

Генетика, 2020, T. 56, № 4, стр. 375-391
Перспективы борьбы со старением мозга: редактирование гена теломеразы в нервных стволовых клетках in vivo
Н. М. Немирович-Данченко 1, *, М. Ю. Ходанович 1, **
1 Томский государственный университет, Научно-исследовательский институт
биологии и биофизики
634050 Томск, Россия
* E-mail: nmn-d@mail.ru
** E-mail: khodanovich@mail.tsu.ru
Поступила в редакцию 12.04.2019
После доработки 15.06.2019
Принята к публикации 23.07.2019
Аннотация
Возрастное укорочение теломер в нервных стволовых клетках сегодня рассматривается в качестве одного из ключевых факторов старения мозга. В связи с этим представляется перспективным поиск способов искусственного удлинения теломер в этих клетках. Подходы, основанные на усилении экспрессии теломеразы в стволовых клетках, показали свою несостоятельность из-за высокого риска возникновения раковых опухолей, неизбежно сопровождающего высокий уровень экспрессии теломеразы. Вместе с тем возможна принципиально иная стратегия повышения теломеразной активности, которая состоит в отмене альтернативного сплайсинга теломеразы. Показано, что только один из 22 сплайс-вариантов теломеразы обладает собственно теломеразной активностью, а другие варианты этого фермента способны его ингибировать и в то же время обладают канцерогенной активностью. Предлагаемая в настоящем обзоре стратегия состоит в редактировании гена теломеразы таким образом, чтобы повысить эффективность удлинения теломер за счет исключения альтернативного сплайсинга. Такой механизм усиления теломеразной активности сохранит безопасный уровень экспрессии теломеразы и исключит повышение вероятности канцерогенеза. Есть основания полагать, что редактирование гена теломеразы в нервных стволовых клетках мозга приведет к усилению нейрогенеза во взрослом мозге и к замедлению возрастных изменений в нервной деятельности. Это перспективное направление исследований в будущем может стать основой для разработки генной терапии, направленной на замедление старения мозга.
Мозг взрослых млекопитающих, включая человека, имеет крайне ограниченный эндогенный регенеративный потенциал. Этот потенциал, прежде всего, связывают с сохранением во взрослом возрасте нейрогенеза – процесса образования новых нейронов, их миграции, созревания и интеграции в нейрональную сеть. Нейрогенез во взрослом мозге животных и человека возможен благодаря сохранению ограниченной популяции нервных стволовых клеток (НСК) [1], его связывают с когнитивными процессами, а также с регенерацией поврежденной нервной ткани [2–12]. Значительное число научных групп во всем мире исследуют возможности регуляции нейрогенеза. Известно, что и без того скудные возможности регенерации мозга существенно снижаются с возрастом, что объясняется, в первую очередь, исчерпанием пула нервных стволовых клеток в нейрогенных нишах [13].
Одной из вероятных причин уменьшения пула НСК в мозге может быть укорочение теломер. Укорочение теломер в стволовых клетках сегодня рассматривается в качестве одного из основных механизмов старения. Теломеры постепенно укорачиваются с каждым новым клеточным делением [14–16]. Достижение теломерами критически малой длины останавливает клеточные деления, и в дальнейшем клетка либо подвергается апоптозу, либо встает на путь клеточного старения [17–21]. Укорочение теломер компенсируется работой фермента теломеразы, которая удлиняет теломеры [22–26]. Во взрослом организме теломераза синтезируется почти исключительно в стволовых клетках, в частности – в НСК [27–32]. По этой причине средняя длина теломер в стволовых клетках выше, чем в дифференцированных [31]. Тем не менее даже в стволовых клетках уровень экспрессии гена теломеразы недостаточно высок, чтобы полностью остановить укорочение теломер. С возрастом теломеры в стволовых клетках становятся все более короткими, что приводит к снижению регенеративной способности тканей организма. На сегодняшний день это рассматривается в качестве основного механизма старения [30, 33–36].
Показано, что процесс укорочения теломер с возрастом происходит в НСК и это ведет к нарушению нейрогенеза [37]. Кроме того, с укорочением теломер коррелирует риск развития возрастных заболеваний ЦНС, таких как болезнь Альцгеймера [38], болезнь Паркинсона [39, 40] и инсульт [41]. С другой стороны, многочисленными работами подтверждено, что воздействия, повышающие пролиферацию и выживание нейро-бластов, интеграцию в сеть молодых нейронов, в свою очередь, улучшают память и замедляют процесс старения мозга [42–45].
К сожалению, уже первые попытки усилить экспрессию гена теломеразы, поместив этот ген под контроль сильного промотора, привели к крайне нежелательным последствиям – развитию раковых опухолей [46–48]. Обнаруженный эффект объяснялся тем, что теломераза повышает активность группы генов, которые потенциально ведут к развитию раковой опухоли. При этом риск канцерогенеза зависит от уровня экспрессии теломеразы. Умеренный уровень экспрессии гена теломеразы, который в норме имеет место во взрослых стволовых клетках, совместим с устойчивой регуляцией клеточных делений, но повышенный уровень ведет к неадекватно интенсивной и неконтролируемой пролиферации и к возникновению рака [49–52].
Частично эту проблему удалось решить в работах с введением гена теломеразы в составе вирусов, не способных к интеграции в геном клетки [53–56]. Этот подход оказался безопасным с точки зрения канцерогенеза, поскольку ДНК вируса, не интегрированная в геном клетки, утрачивается клеткой в процессе последовательных клеточных делений. Однако вместе с исчезновением угрозы рака исчезла и возможность стабильно поддерживать длинные теломеры в делящихся стволовых клетках, поскольку в процессе клеточных делений привнесенный ген теломеразы утрачивался стволовыми клетками и их потомками. Другой подход заключался в усилении экспрессии теломеразы у онкоустойчивых линий мышей [39]. Хотя этот подход был успешно реализован, он по понятным причинам имеет невысокий потенциал трансляции в клинику.
Принципиально иная стратегия, состоящая в том, чтобы повысить не общий уровень экспрессии гена теломеразы, а эффективность удлинения теломер за счет исключения альтернативного сплайсинга, предлагается в настоящей теоретической статье. Здесь рассматриваются предпосылки возникновения этой идеи: данные о роли нейрогенеза и теломеразной активности в возрастных изменениях мозга; данные о связи теломеразной активности с канцерогенезом; результаты попыток преодолеть укорочение теломер с возрастом путем повышения уровня экспрессии теломеразы. Далее в статье описывается новый подход к решению проблемы укорочения теломер, основанный на исключении альтернативного сплайсинга теломеразы, а также современные методы генной инженерии, которые могут быть применены для этого.
1. УСИЛЕНИЕ НЕЙРОГЕНЕЗА ВО ВЗРОСЛОМ МОЗГЕ – СТРАТЕГИЯ, НАПРАВЛЕННАЯ НА ЗАМЕДЛЕНИЕ СТАРЕНИЯ МОЗГА
1.1. Нейрогенез во взрослом мозге – источник пластичности и регенерации нервной ткани
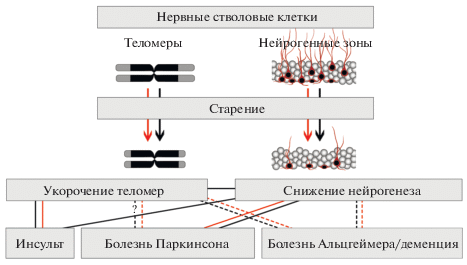
Образование новых нейронов в мозге млекопитающих не ограничивается эмбриональным и ранним постнатальным периодом, а продолжается в течение всей жизни [1]. Во взрослом мозге сохраняются две главные нейрогенные зоны, где присутствуют НСК, – зубчатая извилина гиппокампа и субвентрикулярная зона боковых желудочков. Нейрональные предшественники, появляющиеся в зубчатой извилине, остаются в пределах этой зоны и, становясь нейронами, включаются в местную нейрональную сеть [57–59]. В отличие от них предшественники, образующиеся в субвентрикулярной зоне, мигрируют оттуда в обонятельную луковицу [59], а также, в меньшей степени, в другие зоны мозга, включающие стриатум, кору и гиппокамп [60–63]. При повреждениях мозга, таких как инсульт или черепно-мозговая травма, усиливаются пролиферация в нейрогенных зонах и миграция нейрональных предшественников в зоны повреждения [60, 64–76]. Кроме того, в самих поврежденных зонах, например в коре, могут появляться резидентные нейрональные предшественники [77–80], образующиеся, вероятно, из других типов клеток вследствие репрограммирования под влиянием их микроокружения. Однако экспериментальные исследования на моделях инсульта показали, что только небольшая часть новых нейронов, мигрирующих в зону поражения или образующихся в ней, замещает собой погибшие взрослые нейроны и интегрируется в существующие нейрональные сети [81]. При этом долговременные наблюдения свидетельствуют о дальнейшем снижении нейрогенеза и пула НСК [82]. Нейродегенеративные, демиелинизирующие и психические заболевания также влияют на нейрогенез взрослого мозга. Так, снижение нейрогенеза обнаружено в клинических исследованиях и на моделях таких заболеваиий, как болезнь Альцгеймера [83, 84], болезнь Паркинсона [85], рассеянный склероз [86] и шизофрения [87]. Схематически изменения нейрогенеза при старении и его связь с возрастными неврологическими заболеваниями представлены на рис. 1.
Рис. 1.
Взаимосвязь между укорочением теломер, снижением нейрогенеза и старением мозга. Стрелки и линии обозначают связи и корреляции, обнаруженные для мыши (черный цвет) и человека (красный цвет). Сплошными линиями обозначены твердо установленные отношения, прерывистыми – отношения, подтвержденные только частью исследований. Прерывистая линия со знаком вопроса обозначает предполагаемое отношение, которое пока не исследовано.
Хотя большинство работ, посвященных нейрогенезу во взрослом мозге, было проведено на грызунах, в ряде исследований убедительно продемонстрировано, что интенсивный нейрогенез имеет место и в мозге взрослого человека, хотя данные различных исследований противоречат друг другу. Например, Moreno-Jiménez с соавт. (2019) обнаружили тысячи молодых нейронов в гиппокампе здоровых людей в возрасте от 43 до 87 лет [84]. В то же время в другом недавнем исследовании авторам практически не удалось обнаружить молодых нейронов в гиппокампе у взрослых людей [88]. В этой связи следует подчеркнуть, что выявление молодых нейронов у человека представляет методически значительно более трудную задачу по сравнению с исследованием лабораторных животных. Это обусловлено тем, что ткани мозга человека, как правило, не проходят своевременную и достаточно качественную обработку, необходимую для сохранения антигенов, характерных для молодых нейронов. Moreno-Jiménez с соавт. (2019), а также Kempermann с соавт. (2018) в своих работах убедительно показывают, что различия по выявляемому уровню нейрогенеза существенно зависят от деталей посмертной обработки тканей мозга, и приходят к общему заключению, что фактически очень высокий уровень нейрогенеза у человека сильно занижен во многих исследованиях, применявших менее эффективную обработку тканей или менее избирательно подходивших к первичному отбору материалов [84, 89]. Кроме гиппокампа, в мозге взрослых людей многочисленные нейроны обнаруживаются также в стриатуме, куда они, по всей видимости, мигрируют из субвентрикулярной зоны [90].
