

Генетика, 2020, T. 56, № 5, стр. 525-533
Клинико-генетические характеристики синдрома Нунан и нунан-подобных заболеваний
А. А. Орлова 1, *, Е. Л. Дадали 1, А. В. Поляков 1
1 Медико-генетический научный центр
115478 Москва, Россия
* E-mail: annaf-orlova@yandex.ru
Поступила в редакцию 14.05.2019
После доработки 11.09.2019
Принята к публикации 11.09.2019
Аннотация
Синдром Нунан – группа наследственных аутосомно-доминантных заболеваний, характеризующихся нарушением RAS-MAPK сигнального пути, и приводящих к различным клиническим проявлениям. Распространенность в мире оценивается в один–два на 20 000 новорожденных. В обзоре обсуждаются молекулярно-генетические причины заболевания, особенности клинических проявлений заболевания и способы молекулярно-генетической диагностики.
Синдром Нунан (СН) (OMIM 163 950, 605 275, 609 942, 610 733, 611 553, 613 224, 613 706, 615 355, 616 559, 616 564) – группа генетически гетерогенных заболеваний с идентифицированными генетическими вариантами в девяти генах; для восьми из них описан только аутосомно-доминантный тип наследования, для одного – и доминантный, и рецессивный. Несмотря на генетическую гетерогенность, заболевание имеет классическую фенотипическую картину, характеризующуюся лицевыми дизморфизмами, широким спектром врожденных пороков сердечно-сосудистой системы и низкорослостью. Характерные черты лица включают: широкий лоб, гипертелоризм, высокоарочное небо, а также низкопосаженные уши. Различные пороки сердца встречаются у 90% больных. Наиболее распространенные расстройства – стеноз легочной артерии и гипертрофическая кардиомиопатия, но наблюдаются и другие формы. Дополнительно выделяются следующие признаки: деформации скелета, такие как искривление грудной клетки и позвоночника, шею с кожными складками, крипторхизм и нарушения развития половой системы у мальчиков, иногда встречается умственная отсталость [1].
Изначально СН считали вариантом синдрома Тернера, пока в 1968 г. детский кардиолог Жаклин Нунан не описала данный синдром. Она обследовала 19 больных детей со стенозом легочной артерии, специфичным лицом с гипертелоризмом, птозом, низко посаженными ушами, шейной крыловидной складкой и деформацией грудной клетки, некоторые мальчики имели крипторхизм [2]. После этого пациентам мужского пола начали делать кариотип для уточнения синдрома Тернера, и выявлялся синдром Тернера реже, чем предполагалось [3].
На сегодняшний день в мире распространенность СН оценивается в один–два на 20 000 новорожденных. Хотя в публикациях встречаются цифры от 1 : 1000 до 1 : 2500 новорожденных [4, 5], но данные оценки получены не в результате популяционных исследований [6].
СН является клинически и генетически гетерогенным синдромом, обусловленным мутациями в генах RAS-MAPK сигнального пути. В зависимости от гена и типа мутации клинические проявления могут быть различными.
МОЛЕКУЛЯРНАЯ ПРИЧИНА ЗАБОЛЕВАНИЯ
Синдром Нунан вызывается мутациями в генах RAS-MAPK (RAS-митоген активированные протеинкиназы) сигнального пути, продукты которых обеспечивают передачу сигнала от клеточной мембраны к ядру. Данный каскад участвует в циклах клеточной дифференцировки, росте, ограногенезе, синаптической пластичности и т.д. Разные белки являются ключевыми в различных типах тканей, поэтому проявления синдромов, связанных с мутациями в генах данного пути могут быть различны [7–9].
RAS-MAPK сигнальный путь отвечает за трансдукцию сигнала от внешней клеточной мембраны к ядру в ответ на наличие фактора роста. Сигнальный путь представлен на рис. 1.
Рис. 1.
Схема RAS-MAPK сигнального пути. RAS-MAPK активируется привязыванием лиганда (фактора роста или другого цитокина) к RTK-трансмембранному рецептору с тирозин-киназной активностью. Фосфорилирование внутриклеточного домена трансмембранного рецептора является сигналом для прикрепления специфичного белка-адаптора, в том числе GRB2, CBL, SHP2, SOS1 и дальнейшей активации последующих эффекторных белков. Ключевой элемент RAS-MAPK сигнального пути – семейство протеинов RAS (KRAS, NRAS, HRAS и RIT), которые в активной форме привязаны к мембране и используют ГТФ как кофактор. Активность белков RAS регулируется специфическими белками, делящимися на две группы: факторы замены гуанидина, активирующие связывание ГТФ (например SOS1), и активаторы ГТФазы, индуцирующие гидролиз ГТФ (например NF1). Белки RAS отвечают за активацию каскада митоген-активирующих протеин-киназ (MAPK). Первая киназа этого каскада принадлежит семейству RAF-белков (BRAF или CRAF), активность которых регулируется другими белками, например PP1C-фосфотазой, SHOC2, или белками SPRED1. Белки RAF отвечают за фосфорилирование и активацию киназы MAP2K (также называемой MEK, MAP2K1 или MAP2K2). MAP2K-киназа далее фосфорилирует эффекторные пути киназы – ERK-белки (ERK1/2). ERK-киназы могут перемещаться в ядро, где фосфорилируют специфические факторы транскрипции, регулирующие экспрессию специфичных генов, кодирующих белки вовлеченные в клеточный рост, пролиферацию и дифференцировку [по 10, 49].
