

Генетика, 2020, T. 56, № 2, стр. 127-141
Молекулярные механизмы атаксии первого типа
Е. А. Воловиков 1, А. В. Давиденко 1, М. А. Лагарькова 1, *
1 Федеральный научно-клинический центр физико-химической медицины
Федерального медико-биологического агентства
119435 Москва, Россия
* E-mail: lagar@rcpcm.org
Поступила в редакцию 12.02.2019
После доработки 30.03.2019
Принята к публикации 15.04.2019
Аннотация
Полиглутаминовые (polyQ) заболевания – это тяжелые формы наследственных нейродегенеративных патологий, обусловленные экспансией кодирующих CAG-повторов в определенных генах, что сопровождается патологическим удлинением полиглутаминовых остатков в составе мутантных белков, нарушением белок-белковых взаимодействий и формированием нерастворимых амилоидоподобных включений в нейронах. Терапия этих заболеваний применяется только симптоматическая и она не может повлиять на динамику патологического процесса. Известны девять polyQ заболеваний – спиноцеребеллярные атаксии 1, 2, 3, 6, 7 и 17 типов, спинобульбарная мышечная атрофия, дентаторубро-паллидолюисова атрофия и хорея (болезнь) Гентингтона. Точные механизмы увеличения длины тринуклеотидных повторов сегодня не ясны, по-видимому, экспансия может происходить во время таких процессов как репликация, транскрипция, репарация. Существуют также несколько молекулярных путей, по которым увеличение длины полиглутаминового тракта может приводить к нейродегенерации. Практически полная пенетрантность мутантных аллелей, тяжелая инвалидизация, наступающая спустя 10–15 лет от момента появления симптомов, отсутствие эффективной терапии – все это позволяет рассматривать polyQ заболевания в качестве очень важного объекта исследований, который активно изучается во всем мире. Этот обзор посвящен существующим моделям и поиску молекулярных механизмов одного из polyQ заболеваний – спиноцеребеллярной атаксии первого типа (SCA1), обусловленной экспансией CAG-повторов в гене ATXN1.
Полиглутаминовые (polyQ) заболевания – это группа наследственных нейродегенеративных патологий, возникающих в результате экспансии тринуклеотидных повторов CAG в кодирующей части определенных генов. К ним относятся спиноцеребеллярные атаксии (SCA) 1, 2, 3, 6, 7 и 17 типов, хорея Гентингтона, дентаторубро-паллидолюисова атрофия (DRPLA) и спинобульбарная мышечная атрофия (SBMA). Встречаются эти заболевания с частотой от 1 до 10 на 100 000 в зависимости от конкретной популяции, но, к сожалению, пока неизлечимы [1].
Первым описанным заболеванием, связанным с удлинением полиглутаминового участка, была SBMA. Этим заболеванием страдают практически исключительно мужчины; экспансия триплетов CAG происходит в гене андрогенового рецептора AR. При ней наблюдаются судороги, слабость и атрофия мышц. Нейродегенерации подвержены в первую очередь передние рога спинного мозга и моторные ядра ствола мозга. Присутствуют также нарушения, связанные с нечувствительностью к андрогенам: снижение фертильности, атрофия семенников и т.п. [2, 3].
Болезнь (хорея) Гентингтона – одно из наиболее изученных полиглутаминовых заболеваний. Наиболее характерным симптомом этой болезни является хорея (характерные “танцующие” движения), нередки деменция и различные психические расстройства (раздражительность, депрессия), могут присутствовать расстройства речи, дисфагия и другие нарушения. Патологические изменения затрагивают главным образом полосатое тело, также отмечаются повреждения коры больших полушарий [4].
Спиноцеребеллярные атаксии характеризуются большим разнообразием клинических проявлений. Как следует из названия, общим признаком для них является развитие атаксии (нарушения согласованности движений мышц) вследствие поражения мозжечка. Это же явление характерно и для DRPLA, поэтому эти болезни часто объединяют в одну группу. В целом для SCA типично наличие различных двигательных нарушений, но отсутствие выраженной деменции и психиатрических симптомов [5]. Исключение составляет крайне редкая SCA17, при которой деменция является вторым по частоте встречаемости симптомом после атаксии; также при SCA17 могут развиваться психические расстройства и хорея, из-за чего по клиническим проявлениям это заболевание больше похоже на болезнь Гентингтона, чем на другие спиноцеребеллярные атаксии [5, 6]. У прочих SCA также есть свои характерные признаки: для SCA2 это замедление фиксирующих движений глаз, при SCA7 наблюдается дегенерация сетчатки и т.д., хотя для точной постановки диагноза всегда необходимо проводить генетические тесты [2, 7].
Несмотря на высокую гетерогенность как с клинической, так и с генетической точки зрения, все эти заболевания имеют несколько общих особенностей. Они наследуются как аутосомно-доминантные, за исключением SBMA, наследование которой сцеплено с полом (ген AR находится на X-хромосоме). Первые симптомы болезни, как правило, проявляются в зрелом возрасте (примерно около 40 лет), хотя встречаются случаи, когда болезнь впервые проявляется в пожилом возрасте или у детей. Возраст на момент начала заболевания зависит от количества повторов CAG в мутантном гене: чем их больше, тем раньше начинаются патологические изменения. Отмечается также корреляция между числом повторов и тяжестью протекания заболевания: чем длиннее полиглутаминовый участок, тем сильнее выражены симптомы [2, 8].
Другая особенность полиглутаминовых заболеваний – тенденция к спонтанному изменению длины участка с повторами в ряду поколений, обычно в сторону увеличения. Это приводит к тому, что у детей болезнь зачастую принимает более тяжелую форму, чем у их родителей. Наличие неоднородностей в участке гена, кодирующем полиглутаминовый тракт, таких как триплеты САТ в гене ATXN1, связанном со SCA1, или CAA в гене TBP, связанном со SCA17, повышает его стабильность [2, 6].
Также характерной чертой всех полиглутаминовых заболеваний является формирование внутриклеточных агрегатов, содержащих мутантный белок [2].
Существующая терапия полиглутаминовых заболеваний направлена на замедление их прогрессирования и облегчение симптомов. В настоящее время ведется активная разработка новых методов лечения, таких как заместительная клеточная терапия и различные варианты генной терапии.
Этот обзор посвящен генетическим особенностям и молекулярным механизмам самой распространенной в Российской Федерации атаксии – атаксии первого типа, животным и клеточным моделям для исследования ее патогенеза.
ГЕНЕТИЧЕСКАЯ ПРИЧИНА И ОСНОВНЫЕ КЛИНИЧЕСКИЕ СИМПТОМЫ АТАКСИИ ПЕРВОГО ТИПА
Спиноцеребеллярная атаксия первого типа (SCA1, или СЦА1) впервые была описана в 1950 г. [9]. На нее приходится около 6% всех случаев аутосомно-доминантных церебеллярных атаксий в мире [2], это самый распространенный тип SCA в России [10]. Высокая распространенность заболевания (38.6 на 100 тыс. якутов по сравнению 1–2 : 100 тыс. в мировом населении) в Якутии была оценена как “сибирский очаг” накопления заболевания, крупнейший в мире. Причина SCA-1 – экспансия повторов CAG в гене Ataxin1 (ATXN1). Этот ген расположен в локусе 6p22.3 и содержит девять экзонов [11]. В норме число повторов CAG в участке, кодирующем полиглутаминовую последовательность, составляет от 6 до 44 триплетов; при этом если их более 20, то между ними имеются кодирующие гистидин триплеты CAT. Болезнь развивается, если число триплетов CAG превышает 39 и между ними нет вставок САТ [2, 12].
В ряду поколений наблюдается нестабильность повтора, особенно ярко выраженная при наследовании патогенного аллеля по отцовской линии. По примерным оценкам, изменение длины полиглутаминового тракта при передаче патогенного аллеля по отцовской линии происходит в 82% случаев, а по материнской – в 60% случаев [13]. При этом в среднем при наследовании по отцовской линии за одно поколение длина участка с повторами увеличивается на 1.75 триплета, по материнской же уменьшается на 0.5 триплета [13]. Точные механизмы экспансии пока не ясны; предполагается, что важную роль в этом процессе играет способность одноцепочечных участков ДНК, состоящих из повторяющихся звеньев CAG или CTG, к формированию шпилек в процессах, связанных с расплетанием двойной спирали, – репликации, транскрипции и репарации. Так, например, при репликации образование шпильки на одной из цепей может приводить к “проскальзыванию” ДНК-полимеразы, в результате чего синтезируемая цепь оказывается длиннее или короче матричной [13]. Большое значение в этом аспекте может иметь наличие или отсутствие в участке с повторами триплетов САТ: так, в работе Pearson et al. [14] было продемонстрировано, что наличие таких вставок влияет на вторичную структуру ДНК [14].
