

Генетика, 2019, T. 55, № 8, стр. 863-868
Молекулярные механизмы канцерогенеза, ассоциированного с мутацией в гене MEN1
Д. В. Голиусова 1, *, Н. В. Клементьева 2, **, Н. Г. Мокрышева 2, С. Л. Киселев 1, 3
1 Институт общей генетики им. Н.И. Вавилова Российской академии наук
119991 Москва, Россия
2 Национальный медицинский исследовательский центр эндокринологии
117036 Москва, Россия
3 Медико-генетический научный центр
115522 Москва, Россия
* E-mail: daria.goliusova@mail.ru
** E-mail: nvklementieva@gmail.com
Поступила в редакцию 06.02.2019
После доработки 20.02.2019
Принята к публикации 13.03.2019
Аннотация
Несмотря на последние достижения в области геномики и обнаружение целого ряда генов, вовлеченных в процесс канцерогенеза, в большинстве случаев механизмы опухолевой трансформации той или иной ткани остаются малоизученными. В обзоре представлена характеристика гена MEN1 и его белкового продукта менина, вовлеченных в развитие множественной эндокринной неоплазии 1-го типа (МЭН1), или синдрома Вермера. Рассматриваются предполагаемые молекулярно-генетические механизмы канцерогенеза, ассоциированного с мутацией в гене MEN1, а также существующие и перспективные in vivo и in vitro модели для изучения молекулярных основ патологии. Отсутствие однозначных данных о роли гена MEN1 в инициации и прогрессии опухолевого фенотипа при синдроме МЭН1 позволяет предложить две модели возникновения опухолевой трансформации эндокринных тканей.
Диагностика, профилактика и лечение злокачественных новообразований во многом зависят от понимания молекулярных механизмов развития патологии. В большинстве случаев формированию опухоли на клеточном уровне предшествует возникновение спонтанных или унаследованных мутаций различных генов (генов-супрессоров опухоли, онкогенов и др.). Однако не всегда удается установить четкую корреляцию между наличием определенной мутации и возникновением клона трансформированной клетки, поскольку образование опухоли представляет собой сложный многоэтапный процесс, сопровождающийся комплексным взаимозависимым нарушением структуры генетического и эпигенетического материала клетки. В связи с этим детальное изучение молекулярных основ взаимодействия генов и их продуктов, а также исследование эффектов данного взаимодействия на клеточном и тканевом уровнях необходимо для понимания патогенеза опухолевой прогрессии. Такие подходы в области изучения онкологических заболеваний позволят сформировать системный подход к выявлению опухолей на ранней стадии, а также их профилактике.
Крайне интересным с точки зрения молекулярных механизмов канцерогенеза является синдром Вермера, или множественная эндокринная неоплазия 1-го типа (МЭН1), обусловленная наличием мутаций в гене MEN1 [1]. МЭН1 представляет собой наследственный синдром с высокой пенетрантностью, характеризующийся образованием опухолей в эндокринных железах и других органах. С наибольшей частотой заболевание обнаруживаются в возрасте 20–40 лет, а к 50 годам риск возникновения опухолей достигает почти 100% [2]. Распространенность заболевания колеблется в пределах 1 : 10 000–1 : 30 000 индивидуумов с равным соотношением полов [3]. В большинстве случаев пациенты с МЭН1 предрасположены к развитию опухолей в двух и более эндокринных органах, таких как паращитовидные железы, гипофиз, островки Лангерганса. Реже наблюдается развитие опухолей надпочечников, двенадцатиперстной кишки, желудка, тимуса, бронхов, легких, а также липом, лейомиом, менингиом, коллагеном и рака молочной железы [4, 5]. Наиболее распространенной эндокринопатией при МЭН1 является первичный гиперпаратиреоз [6]. МЭН1 имеет аутосомно-доминантное наследование, соответственно риск развития патологии у потомства составляет 50%. Как правило, у пациентов с МЭН1 наблюдается семейная форма заболевания, а в 8–14% случаев диагностируются de novo мутации, возникающие спонтанно на протяжении всей жизни и увеличивающие риск возникновения спорадических опухолей [7, 8]. Описано более 1300 наследуемых и 200 соматических мутаций гена MEN1 [9]. В целом мутации MEN1 могут быть идентифицированы у 70–95% всех пациентов с синдромом Вермера [10].