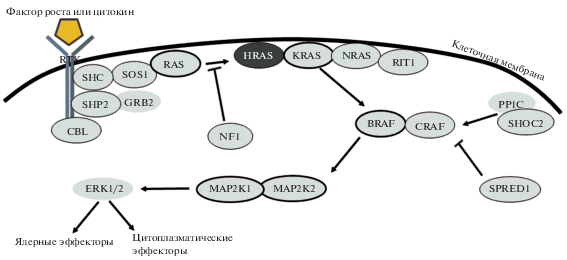
RAS-MAPK сигнальный путь активируется специфическими белками, такими как фактор роста фибробластов (FGF (fibroblast growth factor)), эпидермальный фактор роста (EGF (Epidermal growth factor)), инсулин-подобный фактор роста (IGF (insulin-like growth factor)), гормонами (в том числе гормоном роста), цитокинами, инсулином или клетками иммунной системы. Эти факторы связываются со специфичным трансмембранным рецептором, который после связывания лиганда димеризуется, автофосфорилируется и таким образом активируется. После активации несколько белков-адапторов, такие как GRB2, SHP2, SOS1 захватываются рецептором. Данный процесс приводит к изменению структур белков, особенно SOS1, который становится способным к взаимодействию и активации RAS-белка. Активированный RAS-белок в свою очередь активирует серин-треонин киназу из семейства RAF-киназ (CRAF, BRAF, ARAF), которые также описаны как митоген-активированные протеин-киназы (MAP3K). RAF-киназа фосфорилирует следующие протеины каскада: MEK1 и MAP2K. Фосфорилированная MAP2K активирует эффекторную киназу пути – MAP (экстрацеллюлярные сигнал-регулирующие киназы 1 и 2 (ERK1 и 2). Активированные ERK проходят через ядерную оболочку и фосфорилируют специфичные факторы транскрипции, ответственные за экспрессию некоторых генов [9, 10].
RAS-MAPK сигнальный путь играет важную роль в регуляции клеточной пролиферации, дифференцировке, миграции и апоптозе в эмбриональном и постнатальном развитии [10]. Наследственные мутации в генах, кодирующих белки данного пути, приводящие к заболеваниям, характеризуются постоянной активацией пути, усилению и невозможностью регулировки экспрессии белка, и соответственно всего пути.
Функция регуляции клеточной пролиферации важна не только в процессе развития организма. Активирующие мутации RAS-MAPK генов также ассоциированы с онкогенной трансформацией и процессом канцерогенеза [11].
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ И ГЕНЕТИЧЕСКАЯ ГЕТЕРОГЕННОСТЬ СИНДРОМА НУНАН
В 1994 г. учеными и клиницистами из департамента клинической генетики Нидерландов была предложена схема диагностических критериев синдрома Нунан [12]. Она представлена в табл. 1.
Таблица 1.
| Основные симптомы | Дополнительные симптомы |
|---|---|
| Типичные лицевые дизморфии (увеличение пропорций головы относительно лица, высокий лоб, гипертелоризм, птоз и эпикант, короткий широкий нос с вдавленным корнем и расширенным кончиком, короткая шея, низкосидящие и завернутые назад уши) | Нетипичные лицевые дизморфизмы |
| Стеноз легочной артерии Гипертрофическая обструктивная кардиомиопатия |
Другие дефекты |
| Рост ниже 3 центилей популяции | Рост ниже 10 центилей популяции |
| Деформация грудной клетки | Широкая грудная клетка |
| Первый случай появления синдрома | Подтвержденное наследование |
| Интеллектуальная недостаточность Крипторхизм Лимфатическая дисплазия |
Для постановки диагноза по мнению авторов у пациента должны быть: типичные лицевые дизморфии в сочетании как минимум с одним основным критерием, либо двумя дополнительными. Либо, при отсутствии четких лицевых особенностей, два других основных критерия или три дополнительных.
Выявлен выраженный клинический полиморфизм заболевания. Ядро признаков составляют типичные дизморфии (черты строения лица и шеи) и пороки сердца. Остальные симптомы встречаются реже.
Согласно OMIM существует десять генетических вариантов синдрома Нунан, имеющих характерные фенотипические проявления.
Тип 1. OMIM 163950. Аутосомно-доминантное заболевание, вызванное мутациями в гене PTPN11 (локализация 12q24.13). До 50% случаев диагностированного и подтвержденного молекулярно-генетическими методами синдрома приходятся на мутации в PTPN11 [1, 5]. Ген PTPN11 кодирует белок SHP2, экспрессирующийся практически во всех тканях организма и являющийся одним из первых белков, вовлеченных в RAS-MAPK сигнальный путь. Основной тип патогенных мутаций – миссенс с усилением функции белка, что по сути не дает возможности остановить передачу сигнала (инактивировать молекулу белка-передатчика) при отсутствии на рецепторе сигнальной молекулы.
К наиболее характерным проявлениям СН типа 1 относят (в скобках указана частота встречаемости по разным источникам и популяциям): стеноз легочной артерии (55–88%), низкорослость (56–82%), деформация грудной клетки (17–82%), крипторхизм (43–84%), характерная дизморфия лица (91%), легкое появление синяков (дефицит факторов свертываемости) (17–41%), гипертрофическая кардиомиопатия (6–9%) [1, 5, 13–15].