Молодые нейроны отличаются повышенной пластичностью – как физиологической (изменение функциональных свойств синапсов), так и структурной (образование новых и исчезновение старых синапсов, рост аксонов, изменение формы дендритов) [5–12]. По этой причине нейрогенез играет важную роль в процессах обучения. Обучение усиливает нейрогенез, а также повышает выживаемость и включение в сеть новых нейронов [2–4]. С другой стороны, искусственное снижение или выключение нейрогенеза различными методами (радиоактивное облучение, введение анти-митотических веществ, генетическое выключение) уменьшают способность животных к обучению. Соответственно, улучшение когнитивных способностей вызывается факторами, усиливающими нейрогенез, такими как физические упражнения [42], информационно обогащенная среда [3], умеренное снижение пищевого рациона [43].
1.2. Возрастное ослабление нейрогенеза и старение мозга
С возрастом происходит снижение уровня нейрогенеза в мозге [43–45]. Более того, у пациентов с возрастными нейродегенеративными заболеваниями, такими как болезнь Альцгеймера и Паркинсона, нейрогенез снижается еще в большей степени, чем у здоровых возрастных пациентов [83–85, 91]. Это критически сказывается на функционировании мозга в связи с важной ролью молодых нейронов в регенерации ткани мозга и в обучении. Снижение способности к регенерации, а также снижение уровня пластичности являются характерными проявлениями старения мозга. С другой стороны, внешние факторы, замедляющие возрастное угасание нейрогенеза, одновременно замедляют и снижение когнитивных способностей [42–45]. Кроме того, возрастное ухудшение памяти удается замедлить и даже обратить с помощью трансплантации искусственно полученных нервных стволовых клеток в стареющий мозг [44, 92]. Но этот подход имеет серьезные недостатки, такие как высокая инвазивность и риск развития раковой опухоли [92]. Поэтому сейчас усилия многих научных групп направлены на поиск способов усиления внутреннего нейрогенного потенциала мозга. В качестве потенциальных фармакологических средств, направленных на сохранение или восстановление высокого уровня нейрогенеза в стареющем мозге, исследуются регуляторные молекулы, такие как GDF11, TGF-β, антиоксиданты, такие как α-токоферол, и противовоспалительные препараты, как например индометацин [44]. Введение перечисленных факторов в организм стареющих животных приводит к частичному восстановлению нейрогенного потенциала и к улучшению когнитивных способностей [44]. Эти данные подтверждают, что существенную роль в возрастном снижении нейрогенеза играют нарушение регуляции эндогенной стволовой ниши, повышение уровня свободных радикалов и воспалительные процессы.
Однако перечисленные терапевтические воздействия в лучшем случае только на время замедляют или приостанавливают процесс старения мозга. Наши исследования показали, что первоначальное существенное усиление нейрогенеза, вызванное ишемией мозга, снижается на более длительных сроках наблюдения [82]. Поэтому задача предотвращения старения мозга и восстановления его регенерационного потенциала требует исследования фундаментальных механизмов старения организма. Хотя на данный момент не существует однозначного понимания механизмов старения, в качестве одного из наиболее вероятных механизмов рассматривается укорочение теломер в делящихся клетках.
Таким образом, эндогенная способность мозга к воспроизводству новых нейронов, связанная с существованием в мозге НСК, существенно снижается с возрастом и связана с нейродегенеративными заболеваниями. В свою очередь, способность НСК генерировать новые нейроны существенно зависит от работы теломеразы (рис. 1).
2. ДВОЙСТВЕННАЯ РОЛЬ ТЕЛОМЕРАЗЫ КАК ФАКТОРА, ЗАМЕДЛЯЮЩЕГО СТАРЕНИЕ И ПРОВОЦИРУЮЩЕГО РАК
2.1. Связь укорочения теломер со старением мозга
Белковые комплексы, которые осуществляют репликацию ДНК в эукариотических клетках, не способны эффективно реплицировать концы хромосом [93–96]. Поэтому в ходе последовательных клеточных делений происходит постепенное укорочение концевых участков хромосом – теломер [14–16]. Теломераза способна заново достраивать теломеры [22–26, 97]. Фермент теломераза состоит из двух субъединиц: TERC – молекула РНК, участок которой служит матрицей для синтеза теломерной ДНК; и TERT – обратная транскриптаза, синтезирующая теломерную ДНК на матрице TERC [98–101]. Ген РНК-компонента теломеразы TERC активно экспрессируется в большинстве тканей организма на всех этапах жизни [102]. Однако ген белкового компонента теломеразы TERT (который мы ниже для упрощения будем называть геном теломеразы) высоко активен только на этапе эмбриогенеза [27, 103–105]. Вскоре после рождения уровень экспрессии гена теломеразы резко падает, и в течение всей последующей жизни белок теломераза синтезируется, главным образом, в региональных стволовых клетках [27–32], в частности – в НСК в субвентрикулярной зоне и в гиппокампе [29, 106–108]. По этой причине в региональных стволовых клетках средняя длина теломер существенно выше, чем в дифференцированных клетках [31]. Редким исключением из этого правила являются взрослые нейроны, в которых наблюдается экспрессия теломеразы, с чем, вероятно, связано долгожительство этих клеток [109]. Однако в процессе последующего постнатального развития уровня экспрессии гена теломеразы в стволовых клетках оказывается недостаточно для того, чтобы полностью остановить укорочение теломер [30, 33–35, 110]. В частности, с возрастом происходит укорочение теломер в НСК в субвентрикулярной зоне, которое коррелирует с ослаблением нейрогенеза [31, 108] (рис. 1).
Укорочение теломер в стволовых клетках признано одним из основных механизмов старения [36]. Когда теломеры хромосом достигают критически малой длины, прекращаются клеточные деления, и клетка либо подвергается апоптозу, либо становится на путь клеточного старения с формированием характерного метаболизма и секреторного профиля стареющей клетки [17–21]. Истощение популяций способных к делениям клеток – прежде всего стволовых – приводит к постепенному снижению регенерационного потенциала во всех тканях организма [111]. В то же время стареющие клетки секретируют провоспалительные факторы [112]. В совокупности это определяет снижение качества жизни, характерное для пожилого возраста. Статистические данные демонстрируют сильную корреляцию процента клеток с укороченными теломерами с частотой встречаемости заболеваний, связанных с пожилым возрастом [113]. Кроме того, с укорочением теломер коррелирует риск развития неврологических заболеваний, таких как болезнь Альцгеймера [38], болезнь Паркинсона [39, 40], рассеянный склероз [114] и инсульт [41] (рис. 1).
2.2. Связь теломеразы с канцерогенезом
Осознание роли укорочения теломер при старении дало толчок работам, в которых исследовалась возможность замедлить или остановить старение с помощью искусственного повышения экспрессии гена теломеразы. Однако это направление исследований сразу столкнулось с серьезной проблемой: существенное повышение уровня экспрессии гена теломеразы вызывает развитие рака.
Дело в том, что белок теломераза, помимо его канонической роли в удлинении теломер, выполняет в клетке множество дополнительных функций, которые независимы от теломеразной активности, но могут способствовать канцерогенезу. Прежде всего теломераза взаимодействует с сигнальным путем wnt/β-catenin и участвует в активации ряда генов, ответственных за пролиферацию клеток, в частности генов c-Myc и Cyclin-D1. Кроме того, теломераза непосредственно активирует ген NF-kB и совместно с ним участвует в активации подконтрольных ему генов, которые связаны с возникновением устойчивости к апоптозу и с воспалением. Наконец, теломераза напрямую активирует ген VEGF, отвечающий за усиление роста сосудов. Одновременно по нескольким независимым механизмам теломераза вызывает у клетки устойчивость к факторам, сдерживающим рост, в частности к действию гормона TGF-β [49–52].
Таким образом, теломераза занимает положение одного из центральных регуляторов возникновения и поддержания раковой опухоли. Действительно, примерно 90% всех раковых опухолей сопровождаются повышением экспрессии гена теломеразы [49–52]. В частности, высокая теломеразная активность показана для глиомы [115]. Показано, что искусственное повышение экспрессии гена теломеразы провоцирует образование раковых опухолей, а последующее выключение этого гена приводит к прекращению их развития [46–48].
Необходимо подчеркнуть, что согласно многочисленным данным канцерогенный эффект теломеразы не зависит от удлинения теломер [49, 116]. Более того, у линии мышей с искусственно удлиненными теломерами не наблюдается повышения риска возникновения рака, и из этого следует, что само по себе удлинение теломер не является канцерогенным фактором [117]. Господствующее до недавнего времени мнение, что канцерогенным действием обладает само удлинение теломер, было основано на том, что для длительной и интенсивной пролиферации раковых клеток им необходима постоянная достройка теломер. Действительно, прекращение удлинения теломер в раковых клетках ведет к прекращению клеточных делений и к апоптозу [118–121]. Однако формирование раковой опухоли под действием белка теломеразы происходит еще до того как начинает наблюдаться удлинение теломер [48]. Более того, различные мутантные варианты теломеразы, а также ее альтернативные сплайс-варианты, не обладающие теломеразной активностью, также провоцируют рак, как и активная теломераза [116, 122–126]. В пользу этого свидетельствует и тот факт, что белок теломераза стимулирует пролиферацию в отсутствие РНК-компонента теломеразы TERC, без которого невозможна теломеразная активность [127].