При SCA1 обнаруживается также соматический мозаицизм, выражающийся в неравной длине участка с повторами в клетках, относящихся к разным тканям, в частности в разных участках мозга. Однако прямой корреляции между длиной полиглутаминового тракта в определенном типе клеток и их подверженностью патологическим процессам при SCA1 выявлено не было [13].
Возможные клинические проявления SCA1 включают в себя достаточно широкий спектр симптомов. Наиболее характерными являются прогрессирующая церебеллярная атаксия, дизартрия и бульбарные нарушения, такие как атрофия лицевой и жевательной мускулатуры и дисфагия [12]. Могут также иметь место нистагм, офтальмоплегия, дистония и ряд других нарушений [12, 15]. Количество и степень выраженности симптомов увеличиваются с возрастом, кроме того, как и при других полиглутаминовых заболеваниях, имеется положительная корреляция между числом повторов CAG в мутантном гене и тяжестью течения болезни, а также обратная зависимость между числом повторов и возрастом, в котором были замечены первые признаки заболевания [12].
Как правило, болезнь начинает проявляться в 30–50 лет; средняя продолжительность жизни после появления первых признаков патологии составляет 10–20 лет [15]. Выделяется ювенильная форма SCA1, при которой первые признаки болезни начинаются до достижения пациентом тринадцатилетнего возраста. Такая форма заболевания характеризуется тяжелым течением, быстрым прогрессированием и приводит к ранней гибели [12].
Патологические изменения в тканях головного мозга затрагивают практически все его отделы [16]. Исследование с помощью МРТ и изучение постмортальных срезов мозга пациентов со SCA1 показали наличие атрофии мозжечка, мозжечковых ножек, ствола мозга, лобной, височной и теменной долей коры больших полушарий, III, IV и VI–XII пар черепных нервов [17, 18]. В ряде областей мозга, как затронутых (таламус, мозжечок, неокортекс), так и не затронутых нейродегенерацией (миндалина, гипоталамус), наблюдается астроглиоз (аномальное увеличение числа астроцитов из-за разрушения соседних нейронов) [18].
Согласно данным ЯМР-спектроскопии, концентрация N-ацетиласпартата (NAA), являющегося нейрональным маркером, в ткани мозжечка и моста пациентов со SCA1 была существенно ниже, чем у здоровых испытуемых, а концентрация миоинозитола (глиального маркера) – существенно выше [19, 20]. Снижение концентрации NAA может свидетельствовать о гибели или дисфункции нейронов, а повышение концентрации миоинозитола – о компенсаторном усилении пролиферации глии [20].
ЖИВОТНЫЕ МОДЕЛИ SCA1
Для изучения механизмов патогенеза SCA1 было создано достаточно большое количество животных моделей. Первые трансгенные линии мышей с мутантным ATXN1 опубликованы в 1995 г. [21]. Трансген включал в себя кДНК гена ATXN1 человека, содержащего 30 тринуклеотидных повторов со вставками САТ (PS-30) или 82 повтора без CAT (PS-82), под промотором гена Pcp2/L7 мыши и экспрессировался только в клетках Пуркинье. Фенотип мышей PS-30 не отличался от фенотипа мышей дикого типа, в то время как у мышей PS-82 наблюдались нарушения координации движений, усиливающиеся с возрастом. Кроме того, у мышей с PS-82 наблюдались патологические изменения в ткани мозжечка, включающие гибель клеток Пуркинье [21].
В 1998 г. была получена линия мышей с нокаутом гена ATXN1 путем делеции экзона 8 [22]. Признаков атаксии или каких-либо патологических изменений в структуре мозга у этих животных выявлено не было. Тем не менее имелись некоторые нарушения поведения (более слабо выраженное исследовательское поведение, трудности с ориентацией в пространстве), что свидетельствует о том, что, хотя потеря функциональности ATXN1 не приводит к SCA1, она может негативно влиять на способность к обучению и память [22].
В еще одной модельной линии мышей восьмой экзон гена ATXN1 был заменен на содержащий 154 триплета CAG [23]. У этих мышей наблюдались серьезные нарушения двигательной и когнитивных функций, для них были характерны кифоз, атрофия мышц задних конечностей и преждевременная гибель. Обнаруживались также характерные для SCA1 внутриядерные включения в клетках, расположенных в различных областях мозга, изменение морфологии клеток Пуркинье и нейродегенерация. Стоит отметить, что у полученных ранее аналогичным образом мышей с 78 повторами CAG подобных патологических изменений не наблюдалось, у них наблюдались лишь некоторые изменения в поведении, проявлявшиеся с возрастом [24].
Также были созданы линии мышей, у которых экспрессию мутантного белка можно было контролировать с помощью тетрациклин-индуцибельной системы. В одной из трансгенных линий трансген находился под контролем промотора Pcp2/L7 и содержал 82 повтора CAG. Его экспрессия блокировалась введением аналога тетрациклина доксициклина. Животные имели характерный патологический фенотип. На всех этапах развития болезни (6, 12 или 32 нед. от рождения) прекращение экспрессии трансгена вело к ослаблению признаков патологии; на ранней стадии заболевания неврологические и морфологические характеристики мышей, получавших доксициклин, возвращались к норме, а на поздней наблюдалось только их частичное улучшение [25]. Позднее появилась модельная линия мышей, у которой трансген находился под промотором гена PrP, благодаря чему мутантный белок мог экспрессироваться не только в клетках Пуркинье [26].
Существуют также различные модификации данных модельных систем [27]. Например, для исследования роли фосфорилирования 776-го остатка серина белка ATXN1 в развитии SCA1 были созданы модельные линии мышей, аналогичные описанным в статье Burright et al. [21], но с заменой S776 на аланин [28] или аспарагиновую кислоту [29].
Еще одним модельным объектом, который используется для исследования механизмов SCA1, является дрозофила. В опубликованной в 2000 г. работе Fernandez-Funez et al. [30] были получены трансгенные линии дрозофил, экспрессировавшие ATXN1 человека с 30 или 82 повторами CAG. Было установлено, что повышенная экспрессия ATXN1 с нормальной длиной полиглутаминового тракта ведет к такому же патологическому фенотипу (дегенерация сетчатки), как и экспрессия ATXN1 с 82 остатками глутамина [30]. Позднее аналогичные модельные системы были использованы и в других работах, где было показано наличие роли некоторых белков, к примеру PAK1 [31], а также систем репарации ДНК [32] в патогенезе SCA1.
КЛЕТОЧНЫЕ МОДЕЛИ SCA1
Для моделирования SCA1 широко используются культуры клеток человека и приматов. Большинство из них просты и дешевы в получении и удобны в культивировании. Эксперименты на клеточных культурах позволяют исключить влияние различных систем организма и внешней среды, оставив только вклад особенностей выбранной клеточной линии и стандартизированных условий клеточного инкубатора. Однако эти достоинства также являются и ограничениями метода.
В исследованиях SCA1 широко использовались иммортализованные клеточные линии HeLa, HEK293, COS-1, COS-7, которые являются производными различных типов клеток, что позволяет обобщить полученные на них результаты как повсеместный эффект мутации [33–36]. Для моделирования заболевания эти клетки трансформировали векторами, содержащими мутантный ATXN1. На этих моделях были получены данные о процессах формирования и свойствах телец включений ATXN1 и о взаимодействии мутантного ATXN1 и ядерной мембраны [33, 34]. Все вышеперечисленные виды клеток – трансформированные или раковые клеточные линии, и они накопили большое количество различных мутаций [37, 38]. Такая нестабильность генома не позволяет использовать эти модели для изучения многих регуляторных аспектов развития SCA1, в том числе исследовать проявления заболевания в клетках, являющихся основной мишенью атаксии – в нейронах.
