

Генетика, 2020, T. 56, № 5, стр. 495-513
Геномные и постгеномные технологии в генетике преэклампсии
Е. А. Трифонова 1, 2, *, М. Г. Сваровская 1, 2, В. Н. Сереброва 1, И. Г. Куценко 2, Л. А. Агаркова 2, И. А. Степанов 3, О. В. Жилякова 2, Т. В. Габидулина 2, Е. В. Ижойкина 3, В. А. Степанов 1
1 Научно-исследовательский институт медицинской генетики,
Томский национальный исследовательский медицинский центр Российской академии наук
634050 Томск, Россия
2 Сибирский государственный медицинский университет
634050 Томск, Россия
3 Областной перинатальный центр им. И.Д. Евтушенко
634063 Томск, Россия
* E-mail: ekaterina.trifonova@medgenetics.ru
Поступила в редакцию 30.04.2019
После доработки 05.07.2019
Принята к публикации 08.08.2019
Аннотация
В представленном обзоре рассмотрена роль геномных и постгеномных технологий в раскрытии генетической архитектуры преэклампсии (ПЭ) – тяжелого гипертензивного расстройства беременности, обусловливающего значительный уровень материнской и перинатальной заболеваемости и смертности. Отмечается особая актуальность интегративного анализа геномных, метиломных, транскриптомных и протеомных данных в характеристике молекулярных механизмов ПЭ и идентификации новых генов-кандидатов и мишеней для таргетной терапии этого гестационного осложнения.
Изучение генетической архитектуры многофакторных заболеваний (МФЗ) человека является одной из самых сложных проблем в медицинской генетике. Следует отметить, что наследственная природа широко распространенных заболеваний по-прежнему в значительной степени остается областью terra incognita: полученные к настоящему времени данные об ассоциации полиморфных вариантов генов-кандидатов с развитием МФЗ в большинстве случаев объясняют не более 10–15% их наследуемости [1, 2]. Интенсивно изучаемый в последнее десятилетие феномен “недостающей наследственности” (missing heritability), заключающийся в невозможности на данном этапе полностью объяснить наследственную компоненту многих признаков и патологических состояний, свидетельствует о необходимости анализа ряда факторов, участвующих в модификации доли наследственности в патогенезе МФЗ. Так, существенный вклад в наследственную изменчивость предположительно могут вносить: 1) неохваченные методом GWAS генетические вариации, например изменения генома, варьирующие по числу копий повторов ДНК (copy number variations – CNV), редкие мутантные аллели (популяционная частота которых составляет менее 5%); 2) ген-генные и ген-средовые взаимодействия; 3) экологические и эпигенетические факторы [3].
Завершение проекта “Геном человека” в 2003 г. ознаменовало начало новой эры в медико-биологических исследованиях и стимулировало беспрецедентный технический прогресс в области наук о жизни, включая развитие высокопроизводительных технологий для обнаружения вариабельности генома, метилома и транскриптома, которое сопровождается накоплением огромного объема экспериментальных данных. Анализ полученного массива геномной информации с целью более глубокого понимания молекулярных механизмов многофакторных заболеваний является одной из наиболее актуальных задач современных генетических исследований. В представленном обзоре рассмотрена роль геномных и постгеномных технологий в раскрытии генетической архитектуры преэклампсии (ПЭ) – тяжелого гипертензивного расстройства беременности, обусловливающего значительный уровень материнской и перинатальной заболеваемости и смертности.
На сегодняшний день известно более 30 теорий этиопатогенеза ПЭ, включая и гипотезу генетической детерминированности. Однако ни одна из них не объясняет однозначно и в полной мере многообразие происходящих при данном осложнении беременности морфофункциональных изменений и клинических манифестаций, что, очевидно, связано с многофакторностью этого патологического состояния, и как следствие этого – существенной ролью в предрасположенности наряду с генетическими средовых факторов [4, 5].
ОЦЕНКА РОЛИ НАСЛЕДСТВЕННОСТИ В РАЗВИТИИ ПРЕЭКЛАМПСИИ
Проведенные к текущему времени многочисленные эпидемиологические исследования продемонстрировали значительный вклад наследственной компоненты в структуру предрасположенности к преэклампсии, доля которой в различных популяциях варьирует в зависимости от их географических, социально-экономических и расовых особенностей, что свидетельствует о принадлежности данной патологии к группе многофакторных заболеваний [5–7]. Близнецовые исследования также указывают на существенную роль в подверженности к преэклампсии как генетических, так и средовых факторов (коэффициент наследуемости оценивается от 30 до 54%) [8–10]. Следует отметить, что семейный характер наследования ПЭ отмечался многими авторами [10–13], однако наиболее широкомасштабное исследование в этом направлении было проведено в период с 1967 по 1992 г. норвежскими учеными, которые показали, что женщины, являющиеся дочерями пациенток, беременность которых осложнялась этим заболеванием, имеют двукратный риск развития данной патологии [14]. Кроме того, в этой работе было обнаружено, что наличие родственной связи с больным пробандом ассоциировано с более тяжелым течением ПЭ: в семьях индивидов, рожденных от пациенток с этим гестационным осложнением, гораздо чаще наблюдалась тяжелая форма ПЭ (отношение шансов (OR) составило 3.0 для семей дочерей (95%-ный доверительный интервал – 95%CI: 2.4–3.7) и 1.9 для семей сыновей (95%CI: 1.4–2.5)). На основании полученных данных авторы сделали вывод, что наряду с материнскими генами существенный вклад в генетическую архитектуру ПЭ вносят фетальные гены отцовского происхождения [14]. О возможной роли отцовских генов в структуре подверженности к этому заболеванию свидетельствует также повышенный риск его развития в случае беременности от мужчин, предыдущие супруги которых имели такие гестационные осложнения как ПЭ. Механизмы, опосредующие это наблюдение, предположительно связаны с геномным импринтингом: значимостью отцовских генов в инвазии цитотрофобласта и плацентарном росте наряду с антагонистической функцией материнских генов, подавляющих эти процессы в рамках формирования адаптивного иммунного ответа при беременности [15].
В конце XX в. наибольшую популярность при изучении факторов наследственной предрасположенности к ПЭ бесспорно приобрела стратегия анализа ассоциаций генов-кандидатов по типу случай–контроль. Наиболее крупномасштабное исследование генетической архитектуры ПЭ в рамках вышеописанного подхода, включившее более 1800 человек, было проведено Goddard с коллегами [16], которые изучили 775 однонуклеотидных полиморфных вариантов (SNPs) в 190 генах-кандидатах. Авторы продемонстрировали высокодостоверную ассоциацию (р < 0.01) с данным фенотипом аллельных вариантов пяти локусов: гена рецептора простагландина Е2 типа (PTGER2), гена β1-рецептора интерлейкина 12 (IL12RB1), гена α2-цепи коллагена IV типа (COL4A2), гена α1‑цепи коллагена I типа (COL1A1) и гена интерлейкина 1-альфа (IL1A), однако исследователями не было выявлено связи с ПЭ таких наиболее вероятных генов-кандидатов как ген метилентетрагидрофолатредуктазы (MTHFR) и ген эндотелиальной синтазы оксида азота (NOS3) [16]. К текущему моменту изучено более 400 генов-кандидатов ПЭ, имеющих отношение к тромбофилии и гипофибринолизу, метаболизму фолиевой кислоты, окислительному стрессу, функционированию ренин-ангиотензиновой системы, метаболизму липидов, иммунному ответу и др. (база данных “HuGЕ Navigator”). Однако результаты этих работ нередко являются противоречивыми даже в рамках изучения одной и той же этнической группы, а общепринятых генетических предикторов ПЭ с помощью данного подхода выявить так и не удалось [7, 17, 18]. Важно подчеркнуть, что существенный недостаток подобного анализа ассоциаций отдельных локусов в рамках исследования генетических факторов ПЭ – необходимость точного знания молекулярных механизмов болезни для выбора оптимального гена-кандидата. Решением данной проблемы является использование метода полногеномного анализа ассоциаций (Genome-Wide Association Study, GWAS), позволяющего изучать связь с заболеванием большого массива полиморфных вариантов генов вне зависимости от биологической гипотезы о патофизиологии ПЭ.
ПОЛНОГЕНОМНЫЙ АНАЛИЗ АССОЦИАЦИЙ SNPs С ПРЕЭКЛАМПСИЕЙ
Первый полногеномный анализ ассоциации полиморфных вариантов генов с ПЭ с применением технологии микрочипов был выполнен в 2012 г. в австралийской популяции [19] и продемонстрировал связь с данной патологией 14 генетических маркеров (табл. 1), однако после коррекции на множественные сравнения (поправка Бонферрони) статистически значимую ассоциацию с ПЭ показали только два однонуклеотидных полиморфных варианта – rs7579169 (OR = 1.57) и rs12711941 (OR = 1.56), локализованных на второй хромосоме (2q14.2) на расстоянии 15 тпн от 3'-нетранслируемой области гена ингибина бета (INHBB). Анализ архитектуры неравновесия по сцеплению в регионе ±250 тпн вблизи данных SNPs показал наличие сильного сцепления между этими маркерами (r2 = 0.92) и отсутствие их значимого сцепления (r2 < 0.80) с другими полиморфными вариантами в изучаемом участке генома. Последующее исследование дифференциальной экспрессии генов, расположенных в вышеобозначенном локусе второй хромосомы, в децидуальной ткани пациенток с ПЭ и контрольной группы выявило статистически значимую корреляцию с данной патологией гена EPB41L5, кодирующего мембранный протеин эритроцитов [19].
Таблица 1.