2.3. Искусственное повышение экспрессии гена теломеразы: возможности и ограничения
В первых работах, выполненных на линии трансгенных мышей с повышенной экспрессией гена теломеразы, смертность животных была повышена из-за развития множественных опухолей [46–48, 128]. Тем не менее для таких животных была продемонстрирована повышенная максимальная продолжительность жизни, а в тканях этих животных наблюдалось повышение средней длины теломер [15, 129]. Это мотивировало исследователей искать подходы к тому, чтобы каким-то образом компенсировать канцерогенный эффект теломеразы. Первая удачная работа была проведена Tomas-Loba с соавт. (2008) [39], создавшими трансгенных мышей с геном теломеразы под контролем промотора кератина, которые в то же время были онкоустойчивыми, потому что имели в геноме тройную дозу генов онкосупрессоров p-53, p-16 и Arf. У этих мышей наблюдался повышенный синтез теломеразы в эпителиальных тканях, прежде всего в стволовых клетках эпителия, в связи с чем в клетках эпителия была повышена средняя длина теломер и снижен процент критически коротких теломер. Эти животные жили в среднем примерно на 40% дольше, чем контрольные животные, и у них не наблюдалось повышения вероятности развития рака [39].
Bernardes de Jesus с соавт. (2012) нашли другое решение этой проблемы [53]. Они вводили экспериментальным животным ген теломеразы в составе нереплицирующегося аденоассоциированного вируса. Аденоассоциированные вирусы характеризуются слабой вероятностью интеграции в геном зараженной клетки. Поэтому повышенная экспрессия гена теломеразы у зараженных таким вирусом животных не может существенно поддерживать развитие рака: деление клетки, которое может быть спровоцировано экспрессией теломеразы в зараженной клетке, с высокой вероятностью приводит к потере вирусной ДНК дочерними клетками и не приводит к увеличению копийности вируса. Следовательно, в делящихся клетках экспрессия теломеразы неизбежно затухает. Однократное внутривенное введение такого вируса мышам привело к повышению средней длины теломер в клетках различных органов (в том числе в мозге) и повысило среднюю и максимальную продолжительность жизни на 20% при введении в возрасте одного года и на 13% – при введении в возрасте двух лет. Вместе с тем не наблюдалось повышения риска развития раковых опухолей [53]. Эта же группа в дальнейшем продемонстрировала, что данный метод генной терапии способствует эффективному восстановлению сердца после инфаркта [54]. Кроме того, еще в одной работе было показано, что введение данного вируса с геном теломеразы не ускоряет прогрессию уже имеющейся опухоли в легком. Полученный факт свидетельствует в пользу того, что данный метод терапии не будет повышать риск развития рака даже у тех индивидов, в тканях которых уже имеются предпосылки к канцерогенезу [55].
С точки зрения роли теломеразы в нейрогенезе особенно интересны работы, в которых в гиппокамп взрослых животных вводили не способный к интеграции в геном аденовирус с геном теломеразы. В результате происходило усиление нейрогенеза и повышение когнитивных способностей без повышения риска развития рака [56, 107]. Упомянутые исследования ясно продемонстрировали, что изменение экспрессии гена теломеразы имеет значительный терапевтический потенциал как средство замедления старения и, в частности, замедления старения мозга через поддержание эндогенной популяции НСК.
Тем не менее разработанные подходы имеют очевидные ограничения. Безопасное, с точки зрения развития рака, введение гена теломеразы в составе не способных к интеграции вирусов может обеспечить только временное удлинение теломер в популяции стволовых клеток, поскольку в процессе клеточных делений привнесенный ген теломеразы будет утрачиваться. С другой стороны, встройка в геном стволовых клеток конструкции с геном теломеразы требует дополнительных мер по предотвращению рака. Учитывая эти результаты, можно предложить принципиально иную стратегию удлинения теломер в стволовых клетках, связанную с альтернативным сплайсингом теломеразы, не примененную пока ни в одном из проведенных исследований.
3. ЗНАЧЕНИЕ АЛЬТЕРНАТИВНОГО СПЛАЙСИНГА ТЕЛОМЕРАЗЫ
Альтернативный сплайсинг является одним из источников разнообразия белков у эукариот. Поскольку в эукариотических генах последовательность, кодирующая белок, поделена на экзоны, которые чередуются с интронами, избирательное вырезание интронов и сшивка экзонов могут приводить в конечном счете к разным вариантам аминокислотной последовательности белков, различающимся по функциональным свойствам [130].
В гене теломеразы 16 экзонов и 15 интронов, что создает возможность синтеза очень большого числа вариантов белка теломеразы [131] (рис. 2). Сейчас известно 22 варианта теломеразы, и из них только один вариант обладает теломеразной активностью – способен достраивать теломеры. Это так называемая полноразмерная теломераза, которая содержит все экзоны в правильной последовательности, без вставок интронов [132–138]. Больший процент единиц белка теломеразы в стволовых и раковых клетках приходится на β-вариант, в котором отсутствуют экзоны 7 и 8, а также сдвигается рамка считывания, благодаря чему появляется преждевременный стоп-кодон [135, 137, 139–143]. Доля полноразмерной теломеразы в ряде работ оценивается примерно в 5% [102, 134, 135]. Таким образом, подавляющее большинство единиц белка теломеразы в клетке не способны достраивать теломеры. Кроме того, многие из альтернативных вариантов теломеразы ингибируют каталитическую активность полноразмерного варианта – препятствуют удлинению теломер [141–146]. В частности, ингибитором теломеразной активности является наиболее представленный β-вариант [141–143].
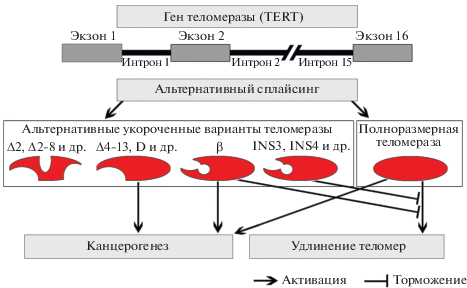
Ингибирование полноразмерной теломеразы ее альтернативными формами осуществляется по множеству механизмов. β-вариант связывается с матрицей TERC и поэтому конкурирует с полноразмерной теломеразой за связывание с TERC [141]. Однако, поскольку TERC в избытке синтезируется в большинстве клеток [102], многие авторы предполагают, что такая конкуренция не должна быть ограничивающим фактором [135, 137]. С другой стороны, было высказано предположение о том, что β-вариант может осуществлять ингибирование и по другому механизму [143]. Показано, что теломераза осуществляет удлинение теломер, действуя в виде димера. В белке теломеразы имеется специальный домен для связывания с другим белком теломеразы. Этот домен присутствует в β-варианте. По этой причине β-вариант может ингибировать активность полноразмерной теломеразы, ассоциируясь с ней и конкурируя с другими единицами полноразмерной формы за ассоциацию [143]. Известно еще несколько вариантов теломеразы, затрудняющих удлинение теломер, в их числе варианты α, INS3 и INS4, которые конкурируют с полноразмерной теломеразой за связывание с теломерной ДНК, и γ-вариант, ингибирующий теломеразную активность по неизвестному механизму [136, 144–146].
На поздних этапах эмбрионального развития происходит сдвиг баланса в сторону большей представленности неактивных ингибирующих форм теломеразы. С этим сдвигом коррелирует начало укорочения теломер в клетках [139]. Во взрослых стволовых клетках небольшой доли теломеразы, с учетом наличия ингибирующих форм, оказывается недостаточно для поддержания стабильной длины теломер. Происходят постепенное укорочение теломер в стволовых клетках и снижение потенциала популяций стволовых клеток к самообновлению и к регенерации соматических тканей [30, 33–35, 110], в частности происходит истощение потенциала НСК в мозге [31, 108].
Хотя только один вариант теломеразы – полноразмерная форма – способна удлинять теломеры, многие из альтернативных форм обладают, как и полноразмерная теломераза, канцерогенным эффектом [49, 126, 141] (рис. 2). В частности, вариант ∆4-13, как и полноразмерная теломераза, стимулирует пролиферацию, а β-вариант подавляет апоптоз [126, 141].
Регуляция сплайсинга теломеразы обладает гомеостатичностью: попытка искусственно сдвинуть баланс в сторону полноразмерной формы может вызвать компенсирующие сдвиги в сплайсинге. Kim (2017) [143] обнаружил в культуре клеток, что введение вектора, экспрессирующего полноразмерную теломеразу (в введенном гене теломеразы были удалены интроны) под контролем доксициклинового промотора, приводит к сдвигу в сплайсинге внутреннего гена теломеразы в сторону ингибирующего β-варианта. В результате введение вектора с геном полноразмерной теломеразы привело не к повышению, а к снижению теломеразной активности [143]. Radan с соавт. (2014) [147] обнаружили, что искусственное подавление сплайсинга, ведущего к образованию вариантов β и α, вопреки ожиданиям, не повышает, а снижает теломеразную активность, что, как предполагают авторы, может быть связано с компенсаторным усилением сплайсинга на других участках теломеразной РНК [147].
4. НОВЫЙ ПОДХОД К УСИЛЕНИЮ ТЕЛОМЕРАЗНОЙ АКТИВНОСТИ: УДАЛЕНИЕ ИНТРОНОВ ИЗ ГЕНА ТЕЛОМЕРАЗЫ
Описанные в предыдущих разделах аспекты биологии теломеразы необходимо учитывать при разработке подходов к искусственному усилению теломеразной активности. В разделе 2.3 были упомянуты работы по введению в организм дополнительного гена теломеразы с целью повышения теломеразной активности и удлинения теломер. Во всех этих исследованиях привнесенный ген теломеразы был лишен интронов и, как следствие, образовывал только полноразмерную теломеразу. Тем не менее даже в работе Tomas-Loba с соавт., где привнесенный ген полноразмерной теломеразы экспрессировался во всех эпителиальных клетках, в них все равно происходило укорочение теломер, хотя оно и существенно замедлилось по сравнению с контролем [39]. Вероятная причина низкой эффективности этой стратегии была в том, что в геноме оставался также и дикий ген теломеразы, экспрессия которого приводила к образованию ингибирующих форм. Присутствие этих форм теломеразы должно было снизить эффективность работы привнесенной теломеразы, особенно если предположить, что избыток полноразмерной формы дал толчок к компенсаторному сдвигу в сплайсинге дикого гена теломеразы, как было показанов работе Kim [143]. Более того, есть основания предполагать, что экспрессия привнесенного гена теломеразы могла усилить экспрессию дикого гена теломеразы. Исследования функциональных связей гена теломеразы с другими генами показывают, что в регуляции активности гена теломеразы имеется много петель с положительной обратной связью, когда ген, который активируется теломеразой, в свою очередь напрямую и через посредников повышает активность гена теломеразы. В частности, такие связи есть между геном теломеразы и генами c-Myc1, β-catenin, NF-kB и VEGF [50]. Таким образом, присутствие в геноме экспериментальных животных дикого гена теломеразы может существенно снижать эффект от работы привнесенного гена.