ГЕН MEN1: ЭКСПРЕССИЯ И ФУНКЦИЯ БЕЛКОВОГО ПРОДУКТА
Ген множественной эндокринной неоплазии 1-го типа – MEN1 – был клонирован в 1997 г. [1, 8]. MEN1 расположен на длинном плече 11-й хромосомы (11q13), состоит из десяти экзонов и кодирует небольшой белок менин (60 кДа), состоящий из 610 аминокислот. Менин является высококонсервативным белком среди различных организмов от нематод до людей, с высоким процентом гомологичности между человеком и мышью (98%). Анализ аминокислотной последовательности не выявил очевидного молекулярного сходства менина с известными консенсусными мотивами, но было обнаружено 28 предполагаемых сайтов фосфорилирования, два из которых подвержены миссенс-мутациям, вызывающим синдром МЭН1 [8]. Поскольку изучение аминокислотной последовательности менина оказалось не очень информативным, основные сведения о функции белка получены из исследований in vitro.
Менин имеет преимущественно ядерную локализацию, но обнаруживается также в цитоплазме и клеточной мембране [11, 12]. Экспрессия белка наблюдается как в эндокринных, так и неэндокринных тканях, включая кору головного мозга, почки, гипофиз, яички, тимус и др. [13]. Менин вовлечен во множество молекулярных взаимодействий в клетке, он участвует в различных сигнальных путях, может оказывать влияние на активность транскрипционных факторов, участвовать в поддержании стабильности генома, ингибировать пролиферацию клеток, а также регулировать экспрессию генов через эпигенетические механизмы, такие как метилирование или ацетилирование гистонов [14]. В настоящее время предполагается, что менин связывает ген-специфические факторы транскрипции с модификаторами хроматина в специфичном для клетки контексте [15]. Было показано, что менин может взаимодействовать с несколькими классами транскрипционных факторов, такими как JunD, Smad, Foxa2, Myc, β-катенин и др. Кроме того, менин способен связывать транскрипционные факторы с гистон-модифицирующими белковыми комплексами из группы Trithorax, регулируя уровень экпрессии генов. Так, в составе MLL1- и MLL2-содержащих комплексов с активностью H3K4me3 метилтрансферазы или HDAC-, PRMT5-, SUV39H1-содержащих комплексов с деацетилирующей активностью менин может активировать или подавлять транскрипцию генов-мишеней [16]. Первыми описанными генами-мишенями менина были гены, кодирующие ингибиторы циклин-зависимых киназ, CDKN2C и CDKN1B. Эти белки являются отрицательными регуляторами клеточного цикла, поэтому менин-опосредованная активация их экспрессии отражает роль менина как супрессора опухоли [17]. Позже было показано, что действие менина в качестве опухолевого супрессора может быть тканеспецифичным. Так, было обнаружено, что мишенью менина также является ген HLXB9. Его продукт Hlxb9 – транскрипционный фактор, который участвует в дифференцировке бета-клеток поджелудочной железы [18]. На мышиных моделях инсулиномы были исследованы эффекты оверэкспрессии и нокдауна менина на регуляцию работы Hlxb9, демонстрирующие менин-опосредованную регуляцию пролиферации бета-клеток и выработку инсулина в инсулиномах [19]. Ряд других исследований подтверждает, что экспрессия различных транскрипционных факторов поджелудочной железы, в том числе Hlxb9, Foxa2, MafA, MafB и Pdx1, является MEN1-зависимой [20–22].