Основные дизморфизмы лица у пациентов с синдромом Нунан включают: увеличение пропорций головы относительно лица, высокий лоб, гипертелоризм, наклонные пальпебральные трещины, птоз и эпикант, короткий широкий нос с вдавленным корнем и расширенным кончиком, усиление рельефа губного желобка, узкие челюсти, короткая шея, низкосидящие и завернутые назад уши с мягкими овальными спиралями [16].
У СН, связанного с мутациями в гене PTPN11, существует аллельный вариант – синдром LEOPARD (OMIM 151100), с частично подобными фенотипическими проявлениями, но со своей характерной особенностью: множественные лентиго по всему телу. Само название синдрома несет в себе основные характеные фенотипические проявления: множественное лентиго (Lentigines), нарушения электрокардиограммы (Electrocardiografic conduction abnormalities), глазной гипертелоризм (Ocular hypertelorism), стеноз легочной артерии (Pulmonic stenosis), изменение строения гениталий (Abnormal genitalia), задержка роста (Retardation of growth), сенсорная глухота (sensorineural Deafness) [17].
Мутации, приводящие к синдрому LEOPARD, характеризуются снижением или потерей активности белка, в отличие от синдрома Нунан с гипер-активностью и отсутствием возможности снизить или прекратить активность киназы [10].
Тип 2. OMIM 605275. Аутосомно-рецессивное заболевание, с характерными для СН типа 1 фенотипическими проявлениями. Заболевание вызывается предполагаемым геном NS2, не картированным на хромосомах. Ген не описан. Но есть несколько описаний семей, где синдром Нунан встречался с аутосомно-рецессивным типом наследования [18–20].
Тип 3. OMIM 609942. Аутосомно-доминантное заболевание, вызываемое мутациями в гене KRAS (локализация 12p12.1). До 2% случаев СН вызваны мутациями в данном гене [21, 22]. Фенотипически данная форма отличается от классической первой. Отмечаются более сильные нарушения когнитивных способностей, задержки психо-речевого развития. У некоторых пациентов отмечены краниосиностозы [23, 24]. Так же как особенности фенотипа отмечаются гипертрофическая кардиомиопатия, невусы, лентиго, кофейные пятна [5, 6, 11, 21].
Ген KRAS кодирует два белка, получаемых альтернативным сплайсингом: KRASA и KRASB. Оба белка являются ГТФ/ГДФ (гуанозинтрифосфат/гуанозиндифосфат) – регулируемыми молекулярными триггерами внутриклеточного сигнального пути. Миссенс мутации в первых, наиболее консервативных экзонах, кодирующих фрагменты обеих форм белка, могут менять сайты связывания с мембраной клетки и процесс перемещения белка по мембране [25]. На мышиных моделях замечено, что KRASB экспрессируется и на эмбриональных стадиях развития, и в постнатальном периоде, изоформа А экспрессируется в ограниченные периоды жизни и точно не экспрессируется в сердце взрослого [26].
Мутации, затрагивающие только изоформу А, не влияют на развитие организма, а мутации, усиливающие функции обеих изоформ или только KRASB, являются патогенными и приводящими к развитию не только синдрома Нунан, но и описаны случаи кардио-фасцио-кутанеозного синдрома, вызванного мутациями в KRAS.
Тип 4. OMIM 610733. Аутосомно-доминантное заболевание, вызванное мутациями в гене SOS1. По разным оценкам, от 10 до 28% случаев СН вызваны мутациями в гене SOS1 [21, 22, 27]. К наиболее часто встречающимся и характерным фенотипическим проявлениям относят: кератозы (50%), дефекты сердечных перегородок (20–25%), птоз (100%), волнистые волосы (88%), макроцефалия (56%), хроническая артралгия (75% взрослых), низкорослость (13–31%), задержки психомоторного развития [21, 28, 29]. Белок SOS1 является фактором изменения гуанина, который активирует РАС-белки, вытесняя ГДФ, что позволяет ГТФ присоединится и активировать белок [30, 31]. Белок SOS1 имеет сложную структуру, некоторые из доменов отвечают за автоингибирование белка. Миссенс мутации в гене SOS1 приводят к изменению аминокислотных последовательностей этих доменов и “отключению” функции автоингибирования, что ведет к постоянной нерегулируемой активности белка [28, 32, 33].
Тип 5. OMIM 611553. Аутосомно-доминантное заболевание, вызываемое мутациями в гене RAF1 (локализация 3p25.2). От 5 до 15% случаев СН вызваны мутациями в данном гене [21, 22]. Белок RAF1 относится к маленькому семейству серин-треонин киназ, выполняющих роль эффекторов белков RAS [34, 35]. RAF1 фосфорилирует и активирует специфические киназы MEK1 и MEK2, которые в свою очередь запускают активацию белков MAPK, ERK1 и ERK2 [7]. В белке RAF1 есть сайт, фосфорилирование которого приводит к автоингибированию белка. Соответственно мутации в данном домене, препятствующие фосфорилированию, приводят к постоянной бесконтрольной работе киназы [9, 21]. Также были выявлены мутации, повреждающие область сегмента активации киназного домена [21].