Задача по специальному подавлению альтернативного сплайсинга теломеразы представляется трудновыполнимой в связи с большим числом интронов в гене теломеразы и существованием компенсаторной регуляции ее сплайсинга [143, 147]. Поэтому наиболее перспективной стратегией представляется редактирование дикого гена теломеразы: замена в геноме животных дикой кодирующей последовательности, содержащей интроны, на кодирующую последовательность с удаленными интронами. Это приведет к тому, что в стволовых клетках будет синтезироваться только полноразмерная теломераза, что при сохранении обычного уровня экспрессии гена теломеразы приведет к повышению эффективности удлинения теломер. Сохранение умеренного уровня экспрессии гена теломеразы означает сохранение низкого риска развития рака. Более того, следует принять во внимание, что развитие рака является не следствием теломеразной активности, а результатом дополнительных функций теломеразы, которые выполняются не только активной теломеразой, но также и многими ее альтернативными сплайс-вариантами. Поэтому следует ожидать, что 100%-ный сдвиг в сторону активной полноразмерной теломеразы не приведет к существенному повышению риска развития раковой опухоли.
Важно также принять во внимание, что эффективная работа теломеразы не означает неконтролируемое удлинение теломер. В эмбриональных стволовых, а также в раковых клетках, где работа теломеразы эффективно удлиняет теломеры, активируются специальные механизмы, предотвращающие возникновение слишком длинных теломер [148, 149]. Эти механизмы пока не полностью изучены, но известно, что в них задействован белок TZAP1, который связывается с теломерной ДНК, конкурируя за связывание с белком комплекса shelterin TRF1 и TRF2 [150, 151]. Поэтому есть основания ожидать, что достижение во взрослых стволовых клетках той же эффективности удлинения теломер, которая имеет место в эмбриональных стволовых и раковых клетках, запустит те же самые стабилизирующие механизмы и в результате во взрослых стволовых клетках будет стабильно поддерживаться оптимальная длина теломер.
Несмотря на приведенные выше аргументы в пользу предлагаемой стратегии, следует подчеркнуть, что речь идет о достаточно сильном вмешательстве в важные и тонкие молекулярно-генетические процессы, детальные механизмы которых не полностью изучены. Поэтому только эксперименты могут показать реальную перспективность или ошибочность данного подхода.
Таким образом, предлагаемая стратегия генной терапии старения мозга состоит в том, чтобы вводить в организм генетические векторы, которые будут в НСК заменять исходный ген теломеразы на ген с удаленными интронами. Есть основания полагать, что это вмешательство существенно повысит эффективность удлинения теломер и даже может привести к стабилизации их длины, что существенно замедлит старение мозга в связи с усилением нейрогенного потенциала.
5. СОВРЕМЕННЫЕ МЕТОДЫ ГЕННОЙ ИНЖЕНЕРИИ
Сформулированная задача соответствует современному уровню генной инженерии. В этом разделе не будет полного описания существующих генно-инженерных методов. Вместо этого мы обсудим подходы, которые могут быть применены для решения сформулированной задачи.
5.1. Типы и серотипы вирусных векторов
В исследованиях центральной нервной системы методами генной инженерии широко используются генетические векторы на основе аденоассоциированных вирусов, аденовирусов, HIV лентивируса и MLV ретровируса. Использование аденоассоциированных векторов предпочтительно, поскольку они признаны наиболее безопасными в получении и использовании, поскольку они вызывают слабый иммунный ответ и с низкой вероятностью интегрируются в геном [152–155]. Недостатком этих вирусов является низкая пакующая способность, но, как будет описано ниже, это не является большим препятствием для их использования в целях редактирования генов in vivo.
Группа аденоассоциированных векторов включает порядка 100 серотипов, но наиболее широко применяются серотипы AAV2/1, AAV2/2, AAV2/4, AAV2/5, AAV2/6, AAV2/8, AAV2/9 [152, 153]. Серотипы различаются по своей способности внедряться в те или иные виды клеток. Одна из целей создания новых серотипов состоит в том, чтобы обеспечить попадание вируса в клетки того или иного типа. К настоящему моменту уже созданы два серотипа аденоассоциированных вирусов, AAV r3.45 и SCH9, которые с высокой специфичностью и эффективностью заражают нервные стволовых клетки в мозге [156–158]. Авторы, создавшие эти серотипы, вводили их животным непосредственно в мозг, интрацеребрально [157, 158]. Пока неизвестно, могут ли эти серотипы проникать через гематоэнцефалический барьер – и, соответственно, можно ли их также использовать для внутривенного введения. Подчеркнем, что внутривенное введение крайне желательно для возможности внедрения нового вида генной терапии в клинику. С другой стороны, существует серотип AAV2/9, который проникает через гематоэнцефалический барьер и с высокой эффективностью заражает клетки мозга после внутривенного введения [154]. К сожалению, он эффективно заражает нейроны, астроциты, но в меньшей степени нервные стволовые клетки [154]. Тем не менее этот серотип тоже может быть использован для редактирования гена теломеразы в мозге – потому что астроциты в мозге способны в некоторых условиях терять дифференцированное состояние и приобретать стволовость [78, 79]. Следовательно, можно допустить, что редактирование гена теломеразы и удлинение теломер в небольшом числе астроцитов приведут к формированию в мозге стволовой популяции, происходящей из бывших астроцитов. В недавней работе Smith с соавт. (2018) [159] удалось выделить серотип аденоассоциированного вируса AAVHSC, который обладает рядом преимуществ для выполнения сформулированной задачи. Во-первых, векторы на основе этого серотипа проникают через гематоэнцефалический барьер и эффективно заражают клетки мозга после внутривенной инъекции [159]. Во-вторых, авторами продемонстрирован новый, простой в исполнении (без помощи эндонуклеаз, см. ниже) способ редактирования генома с помощью вектора этого серотипа.
Следует отметить, что, хотя внутривенное введение вирусных векторов безусловно предпочтительнее для клиники по сравнению с прямым введением в мозг, его минусом является возможность побочного заражения различных клеток вне мозга. Например, серотип AAV2/9, эффективно заражающий нейроны и астроциты в мозге, также заражает широкий спектр клеток в различных органах [152]. Решение данной проблемы в будущем будет определяться разработкой новых высокоспецифичных серотипов вирусных векторов, обеспечивающих специфичность заражения НСК даже после внутривенного введения.
5.2. Методы редактирования генома
Наиболее часто применяемым механизмом редактирования генов является использование сайт-специфичных эндонуклеаз [155]. Сайт-специфичные эндонуклеазы разрезают ДНК в определенном месте, которое они распознают с той или иной степенью специфичности. Из четырех применяемых сайт-специфичных эндонуклеаз – мегануклеазы, ZFN, TALEN и CRISPR/Cas9 – последняя является наиболее легкой в конструировании при высокой специфичности распознавания сайта рестрикции, которая достигается благодаря комплементарному спариванию ДНК целевого сайта с направляющей РНК [155]. Фермент CRISPR/Cas9, полученный из бактерии Streptococcus pyogenes, чаще применяется, но размер его гена слишком велик для встройки в аденоассоциированный вирус [155]. Фермент CRISPR/Cas9, полученный из бактерии Staphylococcus aureus, существенно компактнее и помещается в аденоассоциированный вирус, в связи с чем он более перспективен для редактирования генов in vivo [160].
После того как эндонуклеаза распознает нужный сайт и разрезает молекулу ДНК, в ответ запускаются эндогенные репарационные механизмы, которые заново сшивают образовавшиеся концы. Однако этот процесс может быть модифицирован, если в клетке присутствует экстрахромосомная ДНК, имеющая участки, комплементарные сайту разрыва. В этом случае с некоторой вероятностью репарация происходит вместе со встраиванием этой ДНК. Экстрахромосомной ДНК может быть ДНК аденоассоциированного вируса, несущая генетическую последовательность, которую требуется вставить в данный участок генома. Последовательность, предназначенная для вставки в сайт разрезания эндонуклеазой, в вирусе помещается между двумя участками, комплементарными соответствующим участкам по обе стороны от разрыва в геномной ДНК. Это обеспечивает комплементарное спаривание ДНК вируса с геномной ДНК по обе стороны от разрыва, а затем и встройку целевой последовательности в разрыв [155].
Описанная выше система позволяет эффективно редактировать гены как in vitro, так и in vivo. Однако ее работа в клетке может иметь нежелательные побочные следствия. Во-первых, с некоторой вероятностью возможно образование разрывов ДНК в незапланированных сайтах, с неизвестными последствиями для жизнедеятельности клетки. Во-вторых, разрыв ДНК с некоторой вероятностью может завершаться не встройкой целевой ДНК, а репарацией с возникновением небольших вставок или делеций, что может нарушать функцию того локуса, куда планируется встройка [155].
Альтернативой является редактирование гена без использования эндонуклеаз. Это может быть осуществлено с использованием одной только вирусной ДНК, имеющей частичную комплементарность к определенному участку генома. Эта ДНК должна иметь такую же структуру, как и в методе с эндонуклеазами. Последовательность, предназначенная для встройки в геном, в вирусе помещается между двумя локусами, которые комплементарны двум локусам в геноме. Если в геноме эти локусы следуют сразу один за другим, то результатом может быть встройка целевой последовательности между ними. Если в геноме эти локусы разделены участком, то результатом может быть замена данного участка на целевую последовательность [159, 161–163]. В недавней работе Smith с соавт. (2018) [159] удалось применить эту технологию с использованием AAVHSC серотипа аденоассоциированного вируса, который без помощи эндонуклеаз обеспечивает встройку целевых фрагментов in vivo с беспрецедентной частотой и точностью. Преимущество данного метода, по сравнению с эндонуклеазным, состоит в высокой безопасности (крайне низкая вероятность незапланированных изменений в геноме), а также в простоте выполнения: генно-инженерная система, которую необходимо доставить в клетку, состоит из одного компонента – ДНК аденоассоциированного вируса AAVHSC со встроенной целевой последовательностью. Данный серотип эффективен в отношении широкого спектра тканей и органов (в том числе мозга), эффективен в отношении как делящихся, так и неделящихся клеток. Тем не менее следует иметь в виду, что данный метод пока был применен только одной научной группой, и его эффективность требует дальнейшей проверки.
Таким образом, наиболее предпочтительным методом для редактирования гена теломеразы в нервных стволовых клетках in vivo, особенно с учетом дальнейших перспектив использования в клинике, по-видимому, является описанный выше альтернативный метод редактирования генома без использования эндонуклеаз [159].