ИНДУЦИРОВАННЫЕ ПЛЮРИПОТЕНТНЫЕ СТВОЛОВЫЕ КЛЕТКИ КАК ПЕРСПЕКТИВНЫЙ ПОДХОД К ИССЛЕДОВАНИЯМ НАСЛЕДСТВЕННЫХ НЕЙРОДЕГЕНЕРАТИВНЫХ ЗАБОЛЕВАНИЙ
Открытие технологии генетического репрограммирования соматических клеток человека до плюрипотентного состояния и разработка методов направленной дифференцировки таких индуцированных плюрипотентных стволовых клеток (ИПСК) в определенный тип клеток организма позволили получить более точные клеточные модели заболеваний человека [39]. Для изучения SCA1 особо информативными являются нейроны человека, дифференцированные из ИПСК, полученных из биопсии кожи или клеток крови пациентов – носителей заболевания. На данный момент не существует другой методики получения большого количества живых нейральных клеток пациента. В отличие от иммортализованных линий клеточные модели на основе ИПСК обладают достаточно стабильным геномом, дают возможность изучать патогенез заболевания как на зрелых нейронах, так и на нейрональных предшественниках. Еще одним большим преимуществом ИПСК является возможность направленного получения любого желаемого типа клеток, в том числе и глиальных клеток, что позволяет изучить также вклад глии и нейронально-глиальной коммуникации в развитие заболевания [40, 41]. Уже разработаны протоколы получения из ИПСК мозговых органоидов, что позволяет моделировать заболевания в 3D-культуре различных типов нейрональных и глиальных клеток [42, 43]. Такой подход дает уникальную возможность изучить развитие SCA1 на человеческих клетках с учетом сложности тканевой организации центральной нервной системы. На сегодняшний день существуют несколько таких моделей SCA1 и на них уже получены первые результаты, дополняющие исследования на классических мышиных моделях [44, 45]. Однако у моделей на основе ИПСК существует ряд недостатков. Среди них можно выделить ограничения, накладываемые поздним развитием большинства нейродегенеративных заболеваний, что вынуждает применять различные методы искусственного “состаривания” нейрональных культур. Также стоит отметить высокую стоимость и техническую сложность работы с моделями на основе ИПСК. Тем не менее представляется, что ИПСК – лучшая на сегодняшний день клеточная система для моделирования нейродегенеративных заболеваний, так как эти клетки позволяют изучать процесс развития патологии на разных типах дифференцированных клеток с нормальным кариотипом и при их ко-культивировании, видеть и изучать ранние проявления нейродегенерации. Возможность создавать на основе ИПСК изогенные системы методами геномного редактирования позволяет еще больше расширить их использование для моделирования заболеваний [45а].
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РАЗВИТИЯ ПАТОЛОГИИ
ATXN1 является консервативным геном с одной особенностью: у многих гомологов ATXN1 отсутствует CAG-участок [65]. В геноме человека тоже есть гомолог ATXN1 под названием ATXN1L/BOAT, выполняющий схожие функции, но не имеющий в своем составе CAG-участка [66]. Белок ATXN1 имеет две высокогомологичные изоформы, ATXN1 экспрессируется в большинстве клеток организма и способен осуществлять несколько типов взаимодействий: он обладает РНК-связывающей активностью, может осуществлять белок-белковые и самоассоциативные взаимодействия [67, 68]. В клетке ATXN1 локализуется и в цитоплазме, и в ядре [34, 69]. На посттранскрипционном уровне токсичный эффект экспансии CAG-повторов обусловливается различными механизмами: ослабление или потеря одной из функций белка ATXN1, приобретение новых активностей или усиление уже имеющихся. Так как ATXN1 является участником процессов регуляции экспрессии генов и транспорта РНК, то нарушения в его работе затрагивают функциональность многих молекулярных путей.
В ядре ATXN1 образует фракции массивных растворимых комплексов молекулярной массой около 1.8 МДа и 300 кДа. В этих комплексах с ATXN1 взаимодействует белок CIC, являющийся негативным регулятором транскрипции [70]. Гетеродимеры ATXN1–CIC напрямую регулируют экспрессию генов, связываясь с их промоторами [71]. Одной из функций ATXN1 является транспорт РНК из ядра в цитоплазму [69]. ATXN1 вовлечен в регуляцию биоэнергетики клетки через GSK3b-mTOR сигнальный путь [72]. Также ATXN1 взаимодействует с Notch, Sp1, DRD2, WNT, ANP32A, PQBP1, UBQLN4, ATXN1L, USP7, RBPJ [22, 73–77].
Несмотря на моногенность спиноцеребеллярной атаксии первого типа, экспансия тринуклеотидных повторов в гене ATXN1 вызывает большое количество различных нарушений молекулярных процессов в клетке (см. рис. 1). Еще большую вариативность наблюдаемых проявлений увеличивают дополнительные мутации в гене ATXN1, изменяющие пенетрантность заболевания. Показано также, что для проявления токсичных свойств мутантного белка ATXN1 имеющийся в последовательности гена сигнал ядерной локализации (NLS) должен быть функционален [78]. Еще одним важным звеном является аминокислота в положении 776, фосфорилирование которой вызывает заболевание у носителей нормального аллеля, а аминокислотная замена в этом положении нивелирует эффект мутантного аллеля [28]. Основные молекулярные проявления SCA-1 сведены в табл. 1. Стоит также упомянуть, что в данный момент, по аналогии с некоторыми другими полиглутаминовыми заболеваниями, обсуждается возможная токсичность РНК-продукта мутантного гена ATXN1. Но пока экспериментальные данные скорее опровергают эту теорию [45]. Также открытым остается вопрос о вкладе RAN-трансляции (Repeat-Assosiated Non ATG translation) в развитие SCA1. RAN-трансляция опосредована три- и шестинуклеотидными повторами и может начинаться как с неканонического сайта, так и с самого участка повторов, что зачастую приводит к образованию токсичных белковых продуктов. Данные, полученные на моделях других наследственных заболеваний, связанных с экспансией повторов, свидетельствуют о высокой вероятности наличия такой трансляции и при SCA1 [79, 80].
Рис. 1.
Схема молекулярных путей, вносящих вклад в развитие SCA1, в соответствии с данными литературы.
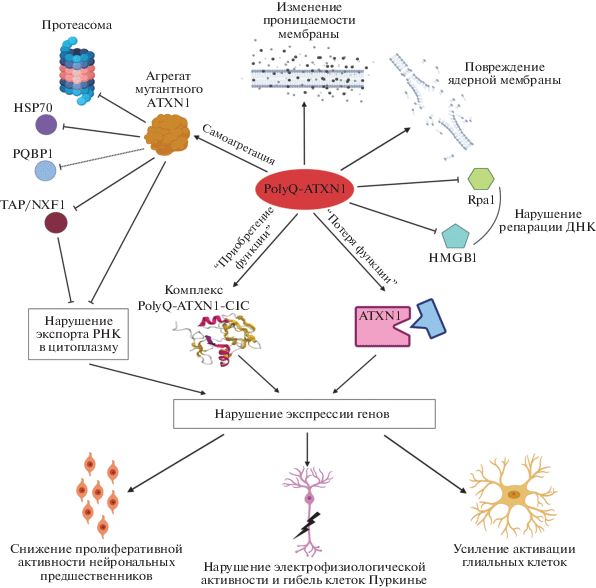
Таблица 1.