ВОЗМОЖНЫЕ МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ МЕХАНИЗМЫ РАЗВИТИЯ МЭН1
В связи с наличием множества разнообразных белков-партнеров менина функция этого белка в онкогенезе остается неясной. На сегодняшний день большинство данных о возможных генетических механизмах развития патологии получено на мышиных моделях [23]. Показано, что частота развития опухолей эндокринных желез у мышей, гетерозиготных по гену MEN1, значительно выше по сравнению с животными дикого типа [24]. У мышей с генотипом Men1+/Men1– развиваются эндокринные опухоли аналогично тем, что наблюдаются у пациентов с синдромом Вермера [24, 25]. Было показано, что нокаут обоих аллелей гена MEN1 во время эмбрионального развития мыши является летальным и приводит к множественным дефектам развития нервной трубки, сердца и печени [26], однако это не свидетельствует о процессах неоплазии. При этом нокаут обоих аллелей в бета-клетках поджелудочной железы или в клетках паращитовидной железы приводит к развитию инсулиномы или аденомы паращитовидной железы соответственно. Тем не менее нокаут MEN1 в гепатоцитах не дает подобного эффекта, что подчеркивает тканеспецифичность экспрессии MEN1 при неопластических процессах.
Интересен тот факт, что в большинстве семейных форм злокачественных новообразований при синдроме Вермера, а также в спорадических опухолях с одним мутантным аллелем MEN1 достоверно обнаруживается потеря гетерозиготности на хромосоме 11q13 [27]. Так, исследования на гетерозиготных линиях мышей с нокаутом гена MEN1 выявили потерю аллеля дикого типа в опухолях животных [26]. Эти данные находят отражение в гипотезе “двойного удара” Альфреда Д. Кнудсона, когда выключение обеих копий гена-супрессора (биаллельная инактивации) приводит к образованию опухоли [28]. Согласно этой теории, мутантный аллель гена MEN1 наследуется от пораженного родителя или возникает de novo на эмбриональном уровне, либо в соматической клетке в случае спорадических опухолей (первый удар). Затем происходит потеря второго аллеля дикого типа уже в соматической клетке (второй удар). В результате, с получением гомозиготного рецессивного состояния в определенной ткани запускается процесс опухолеобразования. С таким подходом логично соотносится высокая частота встречаемости наследственных раков при синдроме Вермера и низкая – спорадических, так как возникновение сразу двух мутаций в одной и той же соматической клетке мало вероятно.
Несмотря на то, что потеря гетерозиготности на хромосоме 11q13 типична для опухолей при синдроме МЭН1 и согласуется с теорией Кнудсона, необходимо учитывать особенности локализации этих опухолей. На сегодняшний день сложно объяснить эндокринную избирательность онкогенеза у пациентов с МЭН1 в условиях убиквитарной экспрессии менина. Кроме того, даже среди эндокринных опухолей наблюдаются сильные различия в генетической природе онкогенеза. Например, для спорадических форм аденомы гипофиза частота обнаружения потери гетерозиготности на хромосоме 11q13 достоверно ниже, чем в случае спорадических опухолей паращитовидной или поджелудочной железы, как было показано в работе Tanaka и коллег. При этом данные молекулярно-генетического исследования исключили наличие точечных мутаций или делеций в гене MEN1, что не укладывалось в теорию “двойного удара” [29]. По всей видимости, различные генетические и/или эпигенетические механизмы (например, трисомия 11-й хромосомы или аберрантная экспрессия других генов, помимо MEN1) могут быть задействованы в развитии эндокринных опухолей у пациентов с синдромом Вермера. Данные in silico анализа также демонстрируют альтернативный путь канцерогенеза, ассоциированного с мутацией в гене MEN1. Так, было предсказано и экспериментально подтверждено, что микроРНК miR-24-1 способна связываться с 3'-нетранслируемой областью мРНК гена MEN1. Обнаруженная отрицательная обратная связь между менином и микроРНК, посттранскрипционно регулирующей его активность, может имитировать “второй удар”, скрывая реальные эффекты воздействия различных внешних или внутренних сигналов (например, гормонального фона или факторов окружающей среды) [30].