ЗАКЛЮЧЕНИЕ
Усиление нейрогенного потенциала взрослого мозга – важнейшее направление исследований в современной биологии и биомедицине. Поскольку укорочение теломер признано одним из главных факторов, ограничивающих регенерационный потенциал организма в целом и мозга – в частности, перспективной задачей представляется искусственное повышение теломеразной активности in vivo. Попытки решить эту задачу через повышение уровня экспрессии гена теломеразы столкнулись с проблемой канцерогенеза. В то же время существует иной путь повышения теломеразной активности – через отмену альтернативного сплайсинга теломеразы. Этот подход в будущем может позволить повышать теломеразную активность в нервных стволовых клетках мозга без повышения риска возникновения рака. Развитие данной стратегии в будущем может стать основой для разработки нового направления в генной терапии.
Авторы благодарят С.В. Кильдяеву за профессиональный перевод аннотации статьи, а также Т.В. Ананьину за подготовку превосходных иллюстраций.
Работа выполнена при поддержке гранта РНФ, проект № 18-15-00229, и государственного задания, проект № 18.2583.2017/4.6.
Настоящая статья не содержит каких-либо исследований с участием в качестве объекта животных.
Настоящая статья не содержит каких-либо исследований с участием в качестве объекта людей.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Abrous D.N., Koehl M., Le Moal M. Adult neurogenesis: from precursers to network and physiology // Physiol. Rev. 2005. V. 85. № 322. P. 523–569. https://doi.org/10.1152/physrev.00055.2003
Kee N., Teixeira C.M., Wang A.H., Frankland P.W. Preferential incorporation of adult-generated granule cells into spatial memory networks in the dentate gyrus // Nat. Neurosci. 2007. V. 10. № 3. P. 355–362. https://doi.org/10.1038/nn1847
Tashiro A., Makino H., Gage F.H. Experience-specific functional modification of the dentate gyrus through adult neurogenesis: A critical period during an immature stage // J. Neurosci. 2007. V. 27. № 12. P. 3252–3259. https://doi.org/10.1523/jneurosci.4941-06.2007
Trouche S., Bontempi B., Roullet P., Rampon C. Recruitment of adult-generated neurons into functional hippocampal networks contributes to updating and strengthening of spatial memory // Proc. Natl Acad. Sci. USA. 2009. V. 106. № 14. P. 5919–5924. https://doi.org/10.1073/pnas.0811054106
Wang S., Scott B.W., Wojtowicz J.M. Heterogenous properties of dentate granule neurons in the adult rat // J. Neurobiol. 2000. V. 42. № 2. P. 248–257.
Schmidt-Hieber C., Jonas P., Bischofberger J. Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus // Nature. 2004. V. 429. № 6988. P. 184–187. https://doi.org/10.1038/nature02553
Zhao C., Teng E.M., Summers Jr. et al. Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus // J. Neurosci. 2006. V. 26. № 1. P. 3–11. https://doi.org/10.1523/jneurosci.3648-05.2006
Ge S., Yang C., Hsu K. et al. A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain // Neuron. 2007. V. 54. № 4. P. 559–566. https://doi.org/10.1016/j.neuron.2007.05.002
Toni N., Teng E.M., Bushong E.A. et al. Synapse formation on neurons born in the adult hippocampus // Nat. Neurosci. 2007. V. 10. № 6. P. 727–734. https://doi.org/10.1038/nn1908
Toni N., Laplagne D.A., Zhao C. et al. Neurons born in the adult dentate gyrus form functional synapses with target cells // Nat. Neurosci. 2008. V. 11. № 8. P. 901–907. https://doi.org/10.1038/nn.2156
Faulkner R.L., Jang M.-H., Liu X.-B. et al. Development of hippocampal mossy fiber synaptic outputs by new neurons in the adult brain // Proc. Natl Acad. Sci. USA. 2008. V. 105. № 37. P. 14157–14162. https://doi.org/10.1073/pnas.0806658105
Kron M.M., Zhang H., Parent J.M. The developmental stage of dentate granule cells dictates their contribution to seizure-induced plasticity // J. Neurosci. 2010. V. 30. № 6. P. 2051–2059. https://doi.org/10.1523/jneurosci.5655-09.2010
Encinas J.M., Michurina T.V., Peunova N. et al. Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. // Cell Stem Cell. Elsevier Inc. 2011. V. 8. № 5. P. 566–579. https://doi.org/10.1016/j.stem.2011.03.010
Blackburn E.H. The end of the (DNA) line // Nat. Struct. Biol. 2000. V. 7. № 10. P. 847–850. https://doi.org/10.1038/79594
Blasco M.A. Telomeres and human disease: Ageing, cancer and beyond // Nat. Rev. Genet. 2005. V. 6. № 8. P. 611–622. https://doi.org/10.1038/nrg1656
Sfeir A.J., Chai W., Shay J.W., Wright W.E. Telomere-end processing: The terminal nucleotides of human chromosomes // Mol. Cell. 2005. V. 18. № 1. P. 131–138. https://doi.org/10.1016/j.molcel.2005.02.035
Dhabhar F.S., Morrow J.D., Adler N.E. et al. Accelerated telomere shortening in response to life stress // Proc. Natl Acad. Sci. USA. 2004. V. 101. № 49. P. 17312–17315. https://doi.org/10.1073/pnas.0407162101
Herbig U., Jobling W.A., Chen B.P.C. et al. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21CIP1, but not p16INK4a // Mol. Cell. 2004. V. 14. № 4. P. 501–513. https://doi.org/10.1016/s1097-2765(04)00256-4
Canela A., Vera E., Klatt P., Blasco M.A. High-throughput telomere length quantification by FISH and its application to human population studies // Proc. Natl Acad. Sci. USA. 2007. V. 104. № 13. P. 5300–5305. https://doi.org/10.1073/pnas.0609367104
Jiang H., Ju Z., Rudolph K.L. Telomere shortening and ageing // Z. Gerontol. Geriatr. 2007. V. 40. № 5. P. 314–324. https://doi.org/10.1007/s00391-007-0480-0
Jesus B.B. De, Blasco M.A. Assessing cell and organ senescence biomarkers // Circ. Res. 2016. V. 111. № 1. P. 97–109. https://doi.org/10.1161/circresaha.111.247866
Greider C.W., Blackburn E.H. Identification of a specific telomere terminal transferase activity in tetrahymena extracts // Cell. 1985. V. 43. № 2. P. 405–413. https://doi.org/10.1016/0092-8674(85)90170-9
Morin G.B. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats // Cell. 1989. V. 59. № 3. P. 521–529. https://doi.org/10.1016/0092-8674(89)90035-4
Hackett J.A., Greider C.W. Balancing instability: Dual roles for telomerase and telomere dysfunction in tumorigenesis // Oncogene. 2002. V. 21. № 4. P. 619–626. https://doi.org/10.1038/sj/onc/1205061
Fajkus J. Detection of telomerase activity by the TRAP assay and its variants and alternatives // Clin. Chim. Acta. 2006. V. 371. № 1–2. P. 25–31. https://doi.org/10.1016/j.cca.2006.02.039
Gallardo F., Laterreur N., Cusanelli E. et al. Live cell imaging of telomerase RNA dynamics reveals cell cycle-dependent clustering of telomerase at elongating telomeres // Mol. Cell. Elsevier Inc. 2011. V. 44. № 5. P. 819–827. https://doi.org/10.1016/j.molcel.2011.09.020
Wright W.E., Piatyszek M.A., Rainey W.E. et al. Telomerase activity in human germline and embryonic tissues and cells // Dev. Genet. 1996. V. 18. № 2. P. 173–179. https://doi.org/10.1002/(sici)1520-6408(1996)18:2< 173::aid-dvg10>3.0.co;2-3
Greenberg R.A., Allsopp R.C., Chin L. et al. Expression of mouse telomerase reverse transcriptase during development, differentiation and proliferation // Oncogene. 1998. V. 16. № 13. P. 1723–1730. https://doi.org/10.1038/sj.onc.1201933
Martin-Rivera L., Herrera E., Albar J.P., Blasco M.A. Expression of mouse telomerase catalytic subunit in embryos and adult tissues // Proc. Natl Acad. Sci. USA. 2002. V. 95. № 18. P. 10471–10476. https://doi.org/10.1073/pnas.95.18.10471
Flores I., Benetti R., Blasco M.A. Telomerase regulation and stem cell behaviour // Curr. Opin. Cell Biol. 2006. V. 18. № 3. P. 254–260. https://doi.org/10.1016/j.ceb.2006.03.003
Flores I., Canela A., Vera E. et al. The longest telomeres: A general signature of adult stem cell compartments // Genes Dev. 2008. V. 22. № 5. P. 654–667. https://doi.org/10.1101/gad.451008
van der Harst P., van Veldhuisen D.J., Samani N.J. Expanding the concept of telomere dysfunction in cardiovascular disease // Arterioscler. Thromb. Vasc. Biol. 2008. V. 28. № 5. P. 807–808. https://doi.org/10.1161/atvbaha.108.164434
Vaziri H., Dragowska W., Allsopp R.C. et al. Evidence for a mitotic clock in human hematopoietic stem cells: Loss of telomeric DNA with age // Proc. Natl Acad. Sci. USA. 1994. V. 91. № 21. P. 9857–9860.