Проявление патологических признаков SCA1 в различных животных и клеточных моделях этого заболевания
| Проявление патологических признаков SCA1 | Тип генетической модели SCA1 | Тип клеток или орган | Метод | Ссылка |
|---|---|---|---|---|
| Изменение экспрессии генов, связанных с ответом на глутамат | Мышь B05[82Q] | Мозжечок | ДНК-микрочип | [46] |
| Снижение в 2 раза числа рецепторов mGlur1 | Мышь 154Q/2Q | Клетки Пуркинье | Количественный иммунохимический анализ | [47] |
| Нарушение электрофизиологической активности клеток Пуркинье у взрослых мышей | Мышь 82Q | Клетки Пуркинье | Локальная фиксация потенциала, кальциевая визуализация, измерение электрической активности на переживающих срезах мозжечка | [48, 49] |
| Изменение количества метаболитов в спиномозжечковой жидкости | Человек | Спиномозговая жидкость | Магнитно-резонансная спектроскопия высокого разрешения | [20] |
| Повышение концентрации глутамина в мозжечке | Мышь 82Q | Мозжечок | Магнитно-резонансная спектроскопия высокого разрешения | [50] |
| Снижение электрофизиологической активности клеток Пуркинье на пресимптоматической стадии | Мышь 82Q | Клетки Пуркинье | Электрофизиологические исследования in vivo | [51] |
| Снижение пролиферативной активности нейрональных предшественников головного мозга | Мышь 154Q/2Q | Нейрональные предшественники (NPC) | Оценка включения бромдизоксиуридина в ДНК | [52] |
| Нарушения глутаматэргического синапса между параллельными волокнами и клетками Пуркинье | Мышь 82Q | Клетки Пуркинье | Пэтч-кламп | [51] |
| Снижение количества транспортера глутамата GLAST в клетках Бергмановой глии | Мышь 82Q | Клетки Бергмановой глии | Вестерн-блот | [53] |
| Повышение числа повреждений ДНК | Мышь 154Q/2Q, дрозофила | Головной мозг мыши | Иммуноцитохимия, ПЦР в реальном времени | [32] |
| Ко-локализация белка Rpa1 с агрегатами ATXN1 | Мышь 154Q/2Q | Клетки Пуркинье | Иммуноцитохимия | [32] |
| Повреждение митохондриальной ДНК | Мышь 154Q/2Q | Нейроны | Иммуноцитохимия, ПЦР-анализ | [54, 55] |
| Нарушение ультраструктуры митохондрий | Мышь [154Q/2Q;HMGB1] |
Нейроны | [54] | |
| Формирование комплексов мутантного ATXN1 с белками Rpa1 и HMGB1 | Мышь 154Q/2Q, клетки HeLa | Иммунопреципитация, иммуноцитохимия | [56, 32 ] | |
| Нарушение динамической активности Rpa1 при повреждениях ДНК | Клетки U20S | Наблюдение in vivo за флуоресцентно меченным Rpa1 | [32] | |
| Аномальный выход из фазы G0 клеточного цикла | Мышь 154Q/2Q | Клетки Пуркинье | Мечение BrDu, иммуноцитохимический анализ | [32] |
| Снижение экспрессии VEGF в нейрональных предшественниках | Мышь 154Q/2Q | Нейрональные предшественники | ПЦР в реальном времени | [52] |
| ATXN1-CIC-комплекс является первопричиной развития заболевания | Мышь 82Q,V591A;S602D; человек |
Клетки Пуркинье (мышь), культура нейронов (человек) |
Функциональные тесты, электрофизиологические тесты, транскриптомный анализ | [45] |
| Блокировка агрегации мутантного ATXN1 белком DUSP18 | Клетки BOSC 23 | Filter retardation assay | [57] | |
| Повышение активации глиальных клеток в постэмбриональном развитии |
Мышь 154Q/2Q | Бергманова глия, микроглия | Количественная иммуноцитохимическая окраска | [58] |
| Увеличение экспрессии проинфламаторных цитокинов | Мышь 154Q/2Q | ПЦР в реальном времени | [58] | |
| Изменение экспрессии микроРНК | Мышь 82Q | Мозжечок | Микрочип ПЦР в реальном времени |
[59] |
| Нарушение целостности ядерной мембраны | Клетки COS-1, мышь 82Q | Клетки Пуркинье | Микроскопия, иммуноцитохимия | [33, 61] |
| Снижение эффективности деградации мутантного ATXN1 | Внеклеточная модель | Анализ скорости деградации мутантного белка клеточным лизатом in vitro | [60] | |
| Образование массивных ядерных включений | Клетки COS7,
COS1, HeLa; мышь 82Q |
Клеточные линии, клетки Пуркинье |
Микроскопия | [33, 34, 61] |
| Нарушение ядерно-цитоплазматического транспорта РНК из-за образования патогенных агрегатов | Клетки COS7 | FRAP-анализ | [34] | |
| Нарушение клеточной локализации протеасомы | Мышь 82Q | Клетки Пуркинье | Иммуноцитохимический анализ | [62] |
| Повышение экспрессии S100B, образование вакуолей с включениями S100B | Мышь 82Q/GFP, клетки HeLa |
Мозжечок | ПЦР в реальном времени, иммуноцитохимический анализ |
[63] |
| Снижение количества рецептора RORa | Мышь 82Q | Клетки Пуркинье | Иммуноцитохимия, Вестерн-блот |
[64] |
МУТАЦИЯ ПРИОБРЕТЕНИЯ ФУНКЦИИ
Из всего спектра различных нарушений, вызываемых экспансией CAG-повторов в гене ATXN1, наибольший вклад в развитие заболевания вносит механизм “приобретения функции” белка ATXN1. Мутантный ATXN1 может взаимодействовать с нехарактерными для него партнерами, а нормальные взаимодействия могут изменять свою силу и характер. Одним из ключевых партнеров ATXN1 является белок Capicua (CIC) – транскрипционный фактор, который негативно регулирует транскрипцию многих генов [70]. 80% клеточного CIC находится в комплексе с ATXN1 [70]. Экспансия CAG-повторов в гене ATXN1 изменяет функциональную активность комплекса ATXN1–CIC, что выражается в значительном изменении экспрессии CIC-регулируемых генов [70]. Двукратное снижение уровня экспрессии CIC улучшает моторные функции, память, поведение и морфологию модельных мышей, а лишение способности мутантного ATXN1 взаимодействовать с CIC ведет к исчезновению всех проявлений SCA1, в том числе и к восстановлению экспрессии более 500 генов [45]. Мутация в гене ATXN1, приводящая к аминокислотной замене S776A, для которой показана способность нивелировать токсичность мутантного аллеля ATXN1, не мешает CIC и ATXN1 взаимодействовать друг с другом, однако при этой мутации CIC пропадает из массивных внутриядерных комплексов ATXN1 [70]. На данный момент именно изменение взаимодействия между ATXN1 и CIC принято считать главной причиной фенотипа, наблюдаемого при SCA1, разнообразие проявлений которого объясняется широким спектром вовлеченных генов [45].
НАРУШЕНИЯ ПЕРЕДАЧИ ГЛУТАМАТНОГО СИГНАЛА
Одним из механизмов возникновения непосредственно атаксического синдрома при SCA1 является нарушение опосредованного рецептором mGlur1 глутаматного сигналинга и, как следствие, синаптической пластичности и моторного обучения. Одним из основных путей передачи возбуждения на клетки Пуркинье (PC) являются параллельные волокна (PF). На синаптической мембране зрелых клеток Пуркинье в местах синапса PC–PF располагаются метаботропные рецепторы глутамата mGlur1 [81]. Активный нейрон параллельного волокна высвобождает глутамат в синаптическую щель, где он взаимодействует с mGlur1, запуская процесс передачи глутаматного сигнала. Из синаптической щели глутамат начинает быстро поглощаться клетками Бергмановой глии при участии переносчика глутамата и аспартата GLAST [82, 83], а остаточные молекулы вновь поглощаются симпортером глутамата и аспартата EAAT4, расположенным на экстрасинаптической мембране клеток Пуркинье [84, 85]. В зрелых клетках Пуркинье SCA1-мышей нарушена экспрессия генов передачи глутаматного сигнала, в том числе более чем в 2 раза снижается количество основного глутаматного рецептора mGluR1, а также симпортеров глутамата EAAT4 и GLAST на мембранах клеток Пуркинье и клеток Бергмановой глии соответственно [46–48, 53]. Как следствие, у клеток Пуркинье модельных животных нарушен ряд электрофизиологических функций, затрагивающих процессы моторного обучения и синаптической пластичности [47, 48]. Экспансия CAG-повторов в гене ATXN1 приводит к нарушению функций нейронов Пуркинье еще до начала их гибели. Направленное лечение аллостерическими модуляторами, основанное на знаниях о нарушениях в глутаматном сигнальном пути, приводит к улучшению состояния модельных животных [47, 48].