Несмотря на большое количество исследований, нет данных, подтверждающих прямую корреляцию между генотипом и фенотипом у пациентов с синдромом Вермера [31]. По всей видимости, эпигенетическая регуляция работы гена MEN1 приводит к тканеспецифичной функции менина и его двойственной роли в формировании опухолевого фенотипа. Так, например, при эстроген-зависимых спорадических формах рака молочной железы менин контролирует пролиферацию именно неопластических клеток, хотя в предшественниках нормальных протоковых клеток менин играет роль супрессора опухолей [32].
Хотя “второй удар” может быть стабильно детектирован в опухоли, зачастую трудно определить, что инициировало процесс трансформации соматической клетки. Стохастические факторы, вероятно, влияют на частоту и время возникновения опухолей у пациентов с МЭН1. Разнообразие клинических проявлений мутантного фенотипа как у членов одной и той же семьи, так и между пациентами с одинаковыми мутациями MEN1 [33] заставляет предположить, что в развитие опухолей вносят свой вклад другие генетические и эпигенетические механизмы, не связанные напрямую с геном MEN1. Таким образом, конечный триггер МЭН1-ассоциированного канцерогенеза остается до конца не изученным.
МОДЕЛИ ДЛЯ ИЗУЧЕНИЯ МОЛЕКУЛЯРНЫХ ОСНОВ ПАТОЛОГИИ
Среди исследователей популярны модели мышей с нокаутом гена MEN1. Наиболее распространенными опухолями у таких мутантных мышей являются пролактинома, инсулинома и аденома околощитовидной железы, обычно наблюдаемые и у пациентов с синдромом Вермера. Несмотря на значительную степень сходства, модели онкогенеза у мышей с МЭН1 не могут полностью воспроизвести ни молекулярно-генетическую, ни клиническую картину, обнаруживаемую у человека. Так, линии мышей с дефектным геном MEN1 были получены путем удаления целых экзонов данного гена, тогда как у пациентов большинство мутаций MEN1 являются точечными [34]. Кроме того, частота различных типов опухолей варьирует: у человека первым и наиболее частым проявлением злокачественной трансформации является первичный гиперпаратиреоз, связанный с новообразованиями в паращитовидной железе, в то время как у мышей – это гиперплазия и опухоли поджелудочной железы [35]. В отличие от пациентов с МЭН1, у мышей с данным заболеванием не развиваются нефункциональные панкреатические нейроэндокринные опухоли [16]. Тем не менее in vivo модели мышей, нокаутных по гену MEN1, используются для поиска и тестирования потенциальных лекарственных препаратов, замедляющих прогрессию МЭН1-ассоциированных опухолей [36].
В качестве другой, более доступной модели выступают опухолевые клеточные линии нейроэндокринного происхождения. Широко используются линии клеток, полученные от пациентов с опухолями поджелудочной железы, такие как BON-1 и QGP-1. Недостатками таких моделей является то, что клетки либо не содержат мутации гена MEN1, либо в исследованиях проводится сравнение гетерогенного материала, полученного от различных пациентов, поэтому неясен вклад индивидуального генома и/или эпигенетической компоненты [37]. Большинство работ, направленных на изучение функции менина и поиск белков-партнеров в условиях in vitro, выполняется на клеточных линиях неэндокринного происхождения, не содержащих мутации гена MEN1, таких как HEK293T или HeLa [38]. При этом убиквитарный характер экспрессии менина не способствует пониманию тканевой избирательности возникновения опухолей у пациентов с МЭН1.