Полиморфные варианты, ассоциированные с преэклампсией по данным GWAS
| № | Хромосома | Полиморфный вариант | Локализация в гене | Ген | Аллели1 | OR2 | 95%-ный доверительный интервал |
|---|---|---|---|---|---|---|---|
| Исследование Johnson и соавт. [19] | |||||||
| 1 | 2 | rs7579169 | м/р | C/T | 1.57 | 1.32–1.87 | |
| 2 | 2 | rs12711941 | » | G/T | 1.56 | 1.31–1.86 | |
| 3 | 15 | rs2453274 | Интрон | CTDSPL2 | C/T | 0.44 | 0.31–0.62 |
| 4 | 13 | rs12431203 | м/р | G/A | 1.73 | 1.38–2.18 | |
| 5 | 21 | rs2826538 | » | T/C | 0.63 | 0.52–0.77 | |
| 6 | 2 | rs9332419 | Интрон | RAB10 | G/A | 0.67 | 0.56–0.80 |
| 7 | 2 | rs4952830 | 5'UTR | ATP6V1E2 | A/G | 0.67 | 0.56–0.80 |
| 8 | 2 | rs6542736 | м/р | G/A | 1.50 | 1.25–1.79 | |
| 9 | 1 | rs6660579 | Интрон | CNIH3 | C/T | 1.81 | 1.39–2.36 |
| 10 | 3 | rs2279720 | Экзон | CHMP2B | G/A | 0.49 | 0.36–0.68 |
| 11 | 3 | rs1044499 | » | CHMP2B | A/C | 0.49 | 0.36–0.68 |
| 12 | 2 | rs11126375 | Интрон | RAB10 | A/G | 0.62 | 0.50–0.77 |
| 13 | 4 | rs7677523 | » | C4orf37 | T/C | 2.31 | 1.58–3.37 |
| 14 | 3 | rs17024019 | м/р | A/G | 0.50 | 0.36–0.68 | |
| Исследование Zhao и соавт. [21], приведены данные для афро-карибской популяции | |||||||
| 1 | 13 | rs11617740 | Интрон | FGF14 | A/G | 16.96 | 5.53–51.97 |
| 2 | 21 | rs2839440 | » | C21orf121 | A/G | 5.31 | 2.70–10.42 |
| 3 | 4 | rs12641856 | м/р | A/G | 15.19 | 4.98–46.35 | |
| 4 | 20 | rs4815879 | Интрон | MCM8 | A/G | 14.57 | 4.81–44.17 |
| 5 | 6 | rs28360974 | м/р | A/G | 10.13 | 3.85–26.67 | |
| 6 | 10 | rs1248993 | » | G/A | 4.27 | 2.28–8.01 | |
| 7 | 7 | rs975369 | » | A/G | 4.55 | 2.34–8.83 | |
| 8 | 20 | rs1556832 | Интрон | ADRA1D | A/G | 4.33 | 2.27–8.27 |
| 9 | 11 | rs11600901 | м/р | A/G | 9.83 | 3.58–27.00 | |
| Исследование Zhao и соавт. [21], приведены данные для европеоидов | |||||||
| 1 | 13 | rs7322722 | Интрон | MYCBP2 | A/G | 2.93 | 1.90–4.52 |
| 2 | 13 | rs624575 | » | MYCBP2 | G/A | 2.90 | 1.88–4.48 |
| 3 | 9 | rs10989019 | » | INVS | G/A | 3.21 | 1.98–5.20 |
| 4 | 9 | rs7020780 | » | INVS | A/G | 3.19 | 1.96–5.17 |
| 5 | 10 | rs10883969 | » | C10orf78 | A/G | 2.97 | 1.88–4.69 |
| 6 | 9 | rs7028939 | » | ERP44 | G/A | 3.17 | 1.95–5.15 |
| 7 | 9 | rs7047693 | » | INVS | G/A | 3.16 | 1.94–5.12 |
| 8 | 9 | rs10988989 | » | INVS | G/A | 3.16 | 1.94–5.12 |
| 9 | 3 | rs17787940 | » | WWTR1 | G/A | 3.17 | 1.95–5.16 |
| 10 | 9 | rs7849323 | » | ERP44 | G/A | 3.15 | 1.94–5.11 |
| 11 | 9 | rs1415927 | » | INVS | G/A | 3.15 | 1.94–5.11 |
| 12 | 21 | rs9976946 | » | RUNX1 | G/A | 5.50 | 2.65–11.39 |
| 13 | 13 | rs6563695 | м/р | A/G | 3.65 | 2.09–6.36 | |
| 14 | 13 | rs9530631 | Интрон | MYCBP2 | G/A | 2.69 | 1.75–4.12 |
| 15 | 13 | rs7337686 | » | MYCBP2 | G/A | 2.68 | 1.74–4.11 |
| Исследование Zhao и соавт. [21], приведены данные для испаноязычной популяции | |||||||
| 1 | 8 | rs17412740 | м/р | A/G | 6.08 | 2.88–12.81 | |
| 2 | 2 | rs17636747 | » | A/G | 8.35 | 3.41–20.46 | |
| 3 | 8 | rs1016646 | » | A/G | 5.68 | 2.63–12.25 | |
| 4 | 16 | rs7199697 | » | G/A | 3.98 | 2.16–7.33 | |
Другое полногеномное исследование ассоциаций, выполненное на относительно небольших по объемам выборках (177 пациенток с ПЭ и 116 женщин с физиологической беременностью), не обнаружило статистически значимых ассоциаций изученных полиморфных маркеров с ПЭ [20]. Причины, объясняющие полученные результаты, авторы связывают с генетической гетерогенностью, экологическими и возрастными эффектами, а также с эпистатическими взаимодействиями. Тем не менее в данной работе была зафиксирована ассоциация с развитием ПЭ делеции 19-й хромосомы (19q13.31), локализованной в гене PSG11, который кодирует трофобластический бета-1-гликопротеин.
Еще один полногеномный анализ ассоциации полиморфных вариантов генов с ПЭ, также проведенный Zhao с коллегами [21], продемонстрировал этническую специфичность генетической архитектуры этого заболевания. Так, в афро-карибской популяции наиболее значимую ассоциацию с ПЭ показали полиморфные варианты генов фактора роста фибробластов 14 (FGF14), ZNF295 антисмысловой РНК 1 (C21orf121), комплекса обслуживания минихромосом 8 (MCM8) и адренорецептора альфа 1D (ADRA1D), в то время как у европеоидов с этим заболеванием были связаны SNPs генов Myc-связывающаго белка-2 (MYCBP2), инверсина (INVS), протеина SWI5-зависимой рекомбинантной репарации (C10orf78), домена WW регулятора транскрипции 1 (WWTR1), белка 44 эндоплазматического ретикулума (ERP44), runt-связанного транскрипционного фактора-1 (RUNX1), а у испаноязычного населения была выявлена ассоциация с аллельными вариантами, расположенными в межгенных регионах вблизи локусов, кодирующих белок, ассоциированный с рецептором TGF-β (TGFBRAP1), опухолевый супрессор F37 (LZTS1) и контактин-ассоциированный белок 4 (CNTNAP4) (табл. 1). Однако после введения поправки Бонферрони все данные ассоциации не были статистически значимыми [21].
Результаты недавно опубликованного масштабного полногеномного ассоциативного исследования, проведенного международным консорциумом ученых, указывают на значимую роль в молекулярных механизмах ПЭ генома плода [22]. Так, при анализе 2658 потомков пациенток с ПЭ и 310 238 индивидов контрольной группы была установлена ассоциация с данной патологией маркера rs4769613 (р-уровень 5.4 × 10–11), локализованного в межгенном регионе генома вблизи локуса FLT1, кодирующего рецептор 1-го типа эндотелиального фактора роста сосудов, растворимая форма которого – sFLT1 является общепризнанным сывороточным маркером ПЭ [23]. Авторы подчеркивают, что ассоциация была наиболее значимой у потомства от пациенток с поздно манифестировавшей ПЭ.
На сегодняшний день полногеномные исследования ассоциации полиморфных вариантов генов с развитием ПЭ являются современным подходом в познании генетической предрасположенности к этой болезни и идентификации новых перспективных генов-кандидатов ПЭ. Однако результаты ассоциативного анализа, несомненно, требуют репликативных исследований на расширенных выборках в различных этнических группах, а также детального изучения структуры неравновесия по сцеплению в регионе генома, показавшем ассоциацию, для идентификации “причинных” аллельных вариантов и интерпретации результатов GWAS с биологической точки зрения. Необходимо отметить, что большинство из подходов, используемых для определения “причинных” аллельных вариантов, сцепленных с SNPs, детектированными в GWAS, сосредоточено на анализе кодирующих или транскрибирующихся регионов генома [3, 24]. Однако, как видно из табл. 1, подавляющее большинство SNPs, выявленных в рассмотренных GWAS (более 95%), находятся в нетранскрибируемых областях генома (интронах и межгенных регионах), и основной механизм их вовлеченности в структуру наследственной предрасположенности к ПЭ предположительно связан с регуляцией экспрессии генов. Вследствие чего при изучении генетической архитектуры ПЭ особую актуальность приобретает использование таких постгеномных технологий как измерение экспрессии генов с помощью гибридизации на микрочипах и высокопроизводительное секвенирование РНК.
ИССЛЕДОВАНИЕ ТРАНСКРИПТОМА ПЛАЦЕНТАРНОЙ ТКАНИ ПРИ ПРЕЭКЛАМПСИИ
Согласно современным представлениям, основополагающую роль в патогенезе ПЭ играет нарушение инвазии цитотрофобласта в спиральные артерии матки c последующим формированием синдрома ишемии-реперфузии и развитием системной эндотелиальной дисфункции [25]. В связи с чем при изучении молекулярных механизмов данного гестационного осложнения одним из наиболее активно разрабатываемых на текущий момент направлений является исследование транскрипционных профилей плацентарной ткани [5].