При синдроме Нунан, вызванном мутациями в гене RAF1, в 75–80% случаев отмечается гипертрофическая кардиомиопатия (при остальных формах СН данное нарушение встречается не чаще, чем в 20% случаев). Особенностью клинических проявлений этого генетического варианта являются: множественные невусы, лентиго, кофейные пятна на коже [13].
Тип 6. OMIM 613224. Аутосомно-доминантное заболевание, вызываемое мутациями в гене NRAS (локализация 1p13.2). Менее чем у 1% больных синдромом Нунан выявлена мутация в данном гене. Из-за низкой частоты встречаемости больных с мутациями в данном гене, четких фенотипических особенностей не установлено, считается что они схожи с фенотипом СН перового типа [36, 37].
Тип 7. OMIM 613706. Аутосомно-доминантное заболевание, вызываемое мутациями в гене BRAF (локализация 7q34). У больных с фенотипом синдрома Нунан мутации в гене BRAF встречаются крайне редко. Мутации в этом гене описаны также для синдрома LEOPARD типа 3, и кардиофасциокутального синдрома. У больных с СН типа 7 описаны характерные фенотипические особенности: низкий рост, долихоцефалия, лицевые дизморфизмы, множественные невусы и лентиго, снижение когнитивных функций, аномалии развития скелета, мышечная гипотония [38, 39]. В отличие от кардиофасциокутального синдрома, гипертрофическая кардиомиопатия отсутствует.
Тип 8. OMIM 615355. Аутосомно-доминантное заболевание, вызываемое мутациями в гене RIT1 (локализация 1q22). Только в 2013 г. были описаны больные с синдромом Нунан и наследственной мутацией в гене RIT1. При мутациях в данном гене гипертрофическая кардиомиопатия встречается в 70% случаев, против 20% в среднем для синдрома Нунан [40]. Остальные фенотипические признаки схожи с таковыми при СН типа 1.
Тип 9. OMIM 616559. Аутосомно-доминантное заболевание, вызванное мутациями в гене SOS2 (локализация 14q21.3). По клинике и фенотипическим проявлением схож с СН типа 4, вызванным мутациями в гене SOS1. Белок SOS2 является гомологом для SOS1, но выполняет дополняющую функцию [41]. Исследования с использованием модельных животных показали, что отсутствие, или нарушение синтеза белка SOS1 приводит к нарушениям развития еще в пренатальном развитии, в то время как подобное нарушение белка SOS2 не приводило к столь тяжелым последствиям [7, 42]. Характерными фенотипическими чертами являются: низкий рост, черепно-лицевые дизморфии, короткая шея, перепонка на шее, нарушения сердечной деятельности, крипторхизм, нарушения свертывания крови [41].
Тип 10. OMIM 616564. Аутосомно-доминантное заболевание, вызванное мутациями в гене LZTR1 (локализация 22q11.21). На данный вариант СН вместе с СН типа 9 (связанным с мутациями в SOS2), приходится около 3% больных. Функциональная роль LZTR1 в каскаде RAS-MAPK до конца не известна, требуются дополнительные исследования, для установления роли данного белка в каскаде [41]. Описаны также аутосомно-рецессивные мутации в гене LZTR1, связанные с синдромом Нунан [43]. Клиническая картина не имеет специфических черт и совпадает с характерной для СН типа 1.
В большинстве случаев фенотипические проявления синдрома развиваются постнатально, но иногда на скрининге первого или второго триместров могут быть отмечены особенности развития, характерные для хромосомной патологии, но при нормальном кариотипе плода. Чаще всего пренатально отмечают многоводие, пороки сердца, плевральный выпот, водянку, кистозную фиброму и другие особенности, характерные для множества генетических нарушений [44, 45]. В постнатальном периоде симптомы развиваются неравномерно и могут улучшаться/ухудшаться с течением времени и ростом организма. Например задержка роста чаще становится видна в пубертатном периоде, до этого момента дети чаще развиваются нормально [46].
В табл. 2 представлены клинико-генетические характеристики десяти типов синдрома Нунан.
Таблица 2.