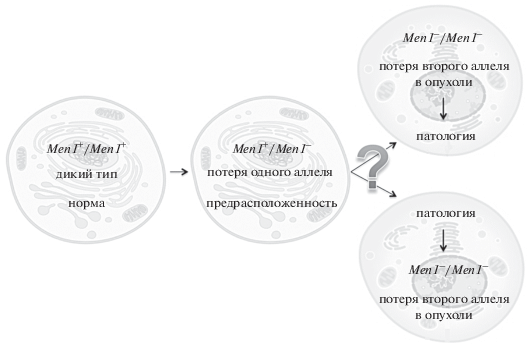
Таким образом, на сегодняшний день нет четких доказательств того, что первично – утрата второго аллеля гена MEN1 и возникновение опухоли по предложенной схеме “двойного удара” именно в эндокринных тканях либо возникновение трансформированного клона, сопряженное с особенностями генетического и эпигенетического состояния ткани, и затем “второй удар” (потеря второго аллеля MEN1), фиксирующий это состояние (рис. 1). Генетические, эпигенетические и экологические факторы, участвующие в проявлении фенотипа синдрома Вермера, остаются неясными в значительной степени именно из-за отсутствия соответствующих клеточных модельных систем заболевания. Современные методы клеточной биологии, в частности, получение изогенных клеточных линий различной специализации, а также возможность целенаправленно инактивировать один или оба аллеля гена в тех или иных соматических клетках, предрасположенных к развитию патологии, позволяют более полно и глубоко изучить вопрос о роли MEN1 в развитии синдрома Вермера, а также разработать модельные системы для поиска средств терапии.
Рис. 1.
Две гипотезы канцерогенеза, ассоциированного с мутациями в гене MEN1. Слева направо: первая мутация в гене MEN1 возникает de novo или наследуется, что означает наличие генетической предрасположенности к развитию синдрома множественной эндокринной неоплазии 1-го типа. Далее показано два потенциальных варианта развития событий – потеря второй, нормальной, копии гена и запуск неопластического процесса либо первичная злокачественная трансформация клетки с последующей инактивацией второго аллеля. Вопрос о причинно-следственных связях в канцерогенезе при синдроме МЭН1 до сих пор остается открытым.
Работа выполнена при финансовой поддержке РФФИ в рамках научного проекта № 19-015-00209-A и в рамках темы государственного задания № 0112-2019-0002.
Настоящая статья не содержит каких-либо исследований с использованием в качестве объектов животных.
Настоящая статья не содержит каких-либо исследований с участием в качестве объектов людей.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Chandrasekharappa S.C., Guru S.C., Manickam P. et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1 // Science. 1997. № 5311 (276). P. 404–407. https://doi.org/10.1126/science.276.5311.404
Dreijerink K.M.A., Beek A.P. van, Lentjes E.G.W.M. et al. Acromegaly in a multiple endocrine neoplasia type 1 (MEN1) family with low penetrance of the disease // Europ. J. Endocrinol. 2005. № 6 (153). P. 741–746. https://doi.org/10.1530/eje.1.02022
White M.L., Doherty G.M. Multiple endocrine neoplasia // Surgical Oncol. Clinics North Amer. 2008. № 2 (17). P. 439–459. https://doi.org/10.1016/j.soc.2007.12.002
Brandi M.L., Gagel R.F., Angeli A. et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2 // J. Clinical Endocrinol. Metabolism. 2001. № 12 (86). P. 5658–5671. https://doi.org/10.1210/jcem.86.12.8070
Asgharian B., Chen Y.-J., Patronas N.J. et al. Meningiomas may be a component tumor of multiple endocrine neoplasia type 1 // Clinical Cancer Res.: An Official J. Amer. Assoc. Cancer Res. 2004. № 3 (10). P. 869–880. https://doi.org/10.1158/1078-0432.CCR-0938-3
Thakker R.V., Newey P.J., Walls G.V. et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1) // J. Clinical Endocrinol. Metabolism. 2012. № 9 (97). P. 2990–3011. https://doi.org/10.1210/jc.2012-1230
Larsson C., Weber G., Kvanta E. et al. Isolation and mapping of polymorphic cosmid clones used for sublocalization of the multiple endocrine neoplasia type 1 (MEN1) locus // Human Genet. 1992. № 2 (89). P. 187–193.