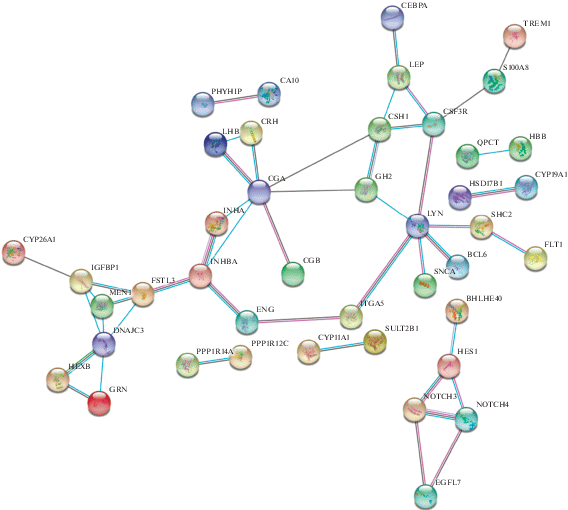
В последнее десятилетие с помощью технологии микрочипов было проведено более 30 независимых исследований, направленных на изучение дифференциальной экспрессии генов плацентарной ткани беременных с преэклампсией и женщин с физиологической беременностью, в которых было выявлено высокодостоверное повышение/снижение экспрессии нескольких сотен генов [26–44] (табл. 2). Примечательно, что, несмотря на выполнение этих работ на материале различного этнического происхождения и на различных типах микроматриц, результаты некоторых из них согласуются в отношении дифференциальной экспрессии ряда генов, играющих, вероятно, важную роль в генетической компоненте ПЭ. При обобщении данных, полученных в вышеобозначенных исследованиях, нами было обнаружено 98 генов, статистически значимо ассоциированных с преэклампсией согласно результатам трех и более транскриптомных исследований плацентарной ткани (на рис. 1 представлена сеть белок-белковых взаимодействий продуктов этих генов, а в табл. 2 дана характеристика наиболее значимых из них).
Таблица 2.
Дифференциально экспрессирующиеся гены плацентарной ткани, ассоциированные с развитием преэклампсии в различных этнических группах
| Ген | Продукт гена | FC | Этнические группы | Изменение уровня экспрессии со ссылкой на источник | |
|---|---|---|---|---|---|
| LEP | Лептин | От 1.5 до 108.9 |
Японцы, китайцы, европеоиды, афроамериканцы, монголоиды, корейцы, жители США, русские | ↑ | [26–35] |
| Н. д. | [36, 37] | ||||
| BCL6 | Транскрипционный фактор – белок цинковых пальцев 51 | От 1.5 до 2.6 |
Европеоиды, афроамериканцы, жители США, монголоиды, японцы, китайцы, русские | ↑ | [26, 27, 31, 33, 34, 38] |
| Н. д. | [37] | ||||
| SIGLEC6 | Иммуноглобулин-подобный лектин 6, связывающий сиаловую кислоту | От 1.5 до 4.5 |
Жители США, европеоиды, китайцы, русские | ↑ | [26, 34, 35, 38] |
| Н. д. | [37] | ||||
| FLT1 | Рецептор сосудистого эндотелиального фактора роста 1 | От 2.41 до 5.98 |
Жители США, японцы, корейцы, шведы, европеоиды | ↑ | [27, 29–32, 34, 39, 40] |
| Н. д. | [36, 37] | ||||
| INHA | Альфа-субъединица ингибина | От 1.9 до 4.04 |
Шведы, жители США, европеоиды | ↑ | [27, 31, 33, 34, 40] |
| Н. д. | [37] | ||||
| INHBA | Бета-субъединица ингибина | От 2.06 до 4.9 |
Европеоиды, японцы, жители США | ↑ | [28, 31–34] |
| Н. д. | [36, 37] | ||||
| CGB | Бета-субъединица хорионического гонадотропина | От 3.32 до 10.2 |
Европеоиды, финны, японцы, жители США | ↑ | [31, 34, 35, 41, 42] |
| Н. д. | [37] | ||||
| ENG | Эндоглин | От 1.97 до 3.9 |
Европеоиды, японцы, жители США | ↑ | [31, 32, 34, 43] |
| Н. д. | [36, 37] | ||||
| PAPPA2 | Ассоциированный с беременностью плазменный препробелок А2 | От 2.38 до 4.9 |
Европеоиды, японцы, жители США | ↑ | [31, 35, 43] |
| Н. д. | [36, 27, 44 ] | ||||
| CRH | Кортиколиберин | От 3.43 до 3.66 |
Европеоиды, финны, японцы, жители США | ↑ | [31, 41] |
| Н. д. | [36, 37, 44] | ||||
Рис. 1.
Сеть белок-белковых взаимодействий продуктов генов, ассоциированных с ПЭ, по данным трех и более транскриптомных исследований плацентарной ткани. Толщина линии между белками отражает доверительный уровень (combined score > 0.9) связи.
Как видно из представленной табл. 2, в большинстве работ максимальные изменения уровня экспрессии в плацентарной ткани зафиксированы для гена LEP, кодирующего лептин – адипоцит-специфический гормон, регулирующий энергетический обмен организма. Интересным представляется тот факт, что данный протеин является одним из ранее описанных сывороточных маркеров ПЭ. Известно, что плацентарный лептин обеспечивает приток питательных веществ к фетоплацентарному комплексу и индуцирует пролиферацию трофобласта путем ингибирования апоптоза [45]. Таким образом, увеличение производства лептина в плаценте может являться компенсаторным механизмом против эндотелиальной дисфункции, наблюдаемой при ПЭ. Наряду с этим было показано участие лептина в активации симпатоадреналовой системы, что способствует возникновению артериальной гипертензии [46]. Кроме того, обнаружена важная иммуномодулирующая функция лептина, которая также может опосредовать роль лептина в патологии беременности [47]. Несмотря на множество работ по изучению экспрессии гена LEP, существует крайне мало исследований, посвященных анализу наследственной вариабельности этого гена и ее роли в изменении уровня транскрипции и структуре подверженности к патологии беременности. Показано, что носители генотипа АА локуса rs2167270 (G19A), расположенного в области промотора гена LEP, имеют повышенный уровень его экспрессии в крови и более высокий риск развития ПЭ и артериальной гипертензии [48, 49]. В чешской популяции была выявлена ассоциация другого полиморфизма – G2548A (rs7799039), локализованного в промоторе гена LEP, с гестационным диабетом [50]. Аналогичные результаты были продемонстрированы и для ПЭ [51]. Необходимо подчеркнуть, что все вышеописанные результаты были получены для европеоидных популяций.
С целью оценить потенциальную биологическую значимость ассоциаций, выявленных в транскриптомных исследованиях молекулярных механизмов ПЭ, с помощью вычислительного метода “GSEA” (GeneSetEnrichmentAnalysis) и базы данных “STRING” мы провели функциональную аннотацию и анализ белковых сетей, включающих идентифицированные гены-кандидаты ПЭ. В представленной работе при проведении функциональной аннотации в ресурсе Molecular Signatures Database (MSigDB) программного обеспечения “GSEA” учитывали категории, имеющие FDR (false discovery rate) <0.00001 (в данном случае FDR представляет собой вероятность неслучайного попадания группы генов в категорию с поправкой на множественные сравнения) [52]. Следует отметить, что большая часть из изученных генов одновременно вовлечена в реализацию нескольких генных онтологий и молекулярных процессов (например, более 70% генов включены минимум в три из описанных категорий). В табл. 3 представлены наиболее значимые биологические пути, процессы и молекулярные функции, обогащенные данными генами-кандидатами ПЭ (полный список всех идентифицированных категорий в ресурсе MSigDB доступен по запросу у авторов). Интересным представляется, что изучаемые гены максимально представлены в такой категории как “GO_IMMUNE_SYSTEM_PROCESS”, связанной с процессами, вовлеченными в развитие или функционирование иммунной и других систем организма, участвующих в защите от чужеродных агентов. Вероятно, этот факт служит доказательством иммуногенетической теории развития ПЭ, которая рассматривает эту патологию как проявление иммунологического конфликта, возникающего на основе генетически обусловленной антигенной неоднородности организма матери и плода [53]. Существует множество моделей, показывающих роль материнского иммунного ответа в развитии ПЭ. Например, риск возникновения этого гестационного осложнения увеличивается при наличии у матери аутоиммунного заболевания. Кроме того, выявлено, что у женщин с ПЭ значительно повышен уровень провоспалительных цитокинов IL-6 и TNF-α и снижен уровень противовоспалительного цитокина IL-10 в сыворотке. В работах по изучению ПЭ на моделях животных было показано, что при введении беременным крысам и бабуинам TNF-α наблюдается повышение концентрации белка в моче, увеличение артериального давления и повышение уровня антиангиогенного фактора s-FLT1 [54]. Дополнительным свидетельством важности иммуногенетических механизмов в развитии данного заболевания является принадлежность ряда изученных генов к категории “GGGAGGRR_MAZ_Q6”, указывающей на наличие в их структуре сайтов связывания для белков семейства транскрипционных факторов Myc, среди мишеней которых описаны гены, участвующие в пролиферации и дифференцировке клеток, апоптозе и регуляции метаболизма [55]. Значимость этих процессов в молекулярных механизмах ПЭ подтверждается также включенностью изученных генов в такие категории как “GO_RECEPTOR_BINDING”, “GO_REGULATION_OF_CELL_PROLIFER-ATION”, “GO_POSITIVE_REGULATION_OF_CELL_COMMUNICATION”, “GO_POSITIVE_REGULATION_OF_RESPONSE_TO_STIMULUS”, “GO_POSITIVE_REGULATION_OF_PROTEIN_METABOLIC_PROCESS” (табл. 3).
Таблица 3.