Типы синдрома Нунан, различия и основные характеристики
| Тип, OMIM | Ген, локация, тип наследования (АД – аутосомно- доминантное, АР – аутосомно- рецессивное) | Типичная картина фенотипа | Отличия от СН типа 1 |
|---|---|---|---|
| Синдром Нунан типа 1, OMIM 163950 | PTPN11, 12q24.13, АД | Стеноз легочной артерии, низкорослость, характерные дизморфизмы лица, деформация грудной клетки, у мальчиков – крипторхидизм | |
| Синдром Нунан типа 2, OMIM 605275 | NS2, неизвестно, АР | Совпадает с картиной типа 1. Семейные случаи | |
| Синдром Нунан типа 3, OMIM 609942 | KRAS, 12p12.1, АД | Гипертрофическая кардиомиопатия, невусы, лентиго | Задержка психо-речевого развития, нарушения когнитивных функций, краниостозы |
| Синдром Нунан типа 4, OMIM 610733 | SOS1, 2p22.1, АД |
Кератозы, дефекты сердечных перегородок, птоз, низкорослость, макроцефалия | Вьющиеся волосы, хроническая артралгия у взрослых, задержки психо-речевого развития |
| Синдром Нунан типа 5, OMIM 611553 | RAF1, 3p25.2, АД |
Фенотип схож с типа 1 | Гипертрофическая кардиомиопатия, множественные невусы, лентиго |
| Синдром Нунан типа 6, OMIM 613224 | NRAS, 1p13.2, АД |
Схож с СН типа 1 | Отличительных особенностей не выявлено |
| Синдром Нунан типа 7, OMIM 613706 | BRAF, 7q34, АД |
Нарушения неонатального роста, низкий рост, долихоцефалия, лицевые дизморфизмы | Множественные невусы и лентиго, снижение когнитивных функций, аномалии развития скелета, мышечная гипотония |
| Синдром Нунан типа 8, OMIM 615355 |
RIT1, 1q22, АД | Схож с СН типа 1 | Гипертрофическая кардиомиопатия |
| Синдром Нунан типа 9, OMIM 616559 | SOS2, 14q21.3, АД | Схож с СН типа 1 | |
| Синдром Нунан типа 10, OMIM 616564 | LZTR1, 22q11.21; 22q11.1-q11.2, описаны и АД и АР типы | Схож с СН типа 1 |
ДРУГИЕ РАСОПАТИИ
Синдром Нунан является самым распространенным заболеванием из группы расопатий. Но, как было сказано выше, с нарушением функционирования RAS-MAPK сигнального пути связано большое количество схожих заболеваний.
К классу расопатий помимо синдрома Нунан относят: синдром Костелло, синдром LEOPARD, кардио-фасцио-кутальный синдром (CFC (Cardiofaciocutaneous syndrome)), синдром капиллярных мальформаций (CM-AVM (Capillary malformations, congenital)), синдром Лейгус, Нейрофиброматоз типа 1, наследственный фиброматоз (hereditary gingival fibromatosis), аутоимунный лимфопролиферативный синдром, синдром Маззанти [10, 47].
Синдром LEOPARD является аллельным вариантом синдрома Нунан, заболевание вызывается мутациями в PTPN11, но отличительной чертой является то, что мутации, вызывающие данный синдром, приводят к резкому снижению или полному отсутствию киназной активности белка [10]. Основные фенотипические проявления следуют из самого названии синдрома: множественное лентиго (Lentigines), нарушения электрокардиограммы (Electrocardiografic conduction abnormalities), глазной гипертелоризм (Ocular hypertelorism), стеноз легочной артерии (Pulmonic stenosis), изменение строения гениталий (Abnormal genitalia), задержка роста (Retardation of growth), сенсорная глухота (sensorineural Deafness) [17]. Характерные пятна проявляются в среднем в возрасте четырех–пяти лет и являются основным критерием манифестации синдрома. Также к типичным отличительным признакам относят гипертрофическую миотрофию, развивающуюся в пренатальном периоде, либо сразу после рождения [17].
Кардио-фасцио-кутальный синдром (CFC) относится к редким расопатиям. Заболевание вызывается мутациями в генах BRAF, MAP2K1 (MEK1 и MEK2) и KRAS. Около 75% пациентов с подтвержденным диагнозом имеют мутации в гене BRAF.
К ярко выраженным клиническим проявлениям синдрома относят: макроцефалию, гипоплазию супраобритальных гребней, изменение структуры волос. Гиперкератоз рук и ног, кофейные пятна и гемангиомы становятся более отчетливыми с возрастом [48].
Большинство пациентов с CFC имеют различные расстройства психического развития, включая задержки психомоторного развития, особенно задержку речи, вследствие отставания интеллектуального развития [49].
Синдром Костелло (СК) – один из самых редких наследственных синдромов класса расопатий. Отсутствие ярких характерных особенностей фенотипа усложняет его диагностику, и только комплекс клинических проявлений позволяет поставить диагноз [50]. Первыми ранними симптомами СК является полигидроамниоз, увеличение веса плода с макроцефалией и тахикардия [51]. Многие дети с СК рождаются раньше срока, но с увеличенным весом в результате водянки. Признаки мышечной гипотонии, возбудимости и хриплый голос описаны у большинства детей с синдромом Костелло. СК в 90% случаев вызван наследственными точковыми мутациями в прото-онкогене HRAS, являющемся одним из регуляторов RAS-MAPK сигнального пути.
К специфичным признакам можно отнести: макроцефалию, грубые черты лица, высокий лоб, короткий нос с уплощенной носовой перегородкой и широким кончиком, полные щеки, низкопосаженные уши, макростомию, полные губы, тонкие и волнистые волосы. Кожа мягкая и рыхлая, гиперпигментированная, на ладонях и ступнях с тенденцией к гиперкератозу в старшем возрасте. Около 75% пациентов имеют патологии сердечно-сосудистой системы такие как: гипертрофия миокарда, стеноз легочной артерии, аритмия (тахикардия предсердий), расширение аорты [50]. Отмечается задержка темпов моторного и психо-речевого развития.
Синдром Костелло связан с повышенным риском развития новообразований. У 75% больных в детском возрасте возникают папилломы вокруг носа и рта, как первичные опухоли [52].