ОБРАЗОВАНИЕ АГРЕГАТОВ МУТАНТНОГО БЕЛКА
Для многих полиглутаминовых нейродегенеративных заболеваний характерно образование цитоплазматических и ядерных белковых телец [86]. При SCA1 тоже образуются агрегаты, содержащие белок ATXN1, однако по своим свойствам они отличаются от таковых у других заболеваний. Домен AXH белка ATXN1 отвечает за различные межмолекулярные взаимодействия, в том числе и за самоассоциацию нормальных форм ATXN1 в функциональные нуклеоплазматические тельца [21]. При нормальной длине CAG-участка такие тельца подвижны, белок, входящий в их состав, имеет высокую динамику обмена с нуклеоплазмой, при делении клетки они диссоциируют, а затем реассоциируют в дочерних клетках в равном количестве, а само образование таких включений способствует осуществлению некоторых функций ATXN1 [33 , 34, 61]. Такие белковые включения встречаются не во всей популяции клеток, однако их наличие само по себе не является признаком патологии [33, 35, 61, 63]. При экспансии CAG-повторов в гене ATXN1 количество включений в клетке становится меньше, но в несколько раз увеличивается их размер [33]. При этом снижается динамика обмена ATXN1 между включениями и нуклеоплазмой, нарушается ядерно-цитоплазматический транспорт ATXN1 [34, 61]. Однако при ингибировании образования телец включений SCA1 прогрессирует быстрее [60]. Мутантный ATXN1, попавший во включения, скорее всего не сможет выйти из его состава за счет понижения динамики белков в составе этих включений. Таким образом, мутантный белок оказывается “связанным” внутри включений и не способен проявить свои токсичные свойства в других частях клетки. Это позволяет рассматривать образование увеличенных ядерных включений не как одну из основных причин патологии, а как один из сдерживающих ее факторов. Но стоит отметить, что во включения ATXN1 также попадают другие белки и РНК, и некоторые из белков при этом существенно изменяют свою нормальную локализацию, что негативно влияет на функционирование клетки [22, 62, 69]. В частности, во включениях мутантного ATXN1 также находятся: 14-3-3 белок, TAP/NXF1, убиквитин, LMP2, CBP, ANP32A, 20s протеасома, P31, PA700, HSP70, HDJ2 и РНК [22, 60, 62, 65, 69].
ПОВРЕЖДЕНИЕ ЯДЕРНОЙ МЕМБРАНЫ
Нарушения структуры ядерной мембраны наблюдаются при некоторых наследственных нейродегенеративных полиглутаминовых заболеваниях, например при хорее Гентингтона [87, 88]. Экспансия CAG-повторов в гене ATXN1 вызывает многочисленные механические нарушения ядерной мембраны, такие как инвагинации и разрывы [33, 35]. ATXN1 может входить в состав ядерной ламины [33]. Опыты на искусственных мембранах показали увеличение хрупкости мембран при взаимодействии с мутантным ATXN1 [35]. От длины полиглутаминового тракта обратнопропорционально зависит сопротивление искусственной мембраны, что делает ее более проницаемой для ионов [35]. Эти данные согласуются со способностью полиглутаминовых пептидов принимать стабильную форму микроспирали, которая при интеграции в мембрану выполняет функции ионного канала [89, 90]. Повреждения ядерной мембраны в клетках, экспрессирующих мутантный ATXN1, могут быть одной из причин повышенного репарационного фона и могут ускорять фенотипические проявления SCA1.
ПОВРЕЖДЕНИЯ ЯДЕРНОЙ И МИТОХОНДРИАЛЬНОЙ ДНК
Есть данные о том, что SCA1 ассоциирована с повышенным количеством повреждений ДНК, в частности двунитевых разрывов в клетках Пуркинье и гранулярных клетках. Повышение экспрессии белков-участников репарации, таких как Rpa1, TP53 (P53), HNF4A, C-MYC, HMGB1, значительно увеличивает продолжительность жизни модельных животных, трансгенных по мутантному ATXN1 [32]. Точные причины увеличения числа повреждений ДНК на данный момент неизвестны, однако можно предположить, что некоторый вклад в этот процесс вносят повреждения ядерной мембраны и снижение концентрации белков репарации за счет их аномальных взаимодействий с мутантным ATXN1. В подтверждение этой теории in vivo показано образование комплексов polyQ–ATXN1–HMGB1 и polyQ–ATXN1–Rpa1, часть из этих комплексов деградирует в протеасомах [32]. У SCA1 мышей перед гибелью клеток Пуркинье наблюдается понижение уровня белка HMGB1, причем снижение HMGB1 наблюдается в клетках, не имеющих ядерных включений мутантного ATXN1, что согласуется с приведенными выше исследованиями [56]. После восстановления репарационного процесса в клетках наблюдается также возвращение к нормальному уровню экспрессии порядка 20% от всех генов, изменивших свою экспрессию при SCA1. Повышение экспрессии Rpa1 или HMGB1 кроме прочего восстанавливает стабильность G0-фазы клеточного цикла нейронов, увеличивает толщину молекулярного слоя, восстанавливает структуру дендритного дерева клеток Пуркинье и структуру дендритных шипиков. При этом повышение экспрессии белков, функции которых направлены на дестабилизацию молекулы ДНК в процессе репарации, приводит к снижению времени жизни модельных животных. К этим белкам относятся BLM, FEN1, LiG1, Chk1.
В митохондриальной ДНК также наблюдается повышенное количество повреждений [54, 55]. На цитологическом уровне эффект этих повреждений выражается в пониженной электронной плотности митохондрий, снижении митохондриального мембранного потенциала, уменьшении количества сукцинатдегидрогеназы и цитохром-с-оксидазы, снижении частоты делений и слияний митохондрий [54].
Способность белков-участников репарации ДНК восстанавливать аномальный фенотип клеток свидетельствует о том, что многие из нарушений, вызванных повреждениями ДНК, носят временный характер.
ИЗМЕНЕНИЕ ЭКСПРЕССИИ микроРНК
При SCA1 в мозжечке изменяется экспрессия десятков микроРНК [59]. У модельных мышей в раннем пресимптоматическом периоде в клетках Пуркинье экспрессия ATXN1 находится на базальном уровне, однако уже в этом периоде можно наблюдать изменение экспрессии некоторых микроРНК. Таким образом, экспансия CAG-повторов в гене ATXN1 способна влиять на общий транскрипционный фон клетки даже при низком количестве белка. В более позднем возрасте наблюдаются корреляции между изменением экспрессии микроРНК и уже известными молекулярными механизмами SCA1. Так, повышается экспрессия miR-150 – негативного регулятора трансляции VEGF, снижение экспрессии которого является одной из причин нарушений при SCA1 [52, 59, 91 ]. Экспрессия ATXN1, в свою очередь, напрямую регулируется несколькими miRNA. Ген ATXN1 имеет длинный 3'UTR размером в 7 тпн с расположенными на нем сайтами связывания miR-101, miR-130a, miR-19a, miR-302, miR-144 и других miRNA, которые способны специфично узнавать мРНК ATXN1 [36, 92]. Ингибирование экспрессии этих miRNA увеличивает токсичность мутантного аллеля ATXN1 [36]. Изменения экспрессии микроРНК при SCA1 могут отражать еще неизвестные молекулярные механизмы, вносящие вклад в развитие заболевания, а также могут послужить индикатором эффективности потенциальной терапии SCA1.
ЗАКЛЮЧЕНИЕ
Изучение молекулярных путей, приводящих к нейродегенеративным заболеваниям у пациента, сопряжено с большими трудностями. Изучать механизмы дегенерации при жизни пациента очень тяжело, так как для изучения доступны только биологические жидкости пациента, а постмортальные образцы тканей представляют собой финальную стадию нейродегенерации. В связи с этим использование животных и клеточных моделей является уникальным способом изучения развития нейродегенеративных заболеваний человека. Их использование внесло серьезный вклад в понимание механизмов, лежащих в основе наследственных нейродегенеративных заболеваний, в том числе атаксии первого типа. Открытие механизмов репрограммирования соматических клеток человека до плюрипотентного состояния дало возможность изучать нейродегенерацию человека на качественно новом уровне. Можно надеяться, что модели на основе ИПСК дополнят арсенал существующих моделей и помогут ответить на ряд фундаментальных нерешенных вопросов: Что является максимально токсичным агентом при полиглутаминовых заболеваниях – мутантный белок или мутантная РНК? Каковы механизмы этой токсичности, особенно РНК-токсичности? Почему polyQ заболевания избирательно повреждают нервную систему, хотя мутантные гены и кодируемые ими белки широко представлены во всех тканях? Какие клеточные органеллы и сигнальные пути наиболее уязвимы для токсического повреждения? Какие пусковые механизмы полиглутаминовых заболеваний характерны для всех polyQ, а какие – зависят от конкретного мутантного гена? Ответив хотя бы на часть этих вопросов, можно найти не только новые связи между первичной структурой ДНК и функциями РНК и белка, но и, возможно, новые мишени для воздействия на полиглутаминовые заболевания.