Наиболее значимые категории, обогащенные изучаемыми генами-кандидатами ПЭ, полученные с помощью ресурса MSigDB
| № | Категория в базе данных | Описание основных процессов категории | Гены, входящие в категорию | FDR q-value |
|---|---|---|---|---|
| 1 | Процессы, протекающие в иммуной системе (GO_IMMUNE_SYSTEM_PROCESS) |
Процессы, связанные с функционированием иммунной и других систем организма, дающие адекватные ответы на потенциальные внутренние или внешние угрозы | LEP, ITGA5, FLT1, NDRG1, INHBA, HES1, INHA, CEBPA, BCL6, LYN, SNCA, NOD1, S100A8, DNAJC3, EBI3, EPAS1, FSTL3, NOTCH4, CSF3R, IFITM2, CFH, TREM1, PROCR, KIF2A | 2.44 × 10–9 |
| 2 | Позитивная регулировка белкового метаболизма (GO_POSITIVE_REGULATION_OF_PROTEIN_METABOLIC_PROCESS) |
Процессы, активирующие или повышающие частоту, скорость или уровень химических реакций и белковых взаимодействий | LEP, ITGA5, FLT1, INHBA, HES1, INHA, CEBPA, BCL6, LYN, SNCA, NOD1, S100A8, DNAJC3, EBI3, ENG, SEPT4, CRH, GBA, SHC2, NOX4, SASH1, ST5, TTK |
2.83 × 10–10 |
| 3 | Регуляция белковых модификаций (GO_REGULATION_OF_PROTEIN_MODIFICATION_PROCESS) |
Процессы, влияющие на частоту, скорость или уровень ковалентной модификации одного или нескольких аминокислотных остатков в белке | LEP, ITGA5, FLT1, INHBA, HES1, INHA, CEBPA, BCL6, LYN, SNCA, NOD1, DNAJC3, ENG, SEPT4, CRH, GBA, SHC2, NOX4, SASH1, ST5, TTK, MEN1, PPP1R14A |
1.36 × 10–9 |
| 4 | Клеточный ответ на воздействие органических веществ (GO_CELLULAR_RESPONSE_TO_ORGANIC_SUBSTANC) | Процессы, приводящие к изменению состояния или активности клетки (движение, секреция, производство ферментов, экспрессия генов и т.д.) в результате воздействия органического вещества | LEP, FLT1, INHBA, CEBPA, LYN, SNCA, NOD1, DNAJC3, EBI3, CSF3R, IFITM2, ENG, IGFBP1, CRH, GBA, NOX4, MEN1, PTGIS, NRIP1, TGFB1I1, CYP11A1, CSH1, GH2 |
3.09 × 10–9 |
| 5 | Позитивная регуляция ответа на какое-либо воздействие (GO_POSITIVE_REGULATION_OF_RESPONSE_TO_STIMULUS) | Процессы, изменяющие состояние или активность клетки (движение, секреция, производство ферментов, генная экспрессия и т.д.) в ответ на какой-либо стимул | LEP, ITGA5, FLT1, INHBA, HES1, INHA, LYN, SNCA, NOD1, S100A8, CFH, ENG, SEPT4, CRH,
GBA, SHC2, NOX4, SASH1, ST5, TTK, MEN1, PTGIS, TGFB1I1 |
5.80 × 10–9 |
| 6 | Позитивная регуляция клеточного взаимодействия (GO_POSITIVE_REGULATION_OF_CELL_COMMUNICATION) | Процессы, повышающие частоту, скорость и степень клеточного взаимодействия (передача сигналов или взаимодействие между клетками, между клеткой и внеклеточным матриксом или между клеткой и окружающей средой) | LEP, ITGA5, FLT1, INHBA, HES1, INHA, LYN, SNCA, NOD1, S100A8, ENG, SEPT4, CRH, GBA,
SHC2, NOX4, SASH1, ST5, TTK, MEN1, PTGIS, TGFB1I1 |
1.36 × 10–9 |
| 7 | Регуляция метаболизма фосфора (GO_REGULATION_OF_PHOSPHORUS_METABOLIC_PROCESS) | Процессы, изменяющие частоту, скорость или уровень метаболизма фосфора или его соединения | LEP, ITGA5, FLT1, INHBA, HES1, INHA, CEBPA, LYN, SNCA, NOD1, DNAJC3, ENG, CRH, GBA,
SHC2, NOX4, SASH1, ST5, TTK, MEN1, PPP1R14A, GPR116 |
2.44 × 10–9 |
| 8 | Позитивная регуляция процессов биосинтеза (GO_POSITIVE_REGULATION_OF_BIOSYNTHETIC_PROCESS) | Процессы, активирующие частоту, скорость и уровень биосинтеза | 22 INHBA, HES1, CEBPA, SNCA, NOD1, S100A8, DNAJC3, EBI3, EPAS1, FSTL3, NOTCH4, ENG, CRH, NOX4, MEN1, NRIP1, HEXB, TGFB1I1, CGA, GPR116, NOTCH3, HBB |
1.07 × 10–8 |
| 9 | (GGGAGGRR_MAZ_Q6) | В категорию входят гены, имеющие один и более повтор GGGAGGRR-мотива, который соответствует сайту связывания транскрипционного фактора V $MAZ_Q6 (v7.4 TRANSFAC) | ITGA5, HES1, INHA, SNCA, EPAS1, FSTL3, BHLHE40, TPBG, SEPT4, ST5, MEN1, GRN, CYP26A1,
CSH1, GH2, NOTCH3, CA10, PHYHIP, HMHA1, SULT2B1, FKBP2, HOXA4 |
4.34 × 10–7 |
| 10 | Репродукция (GO_REPRODUCTION) | Процессы репродукции особей, несущих генетический материал, унаследованный от одного или нескольких родительских организмов | LEP, INHBA, HES1, INHA, CEBPA, BCL6, EPAS1, FSTL3, KRT19, SEPT4, CRH, MEN1, PTGIS, HTRA1, LHB, CYP19A1, NRIP1, HEXB, SPAG4, GRN, PSG6 |
6.89 × 10–10 |
| 11 | Связывание с рецептором (GO_RECEPTOR_BINDING) | Процессы избирательного и нековалентного взаимодействия гормонов, нейротрансмиттеров, лекарственных средств или внутриклеточных переносчиков со специфическими сайтами молекулы рецептора, приводящие к изменению клеточных функций | LEP, ITGA5, INHBA, INHA, LYN, S100A8, EBI3, ENG, IGFBP1, CRH, GBA, SHC2, LHB, NRIP1, GRN, TGFB1I1, CGA, CSH1, GH2, CGB1, PVRL4 | 2.86 × 10–9 |
| 12 | Регуляция клеточной пролиферации (GO_REGULATION_OF_CELL_PROLIFERATION) | Процессы, изменяющиe частоту, скорость и уровень клеточной пролиферации | LEP, FLT1, NDRG1, INHBA, HES1, INHA, CEBPA, BCL6, LYN, EBI3, ENG, CRH, NOX4, TTK, MEN1, HTRA1, TGFB1I1, CGA, NOTCH3, EGFL7 | 1.82 × 10–8 |
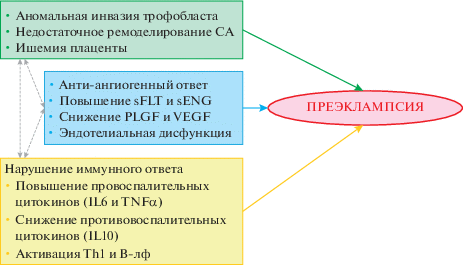
Важно отметить, что по мнению большинства исследователей ни одна из существующих этиопатогенетических теорий ПЭ в отдельности не может объяснить механизмов развития данного осложнения беременности, и вероятно в возникновении ПЭ существенную роль играет сочетание нескольких патологических механизмов, основные из которых представлены на рис. 2. Полученные в настоящем исследовании результаты биоинформатического анализа транскриптомных данных являются подтверждением вышеописанной гипотезы.
Рис. 2.
Патогенетические факторы развития преэклампсии (по [56], с модификацией авторов). Th1 – T-хелперы 1-го типа, B-лф – B-лимфоциты, СА – спиральные артерии, sFLT – растворимая форма рецептора I типа эндотелиального фактора роста сосудов, sENG – растворимый эндоглин, PLGF – плацентарный фактор роста, VEGF – фактор роста эндотелия сосудов, IL6 – интерлейкин 6, IL10 – интерлейкин 10, TNFα – фактор некроза опухоли.
Представленная на рис. 1 сеть белок-белковых взаимодействий, полученная с помощью базы данных “STRING”, подтверждает взаимосвязи между генами, выявленные при анализе биологических путей и процессов. При анализе данной сети, включающей 98 протеинов, кодируемых генами, ассоциированными с преэклампсией согласно результатам трех и более транскриптомных исследований плацентарной ткани, были выявлены только 29 генов, для которых доверительный уровень взаимодействий (combined score) составляет более 0.9: BCL6, CEBPA, CGA, CRH, CSH1, CYP11A1, CYP19A1, DNAJC3, ENG, FLT1, FSTL3, GH2, GRN, HES1, HEXB, HMHA1, HSD17B1, IGFBP1, INHA, INHBA, LEP, LHB, LYN, MEN1, NOTCH3, NOTCH4, SHC2, SNCA, SULT2B1. Продукты некоторых из этих генов, согласно известным на сегодняшний день данным об их функциональных особенностях, вполне могут быть вовлечены в молекулярные механизмы исследуемой патологии. Так, гиперэкспрессирующиеся при ПЭ гены FLT1, ENG, LEP и BCL6 характеризуют такие канонические пути данной патологии как ангиогенез и иммуномодуляция. В то же время для генов LEP, INHA, INHBA, FLT1, ENG показано участие в процессах секреции и клеточной сигнализации [37, 38, 43], которые, вероятно, также вносят свой вклад в патофизиологические механизмы, ответственные за развитие ПЭ. Центральное место в полученный сети генов ПЭ с наибольшим числом взаимодействий занимают гены CGA, DNAJC3 и LYN, кодирующие соответственно альфа-субъединицу ряда гликопротеиновых гормонов (хорионического гонадотропина, лютеинизирующего гормона, фолликулостимулирующего гормона и тиреотропина), белок-ингибитор интерферон-индуцированной протеинкиназы (действует как негативный регулятор активности киназы, ослабляя общий синтез белка в условиях стресса) и тирозин-протеинкиназу, принимающую участие в активации ЕРО-рецептора, регуляции врожденных и адаптивных иммунных реакций и дифференцировке эритронормобластов (база данных “GeneCard”). Примечательно, что полученные данные согласуются с одной из существующих теорий этиопатогенеза ПЭ, в которой важная роль в развитии патологии отведена хронической гипоксии в плаценте и иммунному ответу материнского организма на чужеродные антигены плода [56].