Нейрофиброматоз типа 1 (НФ1) наследуется по аутосомно-доминантному типу, встречается с частотой один на 3000 новорожденных. Характеризуется внутри- и межсемейными полиморфизмами. Основные признаки заболевания: кофейные пятна на коже, выявляются у 95% больных. Они появляются еще в неонатальном периоде, и в первые годы жизни количество таких пятен увеличивается. Типичным видом доброкачественных опухолей нервной системы у больных НФ1 является глиома зрительного нерва, проявляющаяся в различных нарушениях зрения [10, 53].
Для больных НФ1 характерно появление нейрофибром, доброкачественных опухолей на различных участках тела, которые с течением времени могут перерастать в злокачественные. Их форма, размер и количество могут увеличиваться с возрастом [53, 54].
Нейрофиброматоз типа 1 вызывается мутациями в гене NF1, кодирующем белок, выступающий в роли регулятора RAS-MAPK сигнального пути. Белок экспрессируется в основном в клетках нервной системы. Около 50% мутаций появляются de novo, и еще 50% наследуются от родителей. В зависимости от типа мутации и ее влияния на функционирование белка, фенотипическое проявление заболевания может быть различным [53, 54]. Также отмечено, что при микроделеции хромосомы 17, затрагивающей ген NF1, клиническая картина нейрофиброматоза тяжелее, нежели при точковых мутациях в гене [54, 55].
Помимо нейрофиброматоза типа 1, описан нейрофиброматоз типа 2. Несмотря на некоторую фенотипическую схожесть с первым типом, НФ типа 2 не относится к классу расопатий. Белок, продукт экспрессии гена NF2, не участвует в RAS-MAPK сигнальном пути и отвечает за межклеточную адгезию и трансмембранную передачу сигнала [56].
Для некоторых синдромов, входящих в класс расопатий, описаны как наследственные формы, так и соматические мозаичные варианты. Например для синдрома Костелло и нейрофиброматоза типа 1 описаны мозаичные формы, но сильно отличающиеся по фенотипу от наследственных. Также мозаичные формы некоторых расопатий были выявлены при эпидермальных невусах. На данный момент мозаичная форма описана для следующих генов: HRAS, KRAS, NRAS, NF1 [47].
Синдромы Нунан или LEOPARD не встречались в мозаичных формах. Существует единственная статья, где описывается фенотип пациента с предположительно мозаичной формой синдрома LEOPARD, но анализ на мутацию в PTPN11 показал наличие таковой и в пораженных тканях (лентиго на одной руке) и в фибробластах вне пятен [57].
МОЛЕКУЛЯРНАЯ ДИАГНОСТИКА
В 2001 г. был описан ген PTPN11 и доказана его связь с наследственными формами синдрома Нунан [5]. После чего пациентов с выявленным фенотипически СН стали тестировать на мутации в данном гене. Но около 50% случаев оставались без выявленной патогенной мутации. К 2011 г. была показана высокая генетическая гетерогенность синдрома [58]. На настоящий момент выявлено девять генов, приводящих к синдрому Нунан, и еще порядка десяти, приводящих к Нунан-подобным синдромам и подобным фенотипическим состояниям.
По данным HGMD в гене PTPN11 описано более 130 патогенных миссенс-замен, и менее 20 больших/малых делеций или инсерций. Для гена RAF1 описано около 40 патогенных миссенс-вариантов, и всего три больших делеции/инсерции. Для гена SOS1 описано более 60 патогенных вариантов, и только два из них – большие перестройки, остальные варианты – миссенс, либо делеции/инстерции 1–5 букв. Для остальных генов, выходящих в RAS-MAPK сигнальный путь, протяженных делеций и инсерций так же не описано; кроме гена NF1, который не является основным для синдрома Нунан [59].
Для синдрома Нунан описано, что de novo мутации встречаются в 35–70% случаев [21]. Ни в одном гене не описано “горячих” регионов, где бы чаще всего возникали патогенные замены, и что можно было бы изучать в первую очередь.
На данный момент клинический диагноз, поставленный на основании фенотипа, требует подтверждения на молекулярном уровне. Пока генов-кандидатов было немного, можно было использовать секвенирование по Сенгеру. В связи с выраженной генетической гетерогенностью синдрома Нунан наиболее эффективным способом диагностики является использование таргетных панелей генов (NRAS, RIT1, SHOC2, CBL, PTPN11, HRAS, KRAS, SOS2, MAP2K1, SOS1, LZTR1, RAF1, NEK1, BRAF). А при отсутствии патогенных вариантов – полногеномное или полноэкзомное секвенирование для поиска новых генов-кандидатов [8].
Работа выполнена в рамках государственного задания Минобрнауки России.
Настоящая статья не содержит каких-либо исследований с использованием в качестве объекта животных.
Настоящая статья не содержит каких-либо исследований с участием в качестве объекта людей.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Tartaglia M., Kalidas K., Shaw A. et al. PTPN11 mutations in Noonan syndrome: Molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity // Am. J. Hum. Genet. 2002. V. 70. P. 1555–1563.
Noonan J.A. Hypertelorism with Turner phenotype. A new syndrome with associated congenital heart disease // Am. J. Dis. Child. 1968. V. 116. P. 373–380.
Celermajer J.M., Bowdler J.D., Cohen D.H. Pulmonary stenosis in patients with the Turner phenotype in the male // Am. J. Dis. Child. 1968. V. 116. P. 351–358.
