними.
Рис. 1.
Дендрограмма профилей SNPs генов МАСРРЕ12, hsp65 и профилей MATR-VNTR 40 штаммов M. avium subsp. hominissuis. В прямоугольниках слева направо указаны: номер штамма МАН, секвовары АА и NA гена МАСРРЕ12, секвовар гена hsp65, MATR-VNTR-профиль (IP – индивидуальный числовой профиль). Кластеры А, В, С объединяют штаммы МАН с идентичными профилями SNPs генов МАСРРЕ12, hsp65 и MATR-VNTR. Кластеры a, b, c, d, e, f объединяют штаммы МАН с идентичными профилями SNPs генов МАСРРЕ12 и hsp65, но индивидуальными числовыми профилями (IP) MATR-VNTR.
Таким образом, нами установлена относительная консервативность нуклеотидной последовательности конститутивного гена теплового шока hsp65 и полиморфизм гена MACPPE12, а также выявлен доминирующий кластер штаммов, представляющих российскую популяцию МАН.
Вместе с тем сравнительный анализ профилей однонуклеотидных замен генов hsp65 и MACPPE12 позволил выявить различия и сходство между штаммами географически удаленных популяций МАН, что вносит вклад в характеристику глобальной популяции вида M. avium.
Список литературы
Оттен Т.Ф., Васильев А.В. Микобактериоз. СПб: Мед. пресса, 2005. 224 с.
Levy-Frebault V.V., Portaels F. Proposed minimal standards for the genus Mycobacterium and for description of new slowly growing Mycobacterium species // Int. J. Syst. Bacteriol. 1992. V. 42. P. 315–323.
Shinnick T.M., Good R.C. Mycobacterial taxonomy // Europ. J. Clin. Mycrobiol. Infect. Dis. 1994. V. 13. № 11. P. 884–901.
Iwamoto T., Nakajima C., Nishiuchi Y. et al. Genetic diversity of Mycobacterium avium subsp. hominissuis strains isolated from humans, pigs, and human living environment // Infect. Genet. Evol. 2012. V. 12. № 4. P. 846–852.
Falkinham III J.O. Nontuberculous mycobacteria in the environment // Clin. Chest Med. 2002. V. 23. № 3. P. 529–551.
Turenne C.Y., Wallace R., Behr M.A. Mycobacterium avium in the postgenomic era // Clin. Microb. Rev. 2007. V. 20. № 2. P. 205–229.
Rindi L., Garzelli C. Genetic diversity and phylogeny of Mycobacterium avium // Infect. Genet. Evol. 2014. V. 21. № 4. P. 375–383.
Chimara E., Ferrazoli L., Ueky S.Y.M. et al. Reliable identification of mycobacterial species by PCR-restriction enzyme analysis (PRA)-hsp65 in a reference laboratory and elaboration of a sequence-based extended algorithm of PRA-hsp65 patterns // BMC Microbiol. 2008. V. 8. № 48. doi https://doi.org/10.1186/1471-2180-8-48
McNabb A., Eisler D., Adie K. et al. Assessment of partial sequencing of the 65-kilodalton heat shock protein gene (hsp65) for routine identification of Mycobacterium species isolated from clinical sources // J. Clin. Microbiol. 2004. V. 42. № 7. P. 3000–3011.
Telenti A., Marchesi F., Balz M. et al. Rapid identification of mycobacteria to the species level by polymerase chain reaction and restriction enzyme analysis // J. Clin. Microbiol. 1993. V. 31. № 2. P. 175–178.
Sampson S.L. Mycobacterial PE/PPE proteins at the host-pathogen interface // Clin. Dev. Immunol. 2011. doi https://doi.org/10.1155/2011/497203
Mackenzie N., Alexander D.C., Turenne C.Y. et al. Genomic comparison of PE and PPE genes in the Mycobacterium avium complex // J. Clin. Microbiol. 2009. V. 47. P. 1002–1011.
Iwamoto T., Arikawa K., Nakajima C. et al. Intra-subspecies sequence variability of the MACPPE12 gene in Mycobacterium avium subsp. hominissuis // Genet. Evol. 2014. V. 21. P. 479–483.
Старкова Д.А., Оттен Т.Ф., Мокроусов И.В. и др. Генотипическая характеристика штаммов Mycobacterium avium subsp. hominissuis // Генетика. 2013. Т. 49. № 9. С. 1048–1054.
Старкова Д.А., Мокроусов И.В., Вязовая А.А. и др. Геномный полиморфизм штаммов Mycobacterium avium subsp. hominissuis // Мол. генет., микробиол., вирусол. 2014. Т. 29. № 4. С. 14–19.
Van Embden J.D., Cave M.D., Crawford J.T. et al. Strain identification on Mycobacterium tuberculosis by DNA fingerprinting: Recommendations for a standardized methodology // J. Clin. Microbiol. 1993. V. 31. № 2. P. 406–409.
Bartos M., Hlozek P., Svastova P. et al. Identification of members of Mycobacterium avium species by Accu-Probes, serotyping, and single IS900, IS901, IS1245 and IS901-flanking region PCR with internal standards // J. Microbiol. Methods. 2006. V. 64. № 3. P. 333–345.
Inagaki T., Nishimori K., Yagi T. et al. Comparison of a Variable-Number Tandem-Repeat (VNTR) method for typing Mycobacterium avium with Mycobacterial Interspersed Repetitive-Unit–VNTR and IS1245 restriction fragment length polymorphism typing // J. Clin. Microbiol. 2009. V. 47. № 7. P. 2156–2164.
Thibault V.C., Grayon M., Boschiroli M.et al. New Variable-Number Tandem-Repeat markers for typing Mycobacterium avium subsp. paratuberculosis and M. avium strains: Comparison with IS900 and IS1245 restriction fragment length polymorphism typing // J. Clin. Microbiol. 2007. V. 45. № 8. P. 2404–2410.
Turenne C.Y., Semret M., Cousins D.V. et al. Sequencing of hsp65 distinguishes among subsets of the Mycobacterium avium complex // J. Clin. Microbiol. 2006. V. 44. № 2. P. 433–440. doi https://doi.org/10.1128/JCM.44.2.433-440.2006
Kim S.-Y., Jeong B.-H., Park H.Y. et al. Association of ISMav6 with the pattern of antibiotic resistance in Korean Mycobacterium avium clinical isolates but no relevance between their genotypes and clinical features // PLoS One. 2016. V. 11. № 2. doi https://doi.org/10.1371/journal.pone.0148917
Scherrer S., Landolt P., Carroli N., Stephan R. Molecular characterization of Mycobacterium avium subsp. hominissuis of two groups of lymph nodes, being intradermal tuberculin or interferon-gamma test positive and negative, isolated from swiss cattle at slaughter // Front. Vet. Sci. 2018. V. 5. № 32. doi https://doi.org/10.3389/ fvets.2018.00032
Vluggen C., Soetaert K., Duytschaever L. et al. Genotyping and strain distribution of Mycobacterium avium subspecies hominissuis isolated from humans and pigs in Belgium, 2011–2013 // Euro Surveill. 2016. V. 21. № 3. http://dx.doi.org/https://doi.org/10.2807/1560 7917.ES.2016.21.3.30111
Дополнительные материалы отсутствуют.