Herbig U., Ferreira M., Condel L. et al. Cellular senescence in aging primates // Science. 2006. V. 311. № 5765. P. 1257. https://doi.org/10.1126/science.1122446
Seeger F., Dietz K., Hoffmann J. et al. Telomere gap between granulocytes and lymphocytes is a determinant for hematopoetic progenitor cell impairment in patients with previous myocardial infarction // Arterioscler. Thromb. Vasc. Biol. 2008. V. 28. № 5. P. 968–974. https://doi.org/10.1161/atvbaha.107.160846
López-otín C., Blasco M.A., Partridge L., Serrano M. The Hallmarks of Aging // Cell. 2013. V. 153. № 6. P. 1194–1217. https://doi.org/10.1016/j.cell.2013.05.039
Blasco M.A., Marques-Torrejon M.A., Mira H. et al. Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis // J. Neurosci. 2009. V. 29. № 46. P. 14394–14407. https://doi.org/10.1523/jneurosci.3836-09.2009
Forero D.A., González-Giraldo Y., López-Quintero C. et al. Meta-analysis of telomere length in Alzheimer’s disease // J. Gerontol. A. Biol. Sci. Med. Sci. 2016. V. 71. № 8. P. 1069–1073. https://doi.org/10.1093/gerona/glw053
Tomás-Loba A., Flores I., Fernández-Marcos P.J. et al. Telomerase reverse transcriptase delays aging in cancer-resistant mice // Cell. 2008. V. 135. № 4. P. 609–622. https://doi.org/10.1016/j.cell.2008.09.034
Guan J.Z., Maeda T., Sugano M. et al. A percentage analysis of the telomere length in Parkinson’s disease patients // J. Gerontol. A. Biol. Sci. Med. Sci. 2008. V. 63. № 5. P. 467–473. https://doi.org/10.1093/gerona/63.5.467
Jin X., Pan B., Dang X. et al. Relationship between short telomere length and stroke: A meta-analysis // Medicine (Baltimore). 2018. V. 97. № 39. P. 1–7. https://doi.org/10.1097/md.0000000000012489
Ma C.L., Ma X.T., Wang J.J. et al. Physical exercise induces hippocampal neurogenesis and prevents cognitive decline // Behav. Brain Res. 2017. V. 317. P. 332–339. https://doi.org/10.1016/j.bbr.2016.09.067
Fidaleo M., Cavallucci V., Pani G. Nutrients, neurogenesis and brain ageing: From disease mechanisms to therapeutic opportunities // Biochem. Pharmacol. 2017. V. 141. P. 63–76. https://doi.org/10.1016/j.bcp.2017.05.016
Apple D.M., Solano-Fonseca R., Kokovay E. Neurogenesis in the aging brain // Biochem. Pharmacol. 2017. V. 141. P. 77–85. https://doi.org/10.1016/j.bcp.2017.06.116
Katsimpardi L., Lledo P.M. Regulation of neurogenesis in the adult and aging brain // Curr. Opin. Neurobiol. 2018. V. 53. P. 131–138. https://doi.org/10.1016/j.conb.2018.07.006
Gonzalez-Suarez E., Samper E., Ramirez A. et al. Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes // Eur. Mol. Biol. Organ. J. 2001. V. 20. № 11. P. 2619–2630
Artandi S.E., Alson S., Tietze M.K. et al. Constitutive telomerase expression promotes mammary carcinomas in aging mice // Proc. Natl Acad. Sci. USA. 2002. V. 99. № 12. P. 8191. https://doi.org/10.1073/pnas.112515399
Canela A., Martin-Caballero J., Flores J.M., Blasco M.A. constitutive expression of tert in thymocytes leads to increased incidence and dissemination of T-cell lymphoma in lck-tert mice // Mol. Cell. Biol. 2004. V. 24. № 10. P. 4275–4293. https://doi.org/10.1128/mcb.24.10.4275-4293.2004
Low K.C., Tergaonkar V. Telomerase: Central regulator of all of the hallmarks of cancer // Trends Biochem. Sci. 2013. V. 38. № 9. P. 426–434. https://doi.org/10.1016/j.tibs.2013.07.001
Tian Y., He Y., Liu X. et al. Feedback regulation of telomerase reverse transcriptase: new insight into the evolving field of telomerase in cancer // Cell. Signal. 2013. V. 25. № 12. P. 2462–2468. https://doi.org/10.1016/j.cellsig.2013.08.009
Jesus B.B. De, Blasco M.A. Telomerase at the intersection of cancer and aging // Trends Genet. 2014. V. 29. № 9. P. 513–520. https://doi.org/10.1016/j.tig.2013.06.007.telomerase
Terali K., Yilmazer A. New surprises from an old favourite: The emergence of telomerase as a key player in the regulation of cancer stemness // Biochimie. 2016. V. 121. P. 170–178. https://doi.org/10.1016/j.biochi.2015.12.001
Jesus B., Vera E., Schneeberger K. et al. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer // EMBO Mol. Med. 2012. V. 4. № 8. P. 691–704. https://doi.org/10.1002/emmm.201200245
Bär C., Serrano R., Pisano D. et al. Telomerase expression confers cardioprotection in the adult mouse heart after acute myocardial infarction // Nat. Commun. 2014. V. 5. № 1. P. 1–14. https://doi.org/10.1038/ncomms6863
Muñoz-Lorente M.A., Martínez P., Tejera Á. et al. AAV9-mediated telomerase activation does not accelerate tumorigenesis in the context of oncogenic K‑Ras-induced lung cancer // PLoS Genet. 2018. V. 14. № 8. P. 1–25. https://doi.org/10.1371/journal.pgen.1007562
Zhou Q.G., Liu M.Y., Lee H.W. et al. Hippocampal TERT regulates spatial memory formation through modulation of neural development // Stem Cell Reports. 2017. V. 9. № 2. P. 543–556. https://doi.org/10.1016/j.stemcr.2017.06.014
Gage F.H. Mammalian neural stem cells // Science. 2000. V. 287. № 5457. P. 1433–1438. https://doi.org/10.1126/science.287.5457.1433
Duan X., Kang E., Liu C.Y. et al. Development of neural stem cell in the adult brain // Curr. Opin. Neurobiol. 2008. V. 18. № 1. P. 108–115. https://doi.org/10.1016/j.conb.2008.04.001
Imayoshi I., Sakamoto M., Ohtsuka T. et al. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain // Nat. Neurosci. 2008. V. 11. № 10. P. 1153–1161. https://doi.org/10.1038/nn.2185
Nakatomi H., Nakatomi H., Kuriu T. et al. Regeneration of hippocampal pyramidal neurons afetr ischemic brain injury by recruitnt of endogenous neural progenitors // Cell. 2002. V. 110. P. 429–441.
Dayer A.G., Cleaver K.M., Abouantoun T., Cameron H.A. New GABAergic interneurons in the adult neocortex and striatum are generated from different precursors // J. Cell Biol. 2005. V. 168. № 3. P. 415–427. https://doi.org/10.1083/jcb.200407053
Sundholm-Peters N.L., Yang H.K., Goings G.E. et al. Subventricular zone neuroblasts emigrat toward cortical lesions. // J. Neuropathol. Exp. Neurol. 2005. V. 64. № 12. P. 1089–1100.
Shapiro L.A., Ng K., Zhou Q.-Y., Ribak C.E. SVZ-derived newly generated neurons populate several olfactory and limbic forebrain regions // Magn. Reson. Imaging. 2009. V. 31. № 3. P. 477–479. https://doi.org/10.1016/j.immuni.2010.12.017
Jin K., Sun Y., Xie L. et al. Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum // Mol. Cell. Neurosci. 2003. V. 24. № 1. P. 171–189. https://doi.org/10.1016/s1044-7431(03)00159-3
Zhang P., Liu Y., Li J. et al. Ependymal/subventricular zone cells migrate tothe peri-infarct region and differentiate into neurons and astrocytes after focal cerebral ishemia in adult rats // J. First Mil. Med. 2005. V. 4. P. 4–9.
Ramaswamy S., Goings G.E., Soderstrom K.E. et al. Cellular proliferation and migration following a controlled cortical impact in the mouse // Brain Res. 2005. V. 1053. № 1–2. P. 38–53. https://doi.org/10.1016/j.brainres.2005.06.042
Yamashita T., Ninomiya M., Hernandez Acosta P. et al. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum // J. Neurosci. 2006. V. 26. № 24. P. 6627–6636. https://doi.org/10.1523/jneurosci.0149-06.2006
Lan Zhang R., LeTourneau Y., Gregg S.R. et al. Neuroblast division during migration toward the ischemic striatum: A study of dynamic migratory and proliferative characteristics of neuroblasts from the subventricular zone // J. Neurosci. 2007. V. 27. № 12. P. 3157–3162. https://doi.org/10.1523/jneurosci.4969-06.2007
Hou S.W., Wang Y.Q., Xu M. et al. Functional integration of newly generated neurons into striatum after cerebral ischemia in the adult rat brain // Stroke. 2008. V. 39. № 10. P. 2837–2844. https://doi.org/10.1161/strokeaha.107.510982
Liu F., You Y., Li X. et al. Brain injury does not alter the intrinsic differentiation potential of adult neuroblasts // J. Neurosci. 2009. V. 29. № 16. P. 5075–5087. https://doi.org/10.1523/jneurosci.0201-09.2009
Yoshikawa G., Momiyama T., Oya S. et al. Induction of striatal neurogenesis and generation of region-specific functional mature neurons after ischemia by growth factors // J. Neurosurg. 2010. V. 113. № 4. P. 835–850. https://doi.org/10.3171/2010.2.jns09989
Kojima T., Hirota Y., Ema M. et al. Subventricular zone-derived neural progenitor cells migrate along a blood vessel scaffold toward the post-stroke striatum // Stem Cells. 2010. V. 28. № 3. P. 545–554. https://doi.org/10.1002/stem.306
Kreuzberg M., Kanov E., Timofeev O. et al. Increased subventricular zone-derived cortical neurogenesis after ischemic lesion // Exp. Neurol. 2010. V. 226. № 1. P. 90–99. https://doi.org/10.1016/j.expneurol.2010.08.006
Grade S., Weng Y.C., Snapyan M. et al. Brain-derived neurotrophic factor promotes vasculature-associated migration of neuronal precursors toward the ischemic striatum // PLoS One. 2013. V. 8. № 1. https://doi.org/10.1371/journal.pone.0055039