Nora J.J., Nora A.H., Sinha A.K. et al. The Ullrich-Noonan syndrome (Turner phenotype) // Am. J. Dis. Child. 1974. V. 127. P. 48–55.
Tartaglia M., Mehler E.L., Goldberg R. et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome // Nat. Genet. 2001. V. 29. P. 465–468.
Romano A.A., Allanson J.E., Dahlgren J. et al. Noonan Syndrome: Clinical features, diagnosis, and management guidelines // Pediatrics. 2010. V. 126. P. 746–759. https://doi.org/10.1542/peds.2009-3207
Tartaglia M., Zampino G., Gelb B.D. Noonan syndrome: Clinical aspects and molecular pathogenesis // Mol. Syndromol. 2010. V. 1. P. 2–26. https://doi.org/10.1159/000276766
Lepri F.R., Scavelli R., Digilio M.C. et al. Diagnosis of Noonan syndrome and related disorders using target next generation sequencing // BMC Med. Genet. 2014. V. 15. P. 14. https://doi.org/10.1186/1471-2350-15-14
Tidyman W.E., Rauen K.A. The RASopathies: Developmental syndromes of RAS/MAPK pathway dysregulation // Curr. Opin. Genet. Dev. 2009. V. 19. P. 230–236.
Bezniakow N., Gos M.O.E. The rasopathies as an example of RAS/MAPK pathway disturbances – clinical presentation and molecular pathogenesis of selected syndromes* // Dev. Period. Med. 2014. V. 18. P. 285–296.
Schubbert S., Shannon K., Bollag G. Hyperactive RAS in developmental disorders and cancer // Nat. Rev. Cancer. 2007. V. 7. P. 295–308.
van der Burgt I., Berends E., Lommen E. et al. Clinical and molecular studies in a large Dutch family with Noonan syndrome // Am. J. Med. Genet. 1994. V. 53. P. 187–191.
Yoshida R., Hasegawa T., Hasegawa Y. et al. Protein-tyrosine phosphatase, nonreceptor type 11 mutation analysis and clinical assessment in 45 patients with Noonan syndrome // J. Clin. Endocrinol. Metab. 2004. V. 89. P. 3359–3364.
Zenker M., Buheitel G., Rauch R. et al. Genotype-phenotype correlations in Noonan syndrome // J. Pediatr. 2004. V. 144. P. 368–374.
Jongmans M., Otten B., Noordam K. et al. Genetics and variation in phenotype in Noonan syndrome // Horm. Res. 2004. V. 62. Suppl. 3. P. 56–59.
Bhambhani V., Muenke M. Noonan syndrome // Am. Fam. Physician. 2014. V. 89. P. 37–43.
Gelb B.D., Tartaglia M. Noonan syndrome with multiple lentigines // GeneReviews NCBI. 1993.
Abdel-Salam E., Temtamy S.A. Familial Turner phenotype // J. Pediatr. 1969. V. 74. P. 67–72.
Maximilian C., Ioan D.M., Fryns J.P. A syndrome of mental retardation, short stature, craniofacial anomalies with palpebral ptosis and pulmonary stenosis in three siblings with normal parents. An example of autosomal recessive inheritance of the Noonan phenotype? // Genet. Couns. 1992. V. 3. P. 115–118.
van Der Burgt I., Brunner H. Genetic heterogeneity in Noonan syndrome: Evidence for an autosomal recessive form // Am. J. Med. Genet. 2000. V. 94. P. 46–51.
Roberts A.E., Allanson J.E., Tartaglia M. et al. Noonan syndrome // Lancet. 2013. V. 381. P. 333–342. https://doi.org/10.1016/S0140-6736(12)61023-X
Jorge A.A.L., Malaquias A.C., Arnhold I.J.P. et al. Noonan syndrome and related disorders: A review of clinical features and mutations in genes of the RAS/MAPK pathway // Horm. Res. 2009. V. 71. P. 185–193.
Zenker M., Lehmann K., Schulz A.L. et al. Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations // J. Med. Genet. 2007. V. 44. P. 131–135.
Kratz C.P., Zampino G., Kriek M. et al. Craniosynostosis in patients with Noonan syndrome caused by germline KRAS mutations // Am. J. Med. Genet A. 2009. V. 149A. P. 1036–1040.
Friday B.B., Adjei A.A. K-ras as a target for cancer therapy // Biochim. Biophys. Acta. 2005. V. 1756. P. 127–144.
Plowman S.J., Williamson D.J, O’Sullivan M.J. et al. While K-ras is essential for mouse development, expression of the K-ras 4A splice variant is dispensable // Mol. Cell Biol. 2003. V. 23. P. 9245–9250.
Zenker M., Horn D., Wieczorek D. et al. SOS1 is the second most common Noonan gene, but plays no major role in cardio-facio-cutaneous syndrome // J. Med. Genet. 2007. V. 44. P. 651–656.
Tartaglia M., Pennacchio L.A., Zhao C. et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome // Nat. Genet. 2007. V. 39. P. 75–79.
Pierpont E.I., Pierpont M.E., Mendelsohn N.J. et al. Genotype differences in cognitive functioning in Noonan syndrome // Genes Brain Behav. 2009. V. 8. P. 275–282.
Egan S.E., Weinberg R.A. The pathway to signal achievement // Nature. 1993. V. 365. P. 781–783.