Guo S.S., Sawicki M.P. Molecular and genetic mechanisms of tumorigenesis in multiple endocrine neoplasia type-1 // Mol. Endocrinol. 2001. № 10 (15). P. 1653–1664. https://doi.org/10.1210/mend.15.10.0717
Lemos M.C., Thakker R.V. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene // Human Mutat. 2008. № 1 (29). P. 22–32. https://doi.org/10.1002/humu.20605
Marini F., Falchetti A., Luzi E. et al. Multiple Endocrine Neoplasia Type 1 (MEN1) Syndrome // Cancer Syndromes / Eds Riegert-Johnson D.L., Boardman L.A., Hefferon T. et al. Bethesda (MD): National Center Biotechnol. Inform. (US), 2008.
Ikeo Y., Sakurai A., Suzuki R. et al. Proliferation-associated expression of the MEN1 gene as revealed by in situ hybridization: possible role of the menin as a negative regulator of cell proliferation under DNA damage // Laboratory Investigat. 2000. № 6 (80). P. 797–804.
Stewart C., Parente F., Piehl F. et al. Characterization of the mouse Men1 gene and its expression during development // Oncogene. 1998. № 19 (17). P. 2485–2493. https://doi.org/10.1038/sj.onc.1202164
Lemmens I., Merregaert J., Van de Ven W.J. et al. Construction of a 1.2-Mb sequence-ready contig of chromosome 11q13 encompassing the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1 // Genomics. 1997. № 1 (44). P. 94–100.
Lecoq A.-L., Kamenický P., Guiochon-Mantel A. et al. Genetic mutations in sporadic pituitary adenomas–what to screen for? // Nature Rev. Endocrinology. 2015. № 1 (11). P. 43–54. https://doi.org/10.1038/nrendo.2014.181
Matkar S., Thiel A., Hua X. Menin: a scaffold protein that controls gene expression and cell signaling // Trends in Biochem. Sci. 2013. № 8 (38). P. 394–402. https://doi.org/10.1016/j.tibs.2013.05.005
Dreijerink K.M.A., Timmers H.T.M., Brown M. Twenty years of menin: Emerging opportunities for restoration of transcriptional regulation in MEN1 // Endocrine-related Cancer. 2017. № 10 (24). P. T135–T145. https://doi.org/10.1530/ERC-17-0281
Milne T.A., Hughes C.M., Lloyd R. et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors // Proc. Natl Acad. Sci. USA. 2005. № 3 (102). P. 749–754. https://doi.org/10.1073/pnas.0408836102
Scacheri P.C., Davis S., Odom D.T. et al. Genome-wide analysis of menin binding provides insights into MEN1 tumorigenesis // PLoS Genet. 2006. № 4 (2). P. e51. https://doi.org/10.1371/journal.pgen.0020051
Shi K., Parekh V.I., Roy S. et al. The embryonic transcription factor Hlxb9 is a menin interacting partner that controls pancreatic β-cell proliferation and the expression of insulin regulators // Endocrine-related Cancer. 2013. № 1 (20). P. 111–122. https://doi.org/10.1530/erc-12-0077
Zhang H.-L., Li W.-Y., Zhang C.-P. et al. Differentially expressed genes in Men1 knockout and wild‑type embryoid bodies for pancreatic islet development // Mol. Med. Rep. 2011. № 2 (4). P. 301–305. https://doi.org/10.3892/mmr.2011.409
Lu J., Hamze Z., Bonnavion R. et al. Reexpression of oncoprotein MafB in proliferative β-cells and Men1 insulinomas in mouse // Oncogene. 2011. № 31 (31). P. 3647–3654.https://doi.org/10.1038/onc.2011.538
Fontanière S., Tost J., Wierinckx A. et al. Gene expression profiling in insulinomas of Men1 beta-cell mutant mice reveals early genetic and epigenetic events involved in pancreatic beta-cell tumorigenesis // Endocrine-related Cancer. 2006. № 4 (13). P. 1223–1236. https://doi.org/10.1677/erc.1.01294
Agarwal S.K. The future: genetics advances in MEN1 therapeutic approaches and management strategies // Endocrine-related Cancer. 2017. № 10 (24). P. T119–T134. https://doi.org/10.1530/ERC-17-0199
Bertolino P., Tong W.-M., Galendo D. et al. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1 // Mol. Endocrinol. 2003. № 9 (17). P. 1880–1892. https://doi.org/10.1210/me.2003-0154
Crabtree J.S., Scacheri P.C., Ward J.M. et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors // Proc. Natl Acad. Sci. USA. 2001. № 3 (98). P. 1118–1123. https://doi.org/10.1073/pnas.98.3.1118
Harding B., Lemos M.C., Reed A.A.C. et al. Multiple endocrine neoplasia type 1 knockout mice develop parathyroid, pancreatic, pituitary and adrenal tumours with hypercalcaemia, hypophosphataemia and hypercorticosteronaemia // Endocrine-related Cancer. 2009. № 4 (16). P. 1313–1327. https://doi.org/10.1677/ERC-09-0082
Marx S.J. Correction: Molecular genetics of multiple endocrine neoplasia types 1 and 2 // Nature Rev. Cancer. 2005. № 8 (5). P. 663–663. https://doi.org/10.1038/nrc1690
Marx S., Spiegel A.M., Skarulis M.C. et al. Multiple endocrine neoplasia type 1: clinical and genetic topics // Ann. Internal Med. 1998. № 6 (129). P. 484–494.