Saha B., Peron S., Murray K. et al. Cortical lesion stimulates adult subventricular zone neural progenitor cell proliferation and migration to the site of injury // Stem Cell Res. 2013. V. 11. № 3. P. 965–977. https://doi.org/10.1016/j.scr.2013.06.006
Faiz M., Sachewsky N., Gascón S. et al. Adult neural stem cells from the subventricular zone give rise to reactive astrocytes in the cortex after stroke // Cell Stem Cell. 2015. V. 17. № 5. P. 624–634. https://doi.org/10.1016/j.stem.2015.08.002
Ohira K., Furuta T., Hioki H. et al. Ischemia-induced neurogenesis of neocortical layer 1 progenitor cells // Nat. Neurosci. 2010. V. 13. № 2. P. 173–179. https://doi.org/10.1038/nn.2473
Magnusson J.P., Göritz C., Tatarishvili J. et al. A latent neurogenic program in astrocytes regulated by Notch signaling in the mouse // Science. 2014. V. 346. № 6206. P. 237–241. https://doi.org/10.1126/science.346.6206.237
Nato G., Caramello A., Trova S. et al. Striatal astrocytes produce neuroblasts in an excitotoxic model of Huntington’s disease // Development. 2015. V. 142. № 5. P. 840–845. https://doi.org/10.1242/dev.116657
Duan C.L., Liu C.W., Shen S.W. et al. Striatal astrocytes transdifferentiate into functional mature neurons following ischemic brain injury // Glia. 2015. V. 63. № 9. P. 1660–1670. https://doi.org/10.1002/glia.22837
Arvidsson A., Collin T., Kirik D. et al. Neuronal replacement from endogenous precursors in the adult brain after stroke // Nat. Med. 2002. V. 9. № 5. P. 548–553. https://doi.org/10.1038/nm747
Khodanovich M., Kisel A., Kudabaeva M. et al. Effects of fluoxetine on hippocampal neurogenesis and neuroprotection in the model of global cerebral ischemia in rats // Int. J. Mol. Sci. 2018. V. 19. № 1. P. 12–24. https://doi.org/10.3390/ijms19010162
Rodríguez J.J., Verkhratsky A. Neurogenesis in Alzheimer’s disease // J. Anat. 2011. V. 219. № 1. P. 78–89. https://doi.org/10.1111/j.1469-7580.2011.01343.x
Moreno-Jiménez E.P., Flor-garcía M., Terreros-roncal J. et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease // Nat. Med. 2019. V. 25. № 4. P. 554–560. https://doi.org/10.1038/s41591-019-0375-9
Lim J., Bang Y., Choi H.J. Abnormal hippocampal neurogenesis in Parkinson’s disease: relevance to a new therapeutic target for depression with Parkinson’s disease // Arch. Pharm. Res. Pharmaceutical Society of Korea. 2018. V. 41. № 10. P. 943–954. https://doi.org/10.1007/s12272-018-1063-x
Khodanovich M.Y., Pishchelko A.O., Glazacheva V.Y. et al. Plant polyprenols reduce demyelination and recover impaired oligodendrogenesis and neurogenesis in the cuprizone murine model of multiple sclerosis // Phyther. Res. 2019. № 9. P. 1–11. https://doi.org/10.1002/ptr.6327
Reif A., Schmitt A., Fritzen S., Lesch K.P. Neurogenesis and schizophrenia: Dividing neurons in a divided mind? // Eur. Arch. Psychiatry Clin. Neurosci. 2007. V. 257. № 5. P. 290–299. https://doi.org/10.1007/s00406-007-0733-3
Sorrells S.F., Paredes M.F., Cebrian-silla A. et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults // Nature. 2018. V. 555. № 7696. P. 377–381. https://doi.org/10.1038/nature25975.human
Kempermann G., Gage F.H., Aigner L. et al. Human adult neurogenesis: evidence and remaining questions // Cell Stem Cell. 2018. V. 23. № 1. P. 25–30. https://doi.org/10.1016/j.stem.2018.04.004
Ernst A., Alkass K., Bernard S. et al. Neurogenesis in the striatum of the adult human brain // Cell. 2014. V. 156. № 5. P. 1072–1083. https://doi.org/10.1016/j.cell.2014.01.044
Thanseem I., Viswambharan V., Poovathinal S.A., Anitha A. Is telomere length a biomarker of neurological disorders? // Biomark. Med. 2017. V. 11. № 9. P. 799–810. https://doi.org/10.2217/bmm-2017-0032
Grochowski C., Radzikowska E., Maciejewski R. Neural stem cell therapy – Brief review // Clin. Neurol. Neurosurg. 2018. V. 173. P. 8–14. https://doi.org/10.1016/j.clineuro.2018.07.013
Levy M.Z., Allsopp R.C., Futcher A.B. et al. Telomere end-replication problem and cell aging // J. Mol. Biol. 1992. V. 225. № 4. P. 951–960. https://doi.org/10.1016/0022-2836(92)90096-3
Menna P.L., Gomez D.E., Alonso D.F. et al. Telomere structure and telomerase in health and disease // Int. J. Oncol. 2012. V. 41. № 5. P. 1561–1569. https://doi.org/10.3892/ijo.2012.1611
Galati A., Micheli E., Cacchione S. Chromatin structure in telomere dynamics // Front. Oncol. 2013. V. 3. № 3. P. 1–16. https://doi.org/10.3389/fonc.2013.00046
Brazvan B., Ebrahimi-Kalan A., Velaei K. et al. Telomerase activity and telomere on stem progeny senescence // Biomed. Pharmacother. Elsevier. 2018. V. 102. № 2. P. 9–17. https://doi.org/10.1016/j.biopha.2018.02.073
Greider C.W., Blackburn E.H. The telomere terminal transferase of tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity // Cell. 1987. V. 51. № 6. P. 887–898. https://doi.org/10.1016/0092-8674(87)90576-9
Lingner J., Hughes T.R., Shevchenko A. et al. Reverse transcriptase motifs in the catalytic subunit of telomerase // Science. 1997. V. 276. № 5312. P. 561–567. https://doi.org/10.1126/science.276.5312.561
Chang E., Adams R., Chiu C. et al. The RNA component of human telomerase // Science. 2006. V. 269. № 5228. P. 1236–1241. https://doi.org/10.1126/science.7544491
Zvereva M.I., Shcherbakova D.M., Dontsova O.A. Telomerase : structure, functions, and activity regulation // Biochemistry (Mosc.). 2010. V. 75. № 13. P. 1563–1583. https://doi.org/10.1134/S0006297910130055
Sandin S., Rhodes D. Telomerase structure // Curr. Opin. Struct. Biol. 2014. V. 25. P. 104–110. https://doi.org/10.1016/j.sbi.2014.02.003
Yi X. Quantitation of telomerase components and hTERT mRNA splicing patterns in immortal human cells // Nucl. Acids Res. 2002. V. 29. № 23. P. 4818–4825. https://doi.org/10.1093/nar/29.23.4818
Fu W., Killen M., Culmsee C. et al. The catalytic subunit of telomerase is expressed in developing brain neurons and serves a cell survival-promoting function // J. Mol. Neurosci. 2000. V. 14. № 1–2. P. 3–15. https://doi.org/10.1385/jmn:14:1-2:003
Klapper W., Shin T., Mattson M.P. Differential regulation of telomerase activity and TERT expression during brain development in mice // J. Neurosci. Res. 2001. V. 64. № 3. P. 252–260. https://doi.org/10.1002/jnr.1073
Aix E., Gutiérrez-Gutiérrez Ó., Sánchez-Ferrer C. et al. Postnatal telomere dysfunction induces cardiomyocyte cell-cycle arrest through p21 activation // J. Cell Biol. 2016. V. 213. № 5. P. 571–583. https://doi.org/10.1083/jcb.201510091
Caporaso G.L., Lim D.A., Alvarez-Buylla A., Chao M.V. Telomerase activity in the subventricular zone of adult mice // Mol. Cell. Neurosci. 2003. V. 23. № 4. P. 693–702. https://doi.org/10.1016/s1044-7431(03)00103-9
Chen C., Zhu D.-Y., Wu H.-Y. et al. Hippocampal telomerase is involved in the modulation of depressive behaviors // J. Neurosci. 2011. V. 31. № 34. P. 12258–12269. https://doi.org/10.1523/jneurosci.0805-11.2011
Liu M.-Y., Nemes A., Zhou Q.-G. The emerging roles for telomerase in the central nervous system // Front. Mol. Neurosci. 2018. V. 11. № 5. P. 1–12. https://doi.org/10.3389/fnmol.2018.00160
Eitan E., Tamar A., Yossi G. et al. Expression of functional alternative telomerase RNA component gene in mouse brain and in motor neurons cells protects from oxidative stress // Oncotarget. 2016. V. 7. № 48. https://doi.org/10.18632/oncotarget.13049
Wagner W., Bork S., Horn P. et al. Aging and replicative senescence have related effects on human stem and progenitor cells // PLoS One. 2009. V. 4. № 6. P. 1–13. https://doi.org/10.1371/journal.pone.0005846
Flores I., Cayuela M.L., Blasco M.A. Molecular biology: Effects of telomerase and telomere length on epidermal stem cell behavior // Science. 2005. V. 309. № 5738. P. 1253–1256. https://doi.org/10.1126/science.1115025
Campisi J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors // Cell. 2005. V. 120. № 4. P. 513–522. https://doi.org/10.1016/j.cell.2005.02.003
Wang Q., Zhan Y., Pedersen N.L. et al. telomere length and all-cause mortality: a meta-analysis // Ageing Res. Rev. 2018. V. 48. № 8. P. 11–20. https://doi.org/10.1016/j.arr.2018.09.002
Guan J.Z., Guan W.P., Maeda T. et al. Patients with multiple sclerosis show increased oxidative stress markers and somatic telomere length shortening // Mol. Cell. Biochem. 2014. V. 400. № 1–2. P. 183–187. https://doi.org/10.1007/s11010-014-2274-1
Greider C.W., Zhu J.J., Black P.M. et al. Telomerase activity in human gliomas // Neurosurgery. 2004. V. 42. № 5. P. 1120–1124. https://doi.org/10.1097/00006123-199805000-00099
Stewart S.A., Hahn W.C., O’Connor B.F. et al. Telomerase contributes to tumorigenesis by a telomere length-independent mechanism // Proc. Natl Acad. Sci. USA. 2002. V. 99. № 20. P. 12606–12611.