Takai Y., Sasaki T., Matozaki T. Small GTP-binding proteins // Physiol. Rev. 2001. V. 81. P. 153–208.
Roberts A.E., Araki T., Swanson K.D. et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome // Nat. Genet. 2007. V. 39. P. 70–74.
Lepri F., De Luca A., Stella L. et al. SOS1 mutations in Noonan syndrome: Molecular spectrum, structural insights on pathogenic effects, and genotype-phenotype correlations // Hum. Mutat. 2011. V. 32. P. 760–772.
Wellbrock C., Karasarides M., Marais R. The RAF proteins take centre stage // Nat. Rev. Mol. Cell Biol. 2004. V. 5. P. 875–885.
Leicht D.T., Balan V., Kaplun A. et al. Raf kinases: Function, regulation and role in human cancer // Biochim. Biophys. Acta. 2007. V. 1773. P. 1196–1212.
Cirstea I.C., Kutsche K., Dvorsky R. et al. A restricted spectrum of NRAS mutations causes Noonan syndrome // Nat. Genet. 2010. V. 42. P. 27–29.
Denayer E., Peeters H., Sevenants L. et al. NRAS mutations in Noonan syndrome // Mol. Syndromol. 2012. V. 3. P. 34–38.
Sarkozy A., Carta C., Moretti S. et al. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: Molecular diversity and associated phenotypic spectrum // Hum. Mutat. 2009. V. 30. P. 695–702.
Nystrom A.M., Ekvall S., Berglund E. et al. Noonan and cardio-facio-cutaneous syndromes: Two clinically and genetically overlapping disorders // J. Med. Genet. 2008. V. 45. P. 500–506.
Aoki Y., Niihori T., Banjo T. et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome // Am. J. Hum. Genet. 2013. V. 93. P. 173–180.
Yamamoto G.L., Aguena M., Gos M. et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome // J. Med. Genet. 2015. V. 52. P. 413–421.
Wang D.Z., Hammond V.E., Abud H.E. et al. Mutation in Sos1 dominantly enhances a weak allele of the EGFR, demonstrating a requirement for Sos1 in EGFR signaling and development // Genes Dev. 1997. V. 11. P. 309–320.
Johnston J.J., van der Smagt J.J., Rosenfeld J.A. et al. Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants // Genet. Med. 2018. P. 1–11. https://doi.org/10.1038/gim.2017.249
Nisbet D.L., Griffin D.R., Chitty L.S. Prenatal features of Noonan syndrome // Prenat. Diagn. 1999. V. 19. P. 642–647.
Maheshwari M., Belmont J., Fernbach S. et al. PTPN11 mutations in Noonan syndrome type I: Detection of recurrent mutations in exons 3 and 13 // Hum. Mutat. 2002. V. 20. P. 298–304.
Tafazoli A., Eshraghi P., Koleti Z.K. et al. Noonan syndrome – a new survey // Arch. Med. Sci. 2017. V. 13. P. 215–222.
Hafner C., Groesser L. Mosaic RASopathies // Cell Cycle. 2013. V. 12. P. 43–50.
Siegel D.H., McKenzie J., Frieden I.J. et al. Dermatological findings in 61 mutation-positive individuals with cardiofaciocutaneous syndrome // Brit. J. Dermatol. 2011. V. 164. P. 521–529.
Rauen K.A. NIH public access // Annu. Rev. Genomics Hum. Genet. 2013. P. 355–369.
Gripp K.W., Lin A.E. Costello syndrome // GeneReviews NCBI. 1993. https://www.ncbi.nlm.nih.gov/books/NBK1507/
Smith L.P., Podraza J., Proud V.K. Polyhydramnios, fetal overgrowth, and macrocephaly: Prenatal ultrasound findings of Costello syndrome // Am. J. Med. Genet. A. 2009. V. 149A. P. 779–784.
Siegel D.H., Mann J.A., Krol A.L. et al. Dermatological phenotype in Costello syndrome: Consequences of RAS dysregulation in development // Brit. J. Dermatol. 2012. V. 166. P. 601–607.
Boyd K.P., Korf B.R., Theos A. Neurofibromatosis type 1 // J. Am. Acad. Dermatol. 2009. V. 61. P. 1–6.
Friedman J.M. Neurofibromatosis 1 // GeneReviews NCBI. 1993. https://www.ncbi.nlm.nih.gov/books/NBK1109/
Abramowicz A., Gos M. Neurofibromin in neurofibromatosis type 1 – mutations in NF1gene as a cause of disease // Dev. Period Med. 2014. V. 18. P. 297–306.
Curto M., McClatchey A.I. Nf2/Merlin: A coordinator of receptor signalling and intercellular contact // Brit. J. Cancer. 2008. V. 98. P. 256–262.
Writzl K., Hoovers J., Sistermans E.A. et al. LEOPARD syndrome with partly normal skin and sex chromosome mosaicism // Am. J. Med. Genet A. 2007. V. 143A. P. 2612–2615.
Tartaglia M., Gelb B.D., Zenker M. Noonan syndrome and clinically related disorders // Best Pract. Res. Clin. Endocrinol. Metab. 2011. V. 25. P. 161–179.
https://portal.biobase-international.com/hgmd/pro/.
Дополнительные материалы отсутствуют.