Работа финансировалась грантом Российского научного фонда 19-15-00425.
Настоящая статья не содержит каких-либо исследований с использованием в качестве объекта животных.
Настоящая статья не содержит каких-либо исследований с участием в качестве объекта людей.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Иллариошкин С.Н., Руденская Г.Е., Иванова-Смоленская И.А. и др. Наследственные атаксии и параплегии. М.: МЕДпресс-информ, 2006. 416 с.
Stoyas C., La Spada A. The CAG-polyglutamine repeat diseases: a clinical, molecular, genetic, and pathophysiologic nosology // Handb Clin. Neurol. 2018. V. 147. P. 143–170.https://doi.org/10.1016/B978-0-444-63233-3.00011-7
Katsuno M., Tanaka F., Adachi H. et al. Pathogenesis and therapy of spinal and bulbar muscular atrophy (SBMA) // Prog. Neurobiol. 2012. V. 99. № 3. P. 246–256. https://doi.org/10.1016/j.pneurobio.2012.05.007
Ha A.D., Fung V.S. Huntington’s disease // Curr. Opin. Neurol. 2012. V. 25. № 4. P. 491–498. https://doi.org/10.1097/WCO.0b013e3283550c97
Storey E. Genetic cerebellar ataxias // Semin. Neurol. 2014. V. 34. № 3. P. 280–292. https://doi.org/10.1055/s-0034-1386766
Stevanin G., Brice A. Spinocerebellar ataxia 17 (SCA17) and Huntington’s disease-like 4 (HDL4) // Cerebellum. 2008. V. 7. № 2. P. 170–178. https://doi.org/10.1007/s12311-008-0016-1
Zoghbi H.Y., Orr H.T. Glutamine repeats and neurodegeneration // Annu. Rev. Neurosci. 2000. V. 23. P. 217–247. https://doi.org/10.1146/annurev.neuro.23.1.217
Sun Y.M., Zhang Y.B., Wu Z.Y. Huntington’s disease: Relationship between phenotype and genotype // Mol. Neurobiol. 2017. V. 54. № 1. P. 342–348. https://doi.org/10.1007/s12035-015-9662-8
Schut J. Hereditary ataxia: clinical study through six generations // Arch. NeurPsych. 1950. V. 63. P. 535–568.
Конева Л.А., Кучер А.Н., Пузырев В.П. и др. Демографические и клинико-генетические особенности распространенности спиноцеребеллярной атаксии I типа в Усть-Алданском и Абыйском улусах Республики Саха (Якутия) // Матер. науч.-практич. конф. “Актуальные вопросы профилактической медицины”. Улан-Удэ, 2005. С. 97–100.
Banfi S., Servadio A., Chung M. et al. Identification and characterization of the gene causing type 1 spinocerebellar ataxia // Nat. Genet. 1994. V. 7. P. 513–520. https://doi.org/10.1038/ng0894-513
Opal P., Ashizawa T. Spinocerebellar ataxia type 1 // GeneReviews (Internet). 1998.
Kraus-Perrotta C., Lagalwar S. Expansion, mosaicism and interruption: mechanisms of the CAG repeat mutation in spinocerebellar ataxia type 1 // Cerebellum ataxias. 2016. V. 3. P. 20. https://doi.org/10.1186/s40673-016-0058-y
Pearson C., Eichler E., Lorenzetti D. et al. Interruptions in the triplet repeats of SCA1 and FRAXA reduce the propensity and complexity of slipped strand DNA (S-DNA) formation // Biochemistry. 1998. V. 37. № 8. P. 2701–2708. https://doi.org/10.1021/bi972546c
Matilla-Dueñas A., Goold R., Giunti P. Clinical, genetic, molecular, and pathophysiological insights into spinocerebellar ataxia type 1 // The Cerebellum. 2008. P. 106–114. https://doi.org/10.1007/s12311-008-0009-0
Seidel K., Siswanto S., Ewout R. et al. Brain pathology of spinocerebellar ataxias // Acta Neuropathol. 2012. V. 124. P. 1–21. https://doi.org/10.1007/s00401-012-1000-x
Biirk K., Abele M., Fetter M. et al. Autosomal dominant cerebellar ataxia type I: Clinical features and MRI in families with SCA1, SCA2 and SCA3 // Brain. 1996. V. 119. P. 1497–1505. https://doi.org/10.1093/brain/119.5.1497
Rüb U., Bürk K., Timmann D. et al. Spinocerebellar ataxia type 1 (SCA1): new pathoanatomical and clinico-pathological insights // Neuropathol. Applied Neurobiol. 2012. V. 38. P. 665–680. https://doi.org/10.1111/j.1365-2990.2012.01259.x
Guerrini L., Lolli F., Ginestroni A. et al. Brainstem neurodegeneration correlates with clinical dysfunction in SCA1 but not in SCA2. A quantitative volumetric, diffusion and proton spectroscopy MR study // Brain. 2004. V. 127. P. 1785–1795. https://doi.org/10.1093/brain/awh201
Adanyeguh I., Henry P., Nguyen T. et al. In vivo neurometabolic profiling in patients with spinocerebellar ataxia types 1, 2, 3 and 7 // Movement Disorders. 2015. V. 30. № 5. P. 662–670. https://doi.org/10.1002/mds.26181
Burright E., Clark H., Servadio A. et al. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat // Cell. 1995. V. 82. P. 937–948. https://doi.org/10.1016/0092-8674(95)90273-2
Matilla A., Roberson D., Banfi S. et al. Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation // J. Neurosci. 1998. V. 18. P. 5508–5516. https://doi.org/10.1523/JNEUROSCI.18-14-05508.1998
Watase K., Weeber E., Xu B. et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration // Neuron. 2002. V. 34. P. 905–919. https://doi.org/10.1016/S0896-6273(02)00733-X
Lorenzetti D., Watase K., Xu B. et al. Repeat instability and motor incoordination in mice with a targeted expanded CAG repeat in the Sca1 locus // Hum. Mol. Genet. 2000. V. 9. P. 779–785. https://doi.org/10.1093/hmg/9.5.779
Zu T., Duvick L., Kaytor M. et al. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice // J. Neurosci. 2004. V. 24. P. 8853–8861. https://doi.org/10.1523/JNEUROSCI.2978-04.2004
Giovannoni R., Maggio N., Rosaria-Bianco M. et al. Reactive astrocytosis and glial glutamate transporter clustering are early changes in a spinocerebellar ataxia type 1 transgenic mouse model // Neuron Glia Biol. 2007. V. 3. № 4. P. 335–351. https://doi.org/10.1017/S1740925X08000185
Ingram M., Orr H., Clark H. Genetically engineered mouse models of the trinucleotide-repeat spinocerebellar ataxias // Brain Res. Bull. 2012. V. 88. № 1. P. 33–42. https://doi.org/10.1016/j.brainresbull.2011.07.016
Emamian E.S., Kaytor M.D., Duvick L.A. et al. Serine 776 of ataxin-1 is critical for polyglutamine induced disease in SCA1 transgenic mice // Neuron. 2003. V. 38. P. 375–387. https://doi.org/10.1016/S0896-6273(03)00258-7
Duvick L., Barnes J., Ebner B. et al. SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776 // Neuron. 2010. V. 67. P. 929–935. https://doi.org/10.1016/j.neuron.2010.08.022
Fernandez-Funez P., Nino-Rosales M.L., de Gouyon B. et al. Identification of genes that modify ataxin-1-induced neurodegeneration // Nature. 2000. V. 408. № 6808. P. 101–106. https://doi.org/10.1038/35040584
Bondar V. V., Adamski C.J., Onur T.S. et al. PAK1 regulates ATXN1 levels providing an opportunity to modify its toxicity in spinocerebellar ataxia type 1 // Hum. Mol. Genet. 2018. V. 27. № 16. P. 2863–2873. https://doi.org/10.1093/hmg/ddy200
Barclay S.S., Tamura T., Ito H. et al. Systems biology analysis of Drosophila in vivo screen data elucidates core networks for DNA damage repair in SCA1 // Hum. Mol. Genet. 2014. V. 23. № 5. P. 1345–1364. https://doi.org/10.1093/hmg/ddt524