Varela E., Muñoz-Lorente M.A., Tejera A.M. et al. Generation of mice with longer and better preserved telomeres in the absence of genetic manipulations // Nat. Commun. 2016. V. 7. https://doi.org/10.1038/ncomms11739
Harley C.B. Telomerase and cancer therapeutics // Nat. Rev. Cancer. 2008. V. 8. № 3. P. 167–179. https://doi.org/10.1038/nrc2275
Shay J.W., Keith W.N. Targeting telomerase for cancer therapeutics // Br. J. Cancer. 2008. V. 98. № 4. P. 677–683. https://doi.org/10.1038/sj.bjc.6604209
Ruden M., Puri N. Novel anticancer therapeutics targeting telomerase // Cancer Treat. Rev. 2013. V. 39. № 5. P. 444–456. https://doi.org/10.1016/j.ctrv.2012.06.007
Chen Y., Zhang Y. Functional and mechanistic analysis of telomerase: An antitumor drug target // Pharmacol. Ther. 2016. V. 163. P. 24–47. https://doi.org/10.1016/j.pharmthera.2016.03.017
Choi J., Southworth L.K., Sarin K.Y. et al. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program // PLoS Genet. 2008. V. 4. № 1. P. 124–138. https://doi.org/10.1371/journal.pgen.0040010
Zhou L., Zheng D., Wang M., Cong Y.S. Telomerase reverse transcriptase activates the expression of vascular endothelial growth factor independent of telomerase activity // Biochem. Biophys. Res. Commun. 2009. V. 386. № 4. P. 739–743. https://doi.org/10.1016/j.bbrc.2009.06.116
Beck S., Jin X., Sohn Y.W. et al. Telomerase activity-independent function of TERT allows glioma cells to attain cancer stem cell characteristics by inducing EGFR expression // Mol. Cells. 2011. V. 31. № 1. P. 9–15. https://doi.org/10.1007/s10059-011-0008-8
Mukherjee S., Firpo E.J., Wang Y., Roberts J.M. Separation of telomerase functions by reverse genetics // Proc. Natl Acad. Sci. USA. 2011. V. 108. № 50. P. 1363–1371. https://doi.org/10.1073/pnas.1112414108
Hrdlickova R., Nehyba J., Bose H.R. Alternatively spliced telomerase reverse transcriptase variants lacking telomerase activity stimulate cell proliferation // Mol. Cell. Biol. 2012. V. 32. № 21. P. 4283–4296. https://doi.org/10.1128/mcb.00550-12
Sarin K.Y., Cheung P., Gilison D. et al. Conditional telomerase induction causes proliferation of hair follicle stem cells // Nature. 2005. V. 34. № 5. P. 880–886. https://doi.org/10.1038/jid.2014.371
Gonzalez-Suarez E., Flores J.M., Blasco M.A. Cooperation between p53 mutation and high telomerase transgenic expression in spontaneous cancer development // Mol. Cell. Biol. 2002. V. 22. № 20. P. 7291–7301. https://doi.org/10.1128/mcb.22.20.7291-7301.2002
González-Suárez E., Geserick C., Flores J.M., Blasco M.A. Antagonistic effects of telomerase on cancer and aging in K5-mTert transgenic mice // Oncogene. 2005. V. 24. № 13. P. 2256–2270. https://doi.org/10.1038/sj.onc.1208413
Kelemen O., Convertini P., Zhang Z. et al. Function of alternative splicing // Gene. 2017. V. 514. № 1. P. 1–30. https://doi.org/10.1016/j.gene.2012.07.083.function
Wick M., Zubov D., Hagen G. Genomic organization and promoter characterization of the gene encoding the human telomerase reverse transcriptase (hTERT) // Gene. 1999. V. 232. № 1. P. 97–106. https://doi.org/10.1016/s0378-1119(99)00108-0
Kilian A., Bowtell D.D.L., Abud H.E. et al. Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types // Hum. Mol. Genet. 1997. V. 6. № 12. P. 2011–2019. https://doi.org/10.1093/hmg/6.12.2011
Sæbøe-larssen S., Fossberg E., Gaudernack G. Characterization of novel alternative splicing sites in human telomerase reverse transcriptase (hTERT): analysis of expression and mutual correlation in mRNA isoforms from normal and tumour tissues // BMC Mol. Biol. 2006. V. 10. P. 1–10. https://doi.org/10.1186/1471-2199-7-26
Withers J.B., Ashvetiya T., Beemon K.L. Exclusion of exon 2 is a common mRNA splice variant of primate telomerase reverse transcriptases // PLoS One. 2012. V. 7. № 10. e48016. https://doi.org/10.1371/journal.pone.0048016
Bollmann F.M. Physiological and pathological significance of human telomerase reverse transcriptase splice variants // Biochimie. 2013. V. 95. № 11. P. 1965–1970. https://doi.org/10.1016/j.biochi.2013.07.031
Tsukada Y., Hisatomi H., Nagao K. et al. Expression profile of a γ-deletion variant of the human telomerase reverse transcriptase gene // Neoplasia. 2014. V. 5. № 3. P. 193–197. https://doi.org/10.1016/s1476-5586(03)80051-9
Wong M.S., Wright W.E., Shay J.W. Alternative splicing regulation of telomerase: A new paradigm? // Trends Genet. 2014. V. 30. № 10. P. 430–438. https://doi.org/10.1016/j.tig.2014.07.006
Teichroeb J.H., Kim J., Betts D.H. The role of telomeres and telomerase reverse transcriptase isoforms in pluripotency induction and maintenance // RNA Biol. 2016. V. 13. № 8. P. 707–719. https://doi.org/10.1080/15476286.2015.1134413
Ulaner G.A., Hu J.F., Vu T.H. et al. Tissue-specific alternate splicing of human telomerase reverse transcriptase (hTERT) influences telomere lengths during human development // Int. J. Cancer. 2001. V. 91. № 5. P. 644–649. https://doi.org/10.1002/1097-0215(200002)9999:9999< ::aid-ijc1103>3.0.co;2-v
Liu Y., Wu B., Zhong H. et al. Quantification of alternative splicing variants of human telomerase reverse transcriptase and correlations with telomerase activity in lung cancer // PLoS One. 2012. V. 7. № 6. P. 1–9. https://doi.org/10.1371/journal.pone.0038868
Listerman I., Sun J., Francesca S. et al. The major reverse transcriptase-incompetent splice variant of the human telomerase protein inhibits telomerase activity but protects from apoptosis // Cancer Res. 2014. V. 40. № 8. P. 1394–1403. https://doi.org/10.3899/jrheum.121180
Mudge L.-M., Hamilton C.S., Sakoff J.A. et al. Quantification of hTERT splice variants in melanoma by SYBR green real-time polymerase chain reaction indicates a negative regulatory role for the β deletion variant // Neoplasia. 2015. V. 10. № 10. P. 1131–1137. https://doi.org/10.1593/neo.08644
Kim J. Alternative splicing and extra-telomeric role of telomerase reverse transcriptase isoforms in human embryonic stem cells: Dis. Master of science. Univ. Western Ontario, 2017. 148 p.
Yi X., White D.M., Aisner D.L. et al. An alternate splicing variant of the human telomerase catalytic subunit inhibits telomerase activity // Neoplasia. 2000. V. 2. № 5. P. 433–440. https://doi.org/10.1038/sj.neo.7900113
Reddel R.R., Colgin L.M., Wilkinso C. et al. The hTERTα splice variant is a dominant negative inhibitor of telomerase activity // Neoplasia. 2002. V. 2. № 5. P. 426–432. https://doi.org/10.1038/sj.neo.7900112
Gandin V., Rousseau P., Autexier C. et al. Inactive C‑terminal telomerase reverse transcriptase insertion splicing variants are dominant-negative inhibitors of telomerase // Biochimie. 2014. V. 101. P. 93–103. https://doi.org/10.1016/j.biochi.2013.12.023
Radan L., Postovit L.-M., Jewer M. et al. Microenvironmental regulation of telomerase isoforms in human embryonic stem cells // Stem Cells Dev. 2014. V. 23. № 17. P. 2046–2066. https://doi.org/10.1089/scd.2013.0373
Pickett H.A., Cesare A.J., Johnston R.L. et al. Control of telomere length by a trimming mechanism that involves generation of t-circles // EMBO J. 2009. V. 28. № 7. P. 799–809. https://doi.org/10.1038/emboj.2009.42
Rivera T., Haggblom C., Cosconati S., Karlseder J. A balance between elongation and trimming regulates telomere stability in stem cells // Nat. Struct. Mol. Biol. 2017. V. 24. № 1. P. 30–39. https://doi.org/10.1038/nsmb.3335
Li J.S.Z., Denchi E.L. How stem cells keep telomeres in check // Differentiation. 2018. V. 100. № 10. P. 21–25. https://doi.org/10.1016/j.diff.2018.01.004
Li J.S.Z., Fuste J.M., Simavorian T. et al. TZAP: a telomere-associated protein involved in telomere length control // Science. 2018. V. 355. № 6325. P. 638–641. https://doi.org/10.1126/science.aah6752.tzap
Wu Z., Asokan A., Samulski R.J. Adeno-associated virus serotypes: vector toolkit for human gene therapy // Mol. Ther. 2006. V. 14. № 3. P. 316–327. https://doi.org/10.1016/j.ymthe.2006.05.009
Mingozzi F., High K.A. Therapeutic in vivo gene transfer for genetic disease using AAV: Progress and challenges // Nat. Rev. Genet. Nature Publ. Group. 2011. V. 12. № 5. P. 341–355. https://doi.org/10.1038/nrg2988
Kantor B., Bailey R.M., Wimberly K. et al. Methods for gene transfer to the central nervous system // Adv. Genet. 2015. V. 53. № 87. P. 125–197. https://doi.org/10.1016/b978-0-12-800149-3.00003-2
Porteus M.H. Towards a new era in medicine: Therapeutic genome editing // Genome Biol. 2015. V. 16. № 1. P. 1–12. https://doi.org/10.1186/s13059-015-0859-y
Asuri P., Bartel M., Kwon I. et al. An evolved adeno-associated viral variant enhances gene delivery and gene targeting in neural stem cells // Mol. Ther. 2011. V. 19. № 4. P. 667–675. https://doi.org/10.1038/mt.2010.287
Kotterman M.A., Vazin T., Schaffer D. V. Enhanced selective gene delivery to neural stem cells in vivo by an adeno-associated viral variant // Development. 2015. V. 142. № 10. P. 1885–1892. https://doi.org/10.1242/dev.115253
Ojala D.S., Sun S., Santiago-Ortiz J.L. et al. In vivo selection of a computationally designed SCHEMA AAV library yields a novel variant for infection of adult neural stem cells in the SVZ // Mol. Ther. 2018. V. 26. № 1. P. 304–319. https://doi.org/10.1016/j.ymthe.2017.09.006
Smith L.J., Wright J., Clark G. et al. Stem cell-derived clade F AAVs mediate high-efficiency homologous recombination-based genome editing // Proc. Natl Acad. Sci. USA. 2018. V. 115. № 31. P. E7379–E7388. https://doi.org/10.1073/pnas.1802343115
Ran F.A., Cong L., Yan W.X. et al. In vivo genome editing using Staphylococcus aureus Cas9 // Obs. Gynecol. 2015. V. 520. № 7546. P. 186–191. https://doi.org/10.1038/nature14299
Barzel A., Paulk N.K., Shi Y. et al. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice // Nature. 2016. V. 32. № 3. P. 178–182. https://doi.org/10.1055/s-0035-1563737
Barzel A., Muro A.F., Zentilin L. et al. Promoterless gene targeting without nucleases rescues lethality of a Crigler–Najjar syndrome mouse model // EMBO Mol. Med. 2017. V. 9. № 10. P. 1346–1355. https://doi.org/10.15252/emmm.201707601
Funk S.E., Russell D.W., Hirata R.K. et al. Nuclease-free adeno-associated virus-mediated Il2rg gene editing in X-SCID mice // Mol. Ther. 2018. V. 26. № 5. P. 1255–1265. https://doi.org/10.1016/j.ymthe.2018.02.028
Дополнительные материалы отсутствуют.