Следует отметить, что идентифицированные с помощью вышеописанного подхода новые гены-кандидаты ПЭ в большинстве случаев мало изучены и, несомненно, представляют значительный интерес для анализа ассоциации их генетической вариабельности с преэклампсией и рядом других гестационных осложнений. В данном контексте все больший научно-практический интерес представляет направление, посвященное исследованию механизмов регуляции изменений транскрипционного профиля вышеописанных генов. Очевидно, что такой системный подход будет способствовать не только выявлению новых генов-кандидатов ПЭ, но и пониманию их возможной роли в этиопатогенезе этой болезни.
ПОЛНОГЕНОМНЫЙ АНАЛИЗ СЦЕПЛЕНИЯ
Исторически первым широко применяющимся методом полногеномного анализа для идентификации генов предрасположенности к МФЗ стал анализ сцепления (Genome Wide Linkage Studies – GWLS), в рамках которого самыми мощными считаются методы, базирующиеся на известной модели наследования признака, включающей оценку популяционной частоты мутантного аллеля и пенетрантности генотипов [57]. Однако модель наследования может быть и неизвестной, так как существует возможность анализа полученных данных с использованием непараметрических методов. Необходимо отметить, что этот подход является крайне дорогостоящим и трудоемким.
Первый полногеномный рекомбинационный анализ для выявления генов-кандидатов преэклампсии был проведен в 1992 г. в шотландской популяции (табл. 4), однако в результате этого исследования не было получено доказательств существования локуса восприимчивости к ПЭ [58]. Второе сканирование генома с помощью анализа сцепления, проведенное в австралийских семьях, обнаружило кандидатный регион ПЭ на длинном плече 4-й хромосомы [59]. В этой работе исследователи использовали две модели наследования: рецессивный ген с высокой пенетрантностью и доминантный ген с низкой пенетрантностью. Позднее Arngrimsson и соавт. [60], используя непараметрические методы, идентифицировали в качестве локуса предрасположенности к ПЭ участок на 2-й хромосоме – 2p13. Тем не менее после исключения из анализа двух больших семей авторы выявили другой локус этой же хромосомы – 2q23 [60]. Последующие полногеномные сканирования в астралийских, новозеландских, датских и финских семьях с применением аналогичного подхода идентифицировали в качестве локусов восприимчивости к ПЭ участки на различных хромосомах: 2q22-23, 2p25, 4q32, 9p13, 10q, 11q23, 22q [61–64]. Важно подчеркнуть, что в трех из обследованных популяций была картирована ассоциация ПЭ с участком второй хромосомы (2q22-23, 2q25). Используя метод позиционного клонирования в комбинации с биоинформационным подходом, австралийские исследователи в качестве высокоприоритетного позиционного кандидата ПЭ обозначили локус 2q22, включающий область гена рецептора активина 2-го типа – ACVR2A [65]. Кроме того, анализ экспрессионных профилей генов децидуальной ткани показал статистически значимое десятикратное снижение уровня экспрессии этого гена у больных преэклампсией по сравнению с нормотензивными женщинами [66]. Последующий анализ ассоциаций полиморфных вариантов этого гена в австралийской, норвежской и финской популяциях выявил связь ряда полиморфных вариантов гена ACVR2A с развитием ПЭ: rs1424954, rs1364658, rs1895694, rs1424941, rs1014064, rs2161983 и rs3768687 [65–67]. Показано, что аллельные варианты регуляторных областей гена ACVR2A могут влиять на уровень транскрипции данного локуса, обусловливая снижение концентрации кодируемого им протеина, и таким образом играть важную роль в инициации патофизиологических механизмов в плацентарной ткани, вовлеченных в этиологию ПЭ [66]. В различных этнических выборках России также были установлены ассоциации полиморфных вариантов гена ACVR2А с ПЭ: так, в рамках исследования структуры неравновесия по сцеплению в локусе ACVR2А показана ассоциация полиморфизма rs17742342 и двух рисковых гаплотипов данного гена с развитием тяжелой формы ПЭ у русских г. Томска [68], а в популяционной выборке, проживающей на территории Санкт-Петербурга, с помощью метода NGS была выявлена связь с развитием ПЭ 31-го полиморфного варианта гена ACVR2А [69].
Таблица 4.
Результаты полногеномного анализа сцепления при картировании генов предрасположенности к преэклампсии
| № | Популяция | Модель наследования | Число семей | Идентифицированный участок хромосомы | Источник |
|---|---|---|---|---|---|
| 1 | Шотландия | АР | 35 | – | [58] |
| 2 | Австралия | АР, АД | 15 | 4q34 | [59] |
| 3 | Исландия | НП | 124 | 2q23, 2p13 | [60] |
| 4 | Австралия, Новая Зеландия | НП | 34 | 2q22-23, 5q, 11q23, 13q | [61, 64] |
| 5 | Нидерланды | НП | 67 | 10q, 22q | [62] |
| 6 | Финляндия | НП | 15 | 2p25, 4q32, 9p13 | [63] |
Примечательно, что несмотря на достаточно обширные исследования, проведенные в области полногеномного сканирования семейных случаев ПЭ, к настоящему времени выявлено всего лишь два локуса (ACVR2A и STOX1), реплицированных в нескольких популяционных выборках и имеющих подтверждение их роли в генетической архитектуре данного заболевания на уровне транскриптома плацентарной ткани и/или ассоциативных исследований в популяциях в рамках подхода случай–контроль.
Ген STOX1, кодирующий ДНК-связывающий протеин, участвующий в окислительных реакциях и развитии дисфункции трофобласта, был идентифицирован как наиболее вероятный локус подверженности ПЭ в ходе выполнения GWLS, проведенного в голландской популяции, в котором была картирована ассоциация этой патологии с участком 10-й хромосомы 10q [70]. Последующее экзомное секвенирование участка десятой хромосомы 10q22 выявило полиморфный вариант Y153H гена STOX1, ассоциированный с передачей ПЭ по материнской линии в трех поколениях [71]. Важно подчеркнуть, что семьи, включенные в данное исследование, были фенотипически однородной когортой пациентов, имеющих в семейном анамнезе тяжелую раннюю форму преэклампсии, осложненную задержкой внутриутробного роста плода, что позволило предположить авторам “плацентарное происхождение” этого заболевания, по крайней мере в изученной выборке. Последующие проведенные ассоциативные исследования, которые пытались реплицировать полученные данные, не подтвердили значимой роли варианта Y153H гена STOX1 в подверженности к ПЭ, тем не менее результаты, полученные в недавней работе норвежских ученых, свидетельствуют о связи данного варианта с ПЭ при рецидивирующей форме заболевания [5, 72]. Таким образом, вышеописанные результаты, полученные в рамках подхода GWLS и последующих репликативных исследований, свидетельствуют о необходимости при планировании работ, направленных на характеристику генетической архитектуры ПЭ, учитывать объем выборок, форму и особенности клинического течения этого гестационного осложнения, а также роль генома плода.
ТЕХНОЛОГИИ ВЫСОКОПРОИЗВОДИТЕЛЬНОГО СЕКВЕНИРОВАНИЯ В ИЗУЧЕНИИ ГЕНЕТИЧЕСКИХ ФАКТОРОВ ПЭ
Современная методология ассоциативных генетических исследований базируется на использовании технологий высокопроизводительного генетического анализа, прежде всего технологии массового параллельного секвенирования (MPS), которая является наиболее эффективным методом поиска новых маркеров риска МФЗ (SNP, CNV, мРНК, микроРНК). MPS на сегодняшний день рассматривают как наиболее значимую альтернативу методу GWAS с использованием микрочипов [73, 74].
В рамках данного направления хотелось бы обсудить результаты двух недавних полноэкзомных секвенирований (WES) генома при ПЭ. Так, данные, полученные Hansen с коллегами [75], свидетельствуют об отсутствии статистически значимых различий между группой с ПЭ и контрольной выборкой, даже после применения двух различных статистических подходов: “disease association panel” и “gene panel”. Авторы делают заключение о протективной роли генетического разнообразия в отношении развития преэклампсии и ставят под сомнение гипотезу полигенного типа наследования данной патологии [75]. Последующее WES, проведенное в 34 австралийских и новозеландских семьях, продемонстрировало ассоциацию двух новых локусов с развитием ПЭ: rs145743393 (Padj = 0.0032) гена C1orf35 и rs34270076 (Padj = 0.0128) гена QRFPR, кодирующего рецептор пироглутамил-RF-амид-пептидов [76].
На текущий момент проведен также ряд исследований, направленных на изучение особенностей транскриптомных профилей при ПЭ с помощью технологии RNA-seq [77–80]. Так, наиболее интересные результаты получены в работе Tsang и соавт. [77], посвященной характеристике транскриптома едничных клеток (англ. single-cell transcriptomics) плаценты при ПЭ и физиологической беременности с использованием технологии секвенирования “10x Genomics Chromium System”. Исследователи выявили значительную клеточную гетерогенность плаценты по экспрессионным профилям как при нормальной беременности, так и в случае ранней ПЭ, что свидетельствует о необходимости применения одноклеточного аналитического подхода при изучении генетической архитектуры данной патологии на уровне транскриптома. Кроме того, авторы обнаружили существенную роль дисфункции вневорсинчатого трофобласта в этиопатогенезе ранней ПЭ [77].
Опыт отечественных исследователей в использовании платформ MPS для изучения генетических факторов ПЭ на полногеномном и транскриптомном уровнях остается достаточно небольшим, о чем свидетельствует ограниченное количество работ в этой области, сфокусированных преимущественно на анализе роли эпигенетических факторов в развитии этого гестационного осложнения. Данные исследования посвящены изучению вариабельности микроРНК в плаценте и/или плазме женщин с ПЭ и физиологической беременностью и выявили такие потенциальные биомаркеры ПЭ как hsa-miR-4532, hsa-miR-34c-5p, hsa-miR-193b-5p [81] и miR-423-5p [82].
В целом исследования с применением технологии MPS на текущий момент являются одними из наиболее перспективных в области изучения генетической архитектуры ПЭ: развитие методов высокопроизводительного секвенирования, доступность и накопление получаемых генетических данных будут способствовать созданию фундаментальной основы для разработки многокомпонентных моделей оценки генетического риска и характеристики молекулярных механизмов данной патологии.