Skinner P.J., Koshy B.T., Cummings C.J. et al. Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures // Nature. 1997. V. 391. № 6664. P. 307–307. https://doi.org/10.1038/34701
Krol H.A., Krawczyk P.M., Bosch K.S. et al. Polyglutamine expansion accelerates the dynamics of ataxin-1 and does not result in aggregate formation // PLoS One. 2008. V. 3. № 1. e1503. https://doi.org/10.1371/journal.pone.0001503
Mapelli L., Canale C., Pesci D. et al. Toxic effects of expanded ataxin-1 involve mechanical instability of the nuclear membrane // Biochim. Biophys. Acta (BBA) – Mol. Basis of Disease. 2012. V. 1822. № 6. P. 906–917. https://doi.org/10.1016/j.bbadis.2012.01.016
Lee Y., Samaco R.C., Gatchel J.R. et al. miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis // Nat. Neurosci. 2008. V. 11. № 10. P. 1137–1139. https://doi.org/10.1038/nn.2183
Frattini A., Fabbri M., Valli R. et al. High variability of genomic instability and gene expression profiling in different HeLa clones // Sci. Reports. 2015. V. 5. № 1. https://doi.org/10.1038/srep15377
Lin Y.-C., Boone M., Meuris L. et al. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations // Nat. Communicat. 2014. V. 1. https://doi.org/10.1038/ncomms5767
Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors // Cell. 2006. V. 126. № 4. P. 663–676. https://doi.org/10.1016/j.cell.2006.07.024
Muguruma K., Nishiyama A., Kawakami H. et al. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells // Cell Reports. 2015. V. 10. № 4. P. 537–550.https://doi.org/10.1016/j.celrep.2014.12.051
Qian X., Jacob F., Song M.M. et al. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor // Nat. Protocols. 2018. V. 13. № 3. P. 565–580. https://doi.org/10.1038/nprot.2017.152
Gorris R., Fischer J., Erwes K.L. et al. Pluripotent stem cell-derived radial glia-like cells as stable intermediate for efficient generation of human oligodendrocytes // Glia. 2015. V. 63. № 12. P. 2152–2167. https://doi.org/10.1002/glia.22882
Douvaras P., Sun B., Wang M. et al. Directed differentiation of human pluripotent stem cells to microglia // Stem Cell Reports. 2017. V. 8. № 6. P. 1516–1524. https://doi.org/10.1016/j.stemcr.2017.04.023
Lebedeva O.S., Surdina A.V., Bogomiakova M.E. et al. IPSC – based model for the study of molecular mechanisms of spinocerebellar ataxia type 1 // FEBS J. 2017. V. 284. Suppl. 1. P. 141.
Rousseaux M.W.C., Tschumperlin T., Lu H.-C. et al. ATXN1-CIC complex is the primary driver of cerebellar pathology in spinocerebellar ataxia type 1 through a gain-of-function mechanism // Neuron. 2018. V. 97. № 6. P. 1235–1243. https://doi.org/10.1016/j.neuron.2018.02.013 45а. Богомазова А.Н., Васина Е.М., Киселев С.Л. и др. Генетическое репрограммирование клеток: новая технология для фундаментальных исследований и практического использования // Генетика. 2015. Т. 51. № 4. С. 466–478.
Serra H.G., Byam C.E., Lande J.D. et al. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice // Hum. Mol. Genet. 2004. V. 13. № 20. P. 2535–2543. https://doi.org/10.1093/hmg/ddh268
Notartomaso S., Zapulla C., Biagioni F. et al. Pharmacological enhancement of mGlu1 metabotropic glutamate receptors causes a prolonged symptomatic benefit in a mouse model of spinocerebellar ataxia type 1 // Mol. Brain. 2013. V. 6. № 48. https://doi.org/10.1186/1756-6606-6-48
Power E.M., Morales A., Empson R.M. Prolonged type 1 metabotropic glutamate receptor dependent synaptic signaling contributes to spino-cerebellar ataxia type 1 // J. Neurosci. 2016. V. 36. № 18. P. 4910–4916. https://doi.org/10.1523/jneurosci.3953-15.2016
Shuvaev A.N., Hosoi N., Sato Y. et al. Progressive impairment of cerebellar mGluR signalling and its therapeutic potential for cerebellar ataxia in spinocerebellar ataxia type 1 model mice // J. Physiology. 2016. V. 595. № 1. P. 141–164. https://doi.org/10.1113/jp272950
Oz G., Kittelson E., Demirgöz D. et al. Assessing recovery from neurodegeneration in spinocerebellar ataxia 1: Comparison of in vivo magnetic resonance spectroscopy with motor testing, gene expression and histology // Neurobiol. Disease. 2015. V. 74. P. 158–166. https://doi.org/10.1016/j.nbd.2014.11.011
Hourez R., Servais L., Orduz D. et al. Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1 // J. Neurosci. 2011. V. 31. № 33. P. 11795–11807. https://doi.org/10.1523/jneurosci.0905-11.2011
Cvetanovic M., Hu Y.-S., Opal P. Mutant ataxin-1 inhibits neural progenitor cell proliferation in SCA1 // Cerebellum. 2017. V. 16. № 2. P. 340–347. https://doi.org/10.1007/s12311-016-0794-9
Cvetanovic M. Decreased expression of glutamate transporter GLAST in Bergmann glia is associated with the loss of Purkinje neurons in the spinocerebellar ataxia type 1 // Cerebellum. 2014. V. 14. № 1. P. 8–11. https://doi.org/10.1007/s12311-014-0605-0
Ito H., Fujita K., Tagawa K. et al. HMGB1 facilitates repair of mitochondrial DNA damage and extends the lifespan of mutant ataxin-1 knock-in mice // EMBO Mol. Med. 2014. V. 7. № 1. P. 78–101. https://doi.org/10.15252/emmm.201404392
Taniguchi J.B., Kondo K., Fujita K. et al. RpA1 ameliorates symptoms of mutant ataxin-1 knock-in mice and enhances DNA damage repair // Hum. Mol. Genet. 2016. V. 25. № 20. P. 4432–4447. https://doi.org/10.1093/hmg/ddw272
Qi M.-L., Tagawa K., Enokido Y. et al. Proteome analysis of soluble nuclear proteins reveals that HMGB1/2 suppress genotoxic stress in polyglutamine diseases // Nat. Cell Biol. 2007. V. 9. № 4. P. 402–414. https://doi.org/10.1038/ncb1553
Ryu J., Lee D.H. Dual-specificity phosphatase 18 modulates the SUMOylation and aggregation of ataxin-1 // Biochem. Biophys. Res. Communicat. 2018. V. 502. № 3. P. 389–396. https://doi.org/10.1016/j.bbrc.2018.05.178
Cvetanovic M., Ingram M., Orr H., Opal P. Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1 // Neuroscience. 2015. V. 289. P. 289–299. https://doi.org/10.1016/j.neuroscience.2015.01.003
Rodriguez-Lebron E., Liu G., Keiser M. et al. Altered Purkinje cell miRNA expression and SCA1 pathogenesis // Neurobiol. Disease. 2013. V. 54. P. 456–463. https://doi.org/10.1016/j.nbd.2013.01.019
Cummings C.J., Reinstein E., Sun Y. et al. Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice // Neuron. 1999. V. 24. № 4. P. 879–892. https://doi.org/10.1016/s0896-6273(00)81035-1
Stenoien D.L., Mielke M., Mancini M.A. Intranuclear ataxin1 inclusions contain both fast- and slow-exchanging components // Nat. Cell Biol. 2002. V. 4. № 10. P. 806–810. https://doi.org/10.1038/ncb859
Cummings C.J., Mancini M.A., Antalffy B. et al. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1 // Nat. Genet. 1998. V. 19. № 2. P. 148–154. https://doi.org/10.1038/502