Tanaka C., Kimura T., Yang P. et al. Analysis of loss of heterozygosity on chromosome 11 and infrequent inactivation of the MEN1 gene in sporadic pituitary adenomas // J. Clinical Endocrinol. Metab. 1998. № 8 (83). P. 2631–2634. https://doi.org/10.1210/jcem.83.8.4888
Luzi E., Marini F., Giusti F. et al. The negative feedback-loop between the oncomir Mir-24-1 and menin modulates the Men1 tumorigenesis by mimicking the “Knudson’s second hit” // PLoS One. 2012. № 6 (7). P. e39767. https://doi.org/10.1371/journal.pone.0039767
Christakis I., Qiu W., Hyde S.M. et al. Genotype-phenotype pancreatic neuroendocrine tumor relationship in multiple endocrine neoplasia type 1 patients: A 23-year experience at a single institution // Surgery. 2018. № 163(1). P. 212–217. https://doi.org/10.1016/j.surg.2017.04.044
Dreijerink K.M.A., Groner A.C., Vos E.S.M. et al. Enhancer-mediated oncogenic function of the menin tumor suppressor in breast cancer // Cell Rep. 2017. № 10 (18). P. 2359–2372. https://doi.org/10.1016/j.celrep.2017.02.025
Falchetti A. Genetics of multiple endocrine neoplasia type 1 syndrome: what’s new and what’s old // F1000Res. 2017. № 6. P. 73. https://doi.org/10.12688/f1000research.7230.1
Mohr H., Pellegata N.S. Animal models of MEN1 // Endocrine-related Cancer. 2017. № 10 (24). P. T161–T177. https://doi.org/10.1530/ERC-17-0249
Wiedemann T., Pellegata N.S. Animal models of multiple endocrine neoplasia // Mol. Cell. Endocrinol. 2016. (421). P. 49–59. https://doi.org/10.1016/j.mce.2015.07.004
Lopez C.L., Joos B., Bartsch D.K. et al. Chemoprevention with Somatuline© Delays the Progression of Pancreatic Neuroendocrine Neoplasms in a Mouse Model of Multiple Endocrine Neoplasia Type 1 (MEN1) // World J. Surgery. 2019. № 3 (43). P. 831–838. https://doi.org/10.1007/s00268-018-4839-8
Boora G.K., Kanwar R., Kulkarni A.A. et al. Exome-level comparison of primary well-differentiated neuroendocrine tumors and their cell lines // Cancer Genetics. 2015. № 7–8 (208). P. 374–381. https://doi.org/10.1016/j.cancergen.2015.04.002
Pieterman C.R.C., Conemans E.B., Dreijerink K.M.A. et al. Thoracic and duodenopancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1: natural history and function of menin in tumorigenesis // Endocrine-related Cancer. 2014. № 3 (21). P. R121–R142. https://doi.org/10.1530/ERC-13-0482
Дополнительные материалы отсутствуют.