ЗАКЛЮЧЕНИЕ
Многочисленные близнецовые и эпидемиологические исследования продемонстрировали, что ПЭ в большинстве случаев является многофакторным заболеванием с выраженной наследственной предрасположенностью, которая варьирует в зависимости от географических, социально-экономических и расовых особенностей. В конце XX в. анализ генетических причин ПЭ был сфокусирован в области исследований отдельных генов-кандидатов, имеющих отношение к тромбофилии, метаболизму фолиевой кислоты, окислительному стрессу и функционированию ренин-ангиотензиновой системы и др., с использованием подхода случай–контроль. Необходимо отметить, что результаты этих работ показывают крайне низкую репликативность, часто являются противоречивыми даже для одной и той же этнической группы, в связи с чем общепринятых генов восприимчивости к ПЭ выявить так и не удалось.
Исследования, проведенные в рамках подходов GWAS и GWLS, к сожалению, также продемонстрировали весьма скромные результаты в характеристике генетической архитектуры этого гестационного осложнения. Обширные исследования, проводимые с 1992 г. в области полногеномного сканирования семейных случаев ПЭ, на сегодняшний день выявили только два локуса (ACVR2A и STOX1), реплицированных в нескольких популяционных выборках и имеющих подтверждение их роли в генетической архитектуре данного заболевания на уровне транскриптома и/или ассоциативных исследований на геномном уровне в рамках подхода случай–контроль. Немногочисленные данные, полученные на текущий момент в ходе реализации полногеномных ассоциативных исследований полиморфных вариантов генов с развитием ПЭ, не согласуются относительно рисковых генетических маркеров данной патологи и требуют репликации на расширенных клинически подробно охарактеризованных выборках в различных популяциях.
В качестве возможных причин достаточно низкой воспроизводимости вышеописанных результатов можно рассматривать определенный дизайн исследования (например, различия критериев формирования обследуемых групп), этническую специфичность структуры неравновесия по сцеплению идентифицированных генов-кандидатов (и, как следствие, генетической архитектуры ПЭ), малые объемы выборок и (или) методические ошибки. Кроме того, выявленная ассоциация может в действительности касаться не самого гена-кандидата, а другого гена, продукт которого функционально компенсирует эффект мутантного гена (эпистатическое взаимоотношение генов), либо быть связана с ген-генными и/или ген-средовыми взаимодействиями [83]. Важно отметить, что фокусирование исследований в области изучения ген-генных и ген-средовых взаимодействий, которое имеет существенное значение для характеристики структуры наследственной предрасположенности к преэклампсии, сопряжено со значительными проблемами статистического анализа данных (особенно в контексте анализа геномных данных) ввиду большого числа возможных взаимодействий. Данная проблема требует разработки надежных статистических инструментов для изучения эпистатических взаимодействий, а также формирования тщательно охарактеризованных выборок для исследования. Примером таких проспективных когорт беременных женщин могут являться SCOPE (Screening for Pregnancy Endpoints (SCOPE) study) и норвежские биобанки для когортного исследования матери и ребенка.
С развитием постгеномных технологий приоритет в изучении генетических факторов риска ПЭ переместился преимущественно в область транскриптомики плацентарной ткани. В последнее десятилетие с помощью технологии микрочипов было проведено более 30 независимых исследований, направленных на изучение дифференциальной экспрессии генов плацентарной ткани больных преэклампсией и женщин с физиологической беременностью, в которых было выявлено высокодостоверное повышение/снижение экспрессии нескольких сотен генов. Наиболее значимые из этих локусов ассоциированы с такими биологическими путями и молекулярными процессами как функционирование иммунной системы, пролиферация и дифференцировка клеток, апоптоз и регуляция метаболизма, что свидетельствует о существенной роли в этиопатогенезе ПЭ сочетанного воздействия нескольких патологических механизмов. Необходимо отметить, что идентифицированные с помощью вышеописанного подхода новые гены-кандидаты ПЭ в большинстве случаев мало изучены на геномном уровне и, несомненно, представляют значительный интерес для анализа ассоциации их генетической вариабельности с преэклампсией и другими заболеваниями группы больших акушерских синдромов.
В настоящее время традиционное представление о том, что восприимчивость к заболеваниям детерминируется лишь взаимодействием между генами и окружающей средой, дополняется и расширяется новыми данными о ключевой роли эпигенетического репрограммирования [84, 85]. В контексте вопроса о регуляции транскрипционной активности обнаруженных новых генов-кандидатов ПЭ необходимо отметить существенную роль структуры хроматина, определяемую такими обратимыми механизмами как метилирование ДНК и модификация гистонов. Изменение активности вышеописанных процессов обусловливает дерегулирование экспрессии генов и является индуктором развития многих патологических состояний, включая и ПЭ [86, 87]. Зарубежными учеными был проведен ряд исследований, посвященных анализу экспрессионных профилей микроРНК, дифференциального метилирования ДНК плацентарной ткани при ПЭ и физиологической беременности [88–93], однако этот вопрос подробно не рассматривается в нашем обзоре, поскольку заслуживает детального обсуждения в отдельной публикации.
Следует отметить, что в рамках изучения молекулярных механизмов ПЭ все большую научно-практическую значимость приобретает направление исследований, связанное с подходами системной биологии. Так, в недавней работе Than и соавт. [94] проведен комплексный анализ омиксных данных и клинических характеристик пациенток с различными фенотипами преэклампсии. Интеграция результатов протеомных, транскриптомных исследований и данных “виртуальной” жидкостной биопсии позволила авторам разработать мульти-биомаркерные профили для профилактики ПЭ, охарактеризовать на молекулярном уровне основные материнские и плацентарные пути, вовлеченные в формирование отдельных клинических форм и предложить новую парадигму развития этого гестационного осложнения.
Сегодня развитие таких направлений науки как геномика, транскриптомика, протеомика и математическое моделирование, составляющих фундаментальную основу прецизионной медицины, позволяет исследователям пересмотреть свое понимание болезней на молекулярном уровне и разработать новые профилактические мероприятия для конкретного пациента. Современные высокотехнологичные подходы в области медицины в целом и в области акушерства в частности позволяют получать огромные массивы биологических данных, используя значительное количество биологических образцов. Для расшифровки генетической архитектуры ПЭ многообещающим представляется применение “мультиомных” технологий анализа отдельных клеток (single cell analysis), которые бурно развиваются сейчас в США и Европе и заключаются в идентификации изменений генома, эпигенома, транскриптома и протеома на уровне единичной клетки. Эта область представляется крайне интересной, так как позволит охарактеризовать более тонкие молекулярные механизмы патогенеза ПЭ, не выявляемые при профилировании тканей, включающих различные клеточные типы, и идентифицировать потенциальные мишени для таргетной терапии. Дифференцированный подход к лечению различных клинических форм ПЭ на основе выявленных интегративных персональных “омиксных” профилей iPOP (от англ. an integrative Personal “Omics” Profile) – перспективное направление, необходимое для перехода к персонализированной медицине в области акушерства. Таким образом, в настоящее время при исследовании генетической архитектуры ПЭ особую актуальность приобретает комплексный анализ вариабельности генома, метилома, транскриптома и протеома на уровне единичных клеток. Очевидно, что такой системный подход будет способствовать не только выявлению новых генов-кандидатов и мишеней для таргетной терапии ПЭ, но и раскрытию на молекулярном уровне этиопатогенеза данной патологии беременности.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (проект № 18-44-700007).
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики.
От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Пузырев В.П. Медицинская патогенетика // Вавиловский журн. генетики и селекции. 2014. Т. 18. № 1. С. 7–21.
Баранов В.С. Эволюция предиктивной медицины. Старые идеи, новые понятия // Мед. генетика. 2017. Т. 16. № 5. С. 4–9.
Blanco-Gómez A., Castillo-Lluva S., del Mar Sáez-Freire M. et al. Missing heritability of complex diseases: Enlightenment by genetic variants from intermediate phenotypes // Bioessays. 2016. V. 38. № 7. P. 664–673. https://doi.org/10.1002/bies.201600084
Айламазян Э.К. Гестоз: теория и практика / Под ред. Айламазяна Э.К., Мозговой Е.В. М.: МЕДпресс-информ, 2008. 272 с.
Yong H.E.J., Murthi P., Brennecke S.P., Moses E.K. Genetic approaches in preeclampsia // Methods Mol. Biol. 2018. V. 1710. P. 53–72. https://doi.org/10.1007/978-1-4939-7498-6_5
Ananth C.V., Keyes K.M., Wapner R.J. Pre-eclampsia rates in the United States, 1980–2010: age-period-cohort analysis // BMJ. 2013. № 7. P. 347.
Valenzuela F.J., Pérez-Sepúlveda A., Torres M.J. et al. Pathogenesis of preeclampsia: the genetic component // J. Pregnancy. 2012. № 1. P. 1–8.
McDonald S.D., Best C., Lam K. The recurrence risk of severe de novo pre-eclampsia in singleton pregnancies: a population-based cohort // BJOG. 2009. № 12. P. 1578–1584.
Boyd H.A., Tahir H., Wohlfahrt J. et al. Associations of personal and family preeclampsia history with the risk of early-, intermediate- and late-onset preeclampsia // Am. J. Epidemiol. 2013. № 12. P. 1611–1619.
Lardoeyt R., Vargas G., Lumpuy J. et al. Contribution of genome–environment interaction to pre-eclampsia in a Havana Maternity Hospital // MEDICC Review. 2013. V. 15. № 3. P. 22–29.
Nilsson E., Ros H.S., Cnattingius S., Lichtenstein P. The importance of genetic and environmental effects for pre-eclampsia and gestational hypertension: a family study // BJOG. 2004. № 3. P. 200–206.
Arngrimsson R., Bjornsson S., Geirsson R. et al. Genetic and familial predisposition to eclampsia and pre-eclampsia in a defined population // Br. J. Obstet. Gynaecol. 1990. V. 97. P. 762–769.
Berends A.L., Steegers E.A., Isaacs A. et al. Familial aggregation of preeclampsia and intrauterine growth restriction in a genetically isolated population in The Netherlands // Eur. J. Hum. Genet. 2008. V. 16. № 12. P. 1437–1442.