Vig P., Hearst S., Shao Q. et al. Glial S100B protein modulates mutant ataxin-1 aggregation and toxicity: TRTK12 peptide, a potential candidate for SCA1 therapy // Cerebellum. 2011. V. 10. № 2. P. 254–266. https://doi.org/10.1007/s12311-011-0262-5
Serra H.G., Duvick L., Zu T. et al. RORα-mediated Purkinje cell development determines disease severity in adult SCA1 mice // Cell. 2006. V. 127. № 4. P. 697–708. https://doi.org/10.1016/j.cell.2006.09.036
Tsuda H., Jafar-Nejad H., Patel A.J. et al. The AXH domain of ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/senseless proteins // Cell. 2005. V. 122. № 4. P. 633–644. https://doi.org/10.1016/j.cell.2005.06.012
Mizutani A., Wang L., Rajan H. et al. Boat, an AXH domain protein, suppresses the cytotoxicity of mutant ataxin-1 // EMBO J. 2005. V. 24. № 18. P. 3339–3351. https://doi.org/10.1038/sj.emboj.7600785
Yue S., Serra S., Zoghbi H., Orr H. The spinocerebellar ataxia type 1 protein, ataxin-1, has RNA-binding activity that is inversely affected by the length of its polyglutamine tract // Hum. Mol. Genet. 2001. V. 10. № 1. P. 25–30. https://doi.org/10.1093/hmg/10.1.25
De Chiara C., Menon R.P., Dal Piaz F. et al. Polyglutamine is not all: The functional role of the AXH domain in the ataxin-1 protein // J. Mol. Biol. 2005. V. 354. № 4. P. 883–893. https://doi.org/10.1016/j.jmb.2005.09.083
Irwin S., Vandelft M., Pinchev D. et al. RNA association and nucleocytoplasmic shuttling by ataxin-1 // J. Cell Sci. 2005. V. 118(1). P. 233–242. https://doi.org/10.1242/jcs.01611
Lam Y.C., Bowman A.B., Jafar-Nejad P. et al. ATAXIN-1 interacts with the repressor capicua in its native complex to cause SCA1 neuropathology // Cell. 2006. V. 127. № 7. P. 1335–1347. https://doi.org/10.1016/j.cell.2006.11.038
Crespo-Barreto J., Fryer J.D., Shaw C.A. et al. Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis // PLoS Genet. 2010. V. 6. № 7. e1001021. https://doi.org/10.1371/journal.pgen.1001021
Sánchez I., Balagué E., Matilla-Dueñas A. Ataxin-1 regulates the cerebellar bioenergetics proteome through the GSK3β-mTOR pathway which is altered in spinocerebellar ataxia type 1 (SCA1) // Hum. Mol. Genet. 2016. V. 25. № 18. P. 4021–4040. https://doi.org/10.1093/hmg/ddw242
Davidson J.D., Riley B., Burright E. N. et al. Identification and characterization of an ataxin-1-interacting protein: A1Up, a ubiquitin-like nuclear protein // Hum. Mol. Genet. 2000. V. 9. № 15. P. 2305–2312. https://doi.org/10.1093/oxfordjournals.hmg.a018922
Okazawa H., Rich T., Chang A. et al. Interaction between mutant ataxin-1 and PQBP-1 affects transcription and cell death // Neuron. 2002. V. 34. № 5. P. 701–713.https://doi.org/10.1016/s0896-6273(02)00697-9
Hong S., Kim S.-J., Ka S. et al. USP7, a Ubiquitin-specific protease, interacts with ataxin-1, the SCA1 gene product // Mol. Cell. Neurosci. 2002. V. 20. № 2. P. 298–306. https://doi.org/10.1006/mcne.2002.1103
Goold R., Hubank M., Hunt A. et al. Down-regulation of the dopamine receptor D2 in mice lacking ataxin 1 // Hum. Mol. Genet. 2007. V. 16. № 17. P. 2122–2134. https://doi.org/10.1093/hmg/ddm162
Tong X., Gui H., Jin F. et al. Ataxin-1 and brother of ataxin-1 are components of the notch signalling pathway // EMBO Reports. 2011. V. 12. № 5. P. 428–435. https://doi.org/10.1038/embor.2011.49
Klement I.A., Skinner P.J., Kaytor M.D. et al. Ataxin-1 nuclear localization and aggregation // Cell. 1998. V. 95. № 1. P. 41–53. https://doi.org/10.1016/s0092-8674(00)81781-x
Bañez-Coronel M., Ayhan F., Tarabochia A.D. et al. RAN translation in Huntington disease // Neuron. 2015. V. 88. P. 667–677. https://doi.org/10.1016/j.neuron.2015.10.038
Zu T., Gibbens B., Doty N.S. et al. Non-ATG-initiated translation directed by microsatellite expansions // Proc. Natl Acad. Sci. 2010. V. 108. № 1. P. 260–265. https://doi.org/10.1073/pnas.1013343108
Luján R., Nusser Z., Roberts J.D.B. et al. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus // Eur. J. Neurosci. 1996. V. 8. № 7. P. 1488–1500. https://doi.org/10.1111/j.1460-9568.1996.tb01611.x
Lehre K.P., Danbolt N.C. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain // J. Neurosci. 1998. V. 18. P. 8751–8757. https://doi.org/10.1523/JNEUROSCI.18-21-08751.1998
Rothstein J.D., Martin L., Levey A.I. et al. Localization of neuronal and glial glutamate transporters // Neuron. 1994. V. 13. P. 713–725. https://doi.org/10.1016/0896-6273(94)90038-8
Tanaka J., Ichikawa R., Watanabe M. et al. Extra-junctional localization of glutamate transporter EAAT4 at excitatory Purkinje cell synapses // NeuroReport. 1997. V. 8. № 11. P. 2461–2464. https://doi.org/10.1097/00001756-199707280-00010
Furuta A., Martin L., Lin C. et al. Cellular and synaptic localization of the neuronal glutamate transporters excitatory amino acid transporter 3 and 4 // Neuroscience. 1997. V. 81. № 4. P. 1031–1042. https://doi.org/10.1016/s0306-4522(97)00252-2
Havel L.S., Li S., Li X. Nuclear accumulation of polyglutamine disease proteins and neuropathology // Mol. Brain. 2009. V. 2. № 21. https://doi.org/10.1186/1756-6606-2-21
Nekrasov E.D., Vigont V.A., Klyushnikov S.A. et al. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons // Mol. Neurodegeneration. 2016. V. 11. № 1. https://doi.org/10.1186/s13024-016-0092-5
Gasset-Rosa F., Chillon-Marinas C., Goginashvili A. et al. Polyglutamine-expanded Huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport // Neuron. 2017. V. 94. № 1. P. 48–57. https://doi.org/10.1016/j.neuron.2017.03.027
Monoi H., Futaki S., Kugimiya S. et al. Poly-l-glutamine forms cation channels: relevance to the pathogenesis of the polyglutamine diseases // Biophys. J. 2000. V. 78. № 6. P. 2892–2899. https://doi.org/10.1016/s0006-3495(00)76830-5
Hirakura Y., Azimov R., Azimova R., Kagan B.L. Polyglutamine-induced ion channels: a possible mechanism for the neurotoxicity of huntington and other CAG repeat diseases // J. Neurosci. Res. 2000. V. 60. №4. P. 490–494. https://doi.org/10.1002/(SICI)1097-4547(20000515)60: 4<490::AID-JNR7>3.0.CO;2-9
Cvetanovic M., Patel J.M., Marti H.H. et al. Vascular endothelial growth factor ameliorates the ataxic phenotype in a mouse model of spinocerebellar ataxia type 1 // Nat. Med. 2011. V. 17. № 11. P. 1445–1447. https://doi.org/10.1038/nm.2494
Persengiev S., Kondova I., Otting N. et al. Genome-wide analysis of miRNA expression reveals a potential role for miR-144 in brain aging and spinocerebellar ataxia pathogenesis // Neurobiol. Aging. 2011. V. 32. № 12. P. 17–27. https://doi.org/10.1016/j.neurobiolaging.2010.03.014
Дополнительные материалы отсутствуют.