Lie R., Rasmussen S., Brunborg H. Fetal and matemal contributions to risk of preeclampsia: population based study // BML. 1998. V. 316. P. 1343–1347.
Dekker G., Robillard P.Y., Roberts C. The etiology of preeclampsia: the role of the father // J. Reprod. Immunol. 2011. V. 89. P. 126–132.
Goddard K.A., Tromp G., Romero R. et al. Candidate-gene association study of mothers with pre-eclampsia, and their infants, analyzing 775 SNPs in 190 genes // Hum. Heredity. 2007. V. 63. № 1. P. 1–16.
Mütze S., Rudnik–Schöneborn S., Zerres K., Rath W. Genes and the preeclampsia syndrome // J. Perinat. Med. 2008. V. 36. № 1. P. 38–58.
Fong F.M., Sahemey M.K., Hamedi G. et al. Maternal genotype and severe preeclampsia: a HuGE review // Am. J. Epidemiol. 2014. V. 180. № 4. P. 335–345.
Johnson M.P., Brennecke S.P., East C.E. et al. Genome-wide association scan identifies a risk locus for preeclampsia on 2q14, near the inhibin, beta B gene // PLoS One. 2012. V. 7. № 3. P. e33666.
Zhao L., Triche E.W., Walsh K.M. et al. Genome-wide association study identifies a maternal copy-number deletion in PSG11 enriched among preeclampsia patients // BMC Pregnancy Childbirth. 2012. V. 29. № 12. P. e61.
Zhao L., Bracken M.B., De Wan A.T. Genome-wide association study of pre-eclampsia detects novel maternal single nucleotide polymorphisms and copy-number variants in subsets of the Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study cohort // Ann. Hum. Genet. 2013. V. 77. № 4. P. 277–287.
McGinnis R., Steinthorsdottir V., Williams N.O. et al. Variants in the fetal genome near FLT1 are associated with risk of preeclampsia // Nat. Genet. 2017. V. 49. № 8. P. 1255–1260. https://doi.org/10.1038/ng.3895
De Vivo A., Baviera G., Giordano D. et al. Endoglin, PlGF and sFlt-1 as markers for predicting pre-eclampsia // Acta Obstet. Gynecol. Scand. 2008. V. 87. № 8. P. 837–842.
Sadee W., Hartmann K., Seweryn M. et al. Missing heritability of common diseases and treatments outside the protein-coding exome // Hum. Genet. 2014. V. 133. № 10. P. 1199–1215. https://doi.org/10.1007/s00439-014-1476-7
Phipps E.A., Thadhani R., Benzing T., Karumanchi S.A. Pre-eclampsia: pathogenesis, novel diagnostics and therapies // Nat. Rev. Nephrol. 2019. V. 15. № 5. P. 275–289. https://doi.org/10.1038/s41581-019-0119-6
Трифонова Е.А., Габидулина Т.В., Ершов Н.И. и др. Характеристика транскриптома плацентарной ткани у женщин с физиологической беременностью и преэклампсией // Acta Naturae. 2014. Т. 6. № 2(21). С. 77–90.
Enquobahrie D.A., Meller M., Rice K. et al. Differential placental gene expression in preeclampsia // Am. J. Obstet. Gynecol. 2008. V. 199. № 5. P. 566.e1–566.e11. https://doi.org/10.1016/j.ajog.2008.04.020
Hoegh A.M., Borup R., Nielsen F.C. et al. Gene expression profiling of placentas affected by preeclampsia // J. Biomed. Biotechnol. 2010. V. 2010. https://doi.org/10.1155/2010/787545
Lee G.S., Joe Y.S., Kim S.J. et al. Cytokine-related genes and oxidation-related genes detected in preeclamptic placentas // Arch. Gynecol. Obstet. 2010. V. 282. № 4. P. 363–369. https://doi.org/10.1007/s00404-009-1222-x
Meng T., Chen H., Sun M. et al. Identification of differential gene expression profiles in placentas from preeclamptic pregnancies versus normal pregnancies by DNA microarrays // OMICS. 2012. V. 16. № 6. P. 301–311.
Nishizawa H., Ota S., Suzuki M. et al. Comparative gene expression profiling of placentas from patients with severe pre-eclampsia and unexplained fetal growth restriction // Reprod. Biol. Endocrinol. 2011. V. 9:107. https://doi.org/10.1186/1477-7827-9-107
Nishizawa H., Pryor-Koishi K., Kato T. et al. Microarray analysis of differentially expressed fetal genes in placental tissue derived from early and late onset severe pre-eclampsia // Placenta. 2007. V. 28. № 5–6. P. 487–497.
Reimer T., Koczan D., Gerber B. et al. Microarray analysis of differentially expressed genes in placental tissue of pre-eclampsia: up-regulation of obesity-related genes // Mol. Hum. Reprod. 2002. V. 8. № 7. P. 674–680.
Sitras V., Paulssen R.H., Gronaas H. et al. Differential placental gene expression in severe preeclampsia // Placenta. 2009. V. 30. № 5. P. 424–433. https://doi.org/10.1016/j.placenta.2009.01.012
Varkonyi T., Nagy B., Fule T. et al. Microarray profiling reveals that placental transcriptomes of early-onset HELLP syndrome and preeclampsia are similar // Placenta. 2011. V. 32. P. S21–S29. https://doi.org/10.1016/j.placenta.2010.04.014
Buimer M., Keijser R., Jebbink J.M. et al. Seven placental transcripts characterize HELLP-syndrome // Placenta. 2008. V. 29. № 5. P. 444–453. https://doi.org/10.1016/j.placenta.2008.02.007
Winn V.D., Gormley M., Fisher S.J. The impact of preeclampsia on gene expression at the maternal-fetal interface // Pregnancy Hypertens. 2011. V. 1. № 1. P. 100–108. https://doi.org/10.1016/j.preghy.2010.12.001
Xiang Y., Cheng Y., Li X. et al. Up-regulated expression and aberrant DNA methylation of LEP and SH3PXD2A in pre-eclampsia // PLoS One. 2013. V. 8(3):e59753. https://doi.org/10.1371/journal.pone.0059753
Jarvenpaa J., Vuoristo J.T., SaVainen E.R. et al. Altered expression of angiogenesis-related placental genes in pre-eclampsia associated with intrauterine growth restriction // Gynecol. Endocrinol. 2007. V. 23. № 6. P. 351–355.
Centlow M., Wingren C., Borrebaeck C. et al. Differential gene expression analysis of placentas with increased vascular resistance and pre-eclampsia using whole-genome microarrays // J. Pregnancy. 2011. P. 1–12.https://doi.org/10.1155/2011/472354
Heikkilä A., Tuomisto T., Häkkinen S.K. et al. Tumor suppressor and growth regulatory genes are overexpressed in severe early-onset preeclampsia – an array study on case-specific human preeclamptic placental tissue // Acta. Obstet. Gynecol. Scand. 2005. V. 84. № 7. P. 679–689.
Lapaire O., Grill S., Lalevee S. et al. Microarray screening for novel preeclampsia biomarker candidates // Fetal. Diagn. Ther. 2012. V. 31. № 3. P. 147–153. https://doi.org/10.1159/000337325
Tsai S., Hardison N.E., James A.H. et al. Transcriptional profiling of human placentas from pregnancies complicated by preeclampsia reveals disregulation of sialic acid acetylesterase and immune signalling pathways // Placenta. 2011. V. 32. № 2. P. 175–182. https://doi.org/10.1016/j.placenta.2010.11.014
Mayor-Lynn K., Toloubeydokhti T., Cruz A.C. et al. Expression profile of microRNAs and mRNAs in human placentas from pregnancies complicated by preeclampsia and preterm labor // Reprod. Sci. 2011. V. 18. № 1. P. 46–56. https://doi.org/10.1177/1933719110374115
Schanton M., Maymó J.L., Pérez-Pérez A. et al. Invement of leptin in the molecular physiology of the placenta // Reproduction. 2018. V. 155. № 1. P. 1–12. https://doi.org/10.1530/REP-17-0512
Aizawa-Abe M. Pathophysiological role of leptin in obesity-related hypertension // J. Clin. Invest. 2000. V. 105. P. 1243–1252.
Fairfax B.P., Vannberg F.O., Radhakrishnan J. et al. An integrated expression phenotype mapping approach defines common variants in LEP, ALOX15 and CAPNS1 associated with induction of IL-6 // Hum. Mol. Genet. 2010. № 15. P. 720–730.
Fourati M., Mnif M., Kharrat N. et al. Association between Leptin gene polymorphisms and plasma leptin level in three consanguineous families with obesity // Gene. 2013. № 527. P. 75–81.
Ma D., Feitosa M.F., Wilk J.B. et al. Leptin is associated with blood pressure and hypertension in women from the National Heart, Lung, and Blood Institute Family Heart Study // Hypertension. 2009. V. 53. № 3. P. 473–479. https://doi.org/10.1161/HYPERTENSIONAHA.108.118133
Vaskú J.A., Vaskú A., Dostálová Z., Bienert P. Association of leptin genetic polymorphism –2548 G/A with gestational diabetes mellitus // Genes Nutr. 2006. V. 1. № 2. P. 117–123. https://doi.org/10.1007/BF02829953
Sugathadasa B.H., Tennekoon K.H., Karunanayake E.H. et al. Association of 2548 G/A polymorphism in the leptin gene with preeclampsia/pregnancy-induced hypertension // Hypertens. Pregnancy. 2010. V. 29. P. 366–374.
Liberzon A., Birger C., Thorvaldsdóttir H. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection // Cell Syst. 2015. V. 1. № 6. P. 417–425. https://doi.org/10.1016/j.cels.2015.12.004
Yeh C.C., Chao K.C., Huang S.J. Innate immunity, decidual cells, and preeclampsia // Reprod. Sci. 2013. V. 20. № 4. P. 339–353. https://doi.org/10.1177/1933719112450330
Saftlas A.F., Beydoun H., Triche E. Immunogenetic determinants of preeclampsia and related pregnancy disorders: a systematic review // Obstet. Gynecol. 2005. V. 106. № 1. P. 162–172.
Posternak V., Cole M.D. Strategically targeting MYC in cancer // F1000Research. 2016. V. 5. PMID 27081479.https://doi.org/10.12688/f1000research.7879.1
Eiland E., Nzerue C., Faulkner M. Preeclampsia 2012 // J. Pregnancy. 2012. V. 2012. № 586578. https://doi.org/10.1155/2012/586578
Аульченко Ю.С., Аксенович Т.И. Методологические подходы и стратегии картирования генов, контролирующих комплексные признаки человека // Вестник ВОГиС. 2006. № 10. С. 189–202.
Hayward C., Livingstone J., Holloway S. et al. An exclusion map for pre-eclampsia assuming autosomal recessive inheritance // Am. J. Hum. Genet. 1992. V. 50. P. 749–757.
Harrison G.A., Humphrey K.E., Jones N. et al. A genome-wide linkage study of preeclampsia/eclampsia reveals evidence for a candidate region on 4q // Am. J. Hum. Genet. 1997. V. 60. P. 1158–1167.
Arngrımsson R., Sigurõardóttir S., Frigge M.L. et al. A genome-wide scan reveals a maternal susceptibility locus for pre-eclampsia on chromosome 2p13 // Hum. Mol. Genet. 1999. V. 8. P. 1799–1805.
Moses E.K., Lade J.A., Guo G. et al. A genome scan in families from Australia and New Zealand confirms the presence of a maternal susceptibility locus for pre-eclampsia, on chromosome 2 // Am. J. Hum. Genet. 2000. V. 67. P. 1581–1585.
Lachmeijer A.M., Dekker G.A., Pals G. et al. Searching for preeclampsia genes: the current position // Eur. J. Obstet. Gynecol. Reprod. Biol. 2002. V. 5. № 2. P. 94–113.
Laivuori H., Lahermo P., Ollikainen V. et al. Susceptibility loci for preeclampsia on chromosomes 2p25 and 9p13 in Finnish families // Am. J. Hum. Genet. 2003. V. 72. P. 168–177.
Johnson M.P., Fitzpatrick E., Dyer T.D. et al. Identification of two novel quantitative trait loci for pre-eclampsia susceptibility on chromosomes 5q and 13q using a variance components-based linkage approach // Mol. Hum. Reprod. 2007. V. 13. P. 61–67.
Moses E.K., Fitzpatrick E., Freed K.A. et al. Objective prioritization of positional candidate genes at a quantitative trait locus for pre-eclampsia on 2q22 // Mol. Hum. Reprod. 2006. V. 12. № 8. P. 505–512.
Roten L.T., Johnson M.P., Forsmo S. et al. Association between the candidate susceptibility gene ACVR2A on chromosome 2q22 and pre-eclampsia in a large Norwegian population-based study (the HUNT study) // Eur. J. Hum. Genet. 2009. V. 17. № 2. P. 250–257.
Lokki A.I., Klemetti M.M., Heino S. et al. Association of the rs1424954 polymorphism of the ACVR2A gene with the risk of pre-eclampsia is not replicated in a Finnish study population // BMC Res. Notes. 2011. V. 4. P. 545.
Ворожищева А.Ю., Трифонова Е.А., Бутко Ю.К. и др. Роль генетической вариабельности локуса ACVR2A в формировании подверженности преэклампсии // Мед. генетика. 2013. Т. 12. № 10(136). С. 35–40.
Глотов А.С., Вашукова Е.С., Данилова М.М. и др. Секвенирование нового поколения (NGS) для изучения гена ACVR2A у беременных с гестозом // Мол. медицина. 2014. № 5. С. 33–40.
Lachmeijer A.M., Arngrímsson R., Bastiaans E.J. et al. A genome-wide scan for preeclampsia in the Netherlands // Eur. J. Hum. Genet. 2001. V. 9. № 10. P. 758–764.
van Dijk M., Mulders J., Poutsma A. et al. Maternal segregation of the Dutch preeclampsia locus at 10q22 with a new member of the winged helix gene family // Nat. Genet. 2005. V. 37. № 5. P. 514–519.
van Dijk M., Oudejans C.B. STOX1: Key player in trophoblast dysfunction underlying early onset preeclampsia with growth retardation // J. Pregnancy. 2010. V. 2011. № 521826. https://doi.org/10.1155/2011/521826
Hitomi Y., Tokunaga K. Significance of functional disease-causal/susceptible variants identified by whole-genome analyses for the understanding of human diseases // Proc. Jpn Acad. Ser. B Phys. Biol. Sci. 2017. V. 93. № 9. P. 657–676. https://doi.org/10.2183/pjab.93.042
Kalayinia S., Goodarzynejad H., Maleki M., Mahdieh N. Next generation sequencing applications for cardiovascular disease // Ann. Med. 2018. V. 50. № 2. P. 91–109. https://doi.org/10.1080/07853890.2017.1392595
Hansen A.T., Bernth J.M., Hvas A.M., Christiansen M. The genetic component of preeclampsia: A whole-exome sequencing study // PLoS One. 2018. V. 13. № 5. P. e0197217.
Melton P.E., Johnson M.P., Gokhale-Agashe D. et al. Whole-exome sequencing in multiplex preeclampsia families identifies novel candidate susceptibility genes // J. Hypertens. 2019. V. 37. № 5. P. 997–1011. https://doi.org/10.1097/HJH.0000000000002023
Tsang J.C., Vong J.S., Ji L. et al. Integrative single-cell and cell-free plasma RNA transcriptomics elucidates placental cellulardynamics // Proc. Natl Acad. Sci. USA. 2017. V. 114. № 37. P. E7786–E7795. https://doi.org/10.1073/pnas.1710470114
Tong J., Zhao W., Lv H. et al. Transcriptomic profiling in human decidua of severe preeclampsia detected by RNA sequencing // J. Cell Biochem. 2018. V. 119. № 1. P. 607–615. https://doi.org/10.1002/jcb.26221
Zhou W., Wang H., Wu X. et al. The profile analysis of circular RNAs in human placenta of preeclampsia // Experim. Biol. Med. 2018. V. 243. P. 1109–1117. https://doi.org/10.1177/1535370218813525
Sheridan M.A., Yang Y., Jain A. et al. Early onset preeclampsia in a model for human placental trophoblast // Proc. Natl Acad. Sci. USA. 2019. V. 116. № 10. P. 4336–4345. https://doi.org/10.1073/pnas.1816150116
Пакин В.С., Вашукова Е.С., Капустин Р.В. и др. Оценка уровня микроРНК в плаценте при тяжелом гестозе на фоне гестационного сахарного диабета // Журн. акушерства и женских болезней. 2017. Т. 66. № 3. С. 110–115.
Timofeeva A.V., Gusar V.A., Kan N.E. et al. Identification of potential early biomarkers of preeclampsia // Placenta. 2018. V. 61. P. 61–71. https://doi.org/10.1016/j.placenta
Гинтер Е.К. Медицинская генетика. М.: Медицина, 2003. 447 с.
Klein C.J., Benarroch E.E. Epigenetic regulation: basic concepts and relevance to neurologic disease // Neurology. 2014. V. 82. № 20. P. 1833–1840. https://doi.org/10.1212/WNL.0000000000000440
Wang H., Lou D., Wang Z. Crosstalk of genetic variants, allele-specific DNA methylation, and environmental factors for complex disease risk // Front Genet. 2019. № 9. P. 695. https://doi.org/10.3389/fgene.2018.00695
Heard E., Martienssen R.A. Transgenerational epigenetic inheritance: myths and mechanisms // Cell. 2014. V. 157. № 1. P. 95–109. https://doi.org/10.1016/j.cell.2014.02.045
Ariff A., Melton P.E., Brennecke S.P., Moses E.K. Analysis of the epigenome in multiplex pre-eclampsia families identifies SORD, DGKI, and ICA1 as novel candidate risk genes // Front Genet. 2019. V. 10. P. 227. https://doi.org/10.3389/fgene.2019.00227
Bourque D.K., Avila L., Peñaherrera M. et al. Decreased placental methylation at the H19/IGF2 imprinting control region is associated with normotensive intrauterine growth restriction but not preeclampsia // Placenta. 2010. V. 31. № 3. P. 197–202. https://doi.org/10.1016/j.placenta.2009.12.003
Ruebner M., Strissel P.L., Ekici A.B. et al. Reduced syncytin-1 expression levels in placental syndromes correlates with epigenetic hypermethylation of the ERVW-1 promoter region // PLoS One. 2013. V. 8. № 2. P. e56145. https://doi.org/10.1371/journal.pone.0056145
Choi S.Y., Yun J., Lee O.J. et al. MicroRNA expression profiles in placenta with severe preeclampsia using a PNA-based microarray // Placenta. 2013. V. 34. № 9. P. 799–804. https://doi.org/10.1016/j.placenta.2013.06.006
Ching T., Song M.A., Tiirikainen M. et al. Genome-wide hypermethylation coupled with promoter hypomethylation in the chorioamniotic membranes of early onset pre-eclampsia // Mol. Hum. Reprod. 2014. V. 20. № 9. P. 885–904. https://doi.org/10.1093/molehr/gau046
Anton L., Brown A.G., Bartolomei M.S., Elovitz M.A. Differential methylation of genes associated with cell adhesion in preeclamptic placentas // PLoS One. 2014. V. 9. № 6. P. e100148. https://doi.org/10.1371/journal.pone.0100148
Wang Y., Lumbers E.R., Arthurs A.L. et al. Regulation of the human placental (pro)renin receptor-prorenin-angiotensin system by microRNAs // Mol. Hum. Reprod. 2018. V. 24. № 9. P. 453–464. https://doi.org/10.1093/molehr/gay031
Than N.G., Romero R., Tarca A.L. et al. Integrated systems biology approach identifies novel maternal and placental pathways of preeclampsia // Front Immunol. 2018. V. 9. P. 1661.https://doi.org/10.3389/fimmu.2018.01661
Дополнительные материалы отсутствуют.