

Генетика, 2021, T. 57, № 10, стр. 1103-1120
Частная история нейрогенетики: ген swiss cheese и его ортологи
П. А. Мелентьев 1, Е. В. Рябова 1, С. В. Саранцева 1, *
1 Петербургский институт ядерной физики им. Б.П. Константинова Национального исследовательского центра
“Курчатовский институт”
188300 Гатчина, Ленинградская область, Россия
* E-mail: Sarantseva_SV@pnpi.nrcki.ru
Поступила в редакцию 28.10.2020
После доработки 11.01.2021
Принята к публикации 04.02.2021
Аннотация
Один из вопросов генетики посвящен роли наследственности в определении поведенческих признаков человека и животных. Как считалось долгие годы, поведение определяется как действием среды, так и плейотропным действием множества генов, и потому дискретные наследственные факторы поведения являются неуловимыми. Целенаправленное получение и исследование мутантов дрозофилы с измененным поведением, начатые более полувека назад Сеймуром Бензером, заложили основу направления, названного нейрогенетикой. Дальнейшее установление связи мутаций с различными фенотипами позволило выявить роль генов в детерминации функций нервной системы и в соответствующей регуляции поведения. Один из таких генов был открыт благодаря новаторским подходам Сеймура Бензера и его последователей, в частности Мартина Хайзенберга, в ходе исследований мутантов дрозофилы – ген swiss cheese, контролирующий жизнеспособность нервных и глиальных клеток. Многочисленные исследования этого эволюционно-консервативного гена и его ортологов, вызванные интересом к анализу генетического контроля жизнедеятельности клеток нервной ткани, расшифровке биологических механизмов действия фосфорорганических ядов и изучению некоторых наследственных синдромов человека, позволили выявить аспекты роли гена swiss cheese и его ортологов в метаболизме клеток, функционировании нервной системы, регуляции поведения.
ВВЕДЕНИЕ: К 100-ЛЕТИЮ СЕЙМУРА БЕНЗЕРА (1921–2007)
История науки знает множество примеров блестящих личностей, определивших развитие той или иной дисциплины, которые благодаря своей особой проницательности и трудоспособности открыли закономерности, кажущиеся теперь совершенно обыденными явлениями. Среди таких личностей в истории биологии XX в. встречаются физики, мышление которых находило удачные редукционистские подходы, представляющие сложные биологические процессы в виде простых “атомарных” моделей. Одним из таких ученых является “основатель нейрогенетики” Сеймур Бензер, 100-летие со дня рождения которого приходится на 2021-й год. Во время Второй мировой войны он работал над разработкой германиевых полупроводников в университете Пёрдью. Затем стал интересоваться молекулярной биологией: стажировался в Cold Spring Harbor, в Калифорнийском техническом институте, в институте Пастера. Вернувшись в Пёрдью, выполнил работу по изучению “тонкой структуры” локуса rII у бактериофага Т4: картировал тысячи мутаций, рассчитывал рекомбинационные расстояния и вычислял реальное расстояние в нуклеотидах. Так он доказал, что фенотипически проявляющиеся мутации соответствуют изменениям нескольких нуклеотидов. Тем самым, сопоставив функциональный и рекомбинационный тесты на аллелизм, он первый показал связь между классической формальной генетикой и моделью ДНК по Уотсону и Крику [1–5]. Далее Бензер решил заняться еще одной нерешенной проблемой генетики, а именно проблемой генетической детерминации форм поведения. К середине XX в. уже было проведено достаточно много исследований по изучению наследуемости различных поведенческих признаков, например таланта и характера у человека, специфического поведения у “вальсирующих” мышей, различных форм поведения у крыс. Такие исследования, как правило, заканчивались выводом, содержание которого можно передать следующим тезисом: “Поведенческие признаки определяются множеством (генетических) факторов” [6]. Такой результат не удовлетворял Бензера, поэтому он хотел найти элементарные факторы, определяющие признаки поведения, и выбрал в качестве объекта изучения плодовую мушку Drosophila melanogaster. Благодаря работам лабораторий Вильяма Касла и Томаса Ханта Моргана в первые два десятилетия XX в. было изучено несколько форм поведения дрозофилы: фото-, анемо- и геотаксис, реакция на обонятельные стимулы, половое поведение. Эксперименты второй четверти XX в. по изучению наследования признаков при постановке разнообразных скрещиваний дрозофил и селекции того или иного поведенческого признака в ряду поколений приводили все к тому же выводу о генетической многофакторности, определяющей поведение. Например, Джерри Хирш утверждал, что поведенческие признаки определяются как наследственными факторами, так и действием окружающей среды. А существующие в инбредных линиях дрозофилы индивидуальные отличия детерминированы, тем не менее, генетической гетерогенностью популяции [7]. Хотя некоторые исследователи допускали, что поведение может определяться даже одним геном, но не имели достаточных доказательств, чтобы подтвердить свою гипотезу [8]. Эти и другие работы проводили, используя уже известных мутантов white, brown, eyeless, Bar, yellow, ebony, cut, vestigial, tan, и обнаруживали различные особенности поведения мутантных мух, объясняя это плейотропным действием изучаемых генов [6]. Такие работы ничего не говорили о роли нервной системы в регуляции поведения. И потому идея Бензера заключалась в возможности “микрохирургического препарирования нервной системы с помощью изучения мутантов”, которую он воплотил впервые в 1967 г. в Калифорнийском техническом институте: “Complex as it is, much of the vast network of cellular functions has been successfully dissected, on a microscopic scale, by the use of mutants in which one element is altered at a time” [9]. Тогда в практику вошел метод химически индуцированного мутагенеза: самцов дрозофилы пересаживали на среду, содержащую смесь сахарозы и этилметансульфоната, и инкубировали в течение 24 ч при температуре 25°С [10, 11]. Затем каждого самца отсаживали отдельно к виргинным самкам со сцепленными X-хромосомами (генотип XXY), получая в первом поколении самцов, унаследовавших свою мутантную X-хромосому от отца, а нормальную Y-хромосому – от матери. Таким образом выводили линии, в которых самцы несли мутации в X-хромосоме [9]. И проводили скрининг этих мутантов на наличие каких-либо поведенческих, электрофизиологических или морфологических девиаций, для чего разрабатывали соответствующие специальные методы анализа, в частности метод “противоточного разделения”, принцип которого Бензер позаимствовал из хроматографии. Для этого он использовал сдвоенные стаканчики, куда помещал мух и скидывал их на дно, и в течение минуты позволял мухам подниматься вверх, к источнику света. Затем нижний стаканчик закрывал и брал новый; повторял такую процедуру 15 раз. Наконец, строил график зависимости числа оставшихся мух в нижнем стаканчике от порядкового номера стаканчика [9]. Метод противоточного разделения может быть использован для разделения популяции мух на группы в зависимости от ответа особей на различные стимулы, такие как: земное притяжение, запах, звук, видимые образы. С помощью принципиально нового подхода в лаборатории Бензера или при его участии были получены и охарактеризованы различные мутанты с измененными характеристиками: period (циркадные ритмы), dunce (обучаемость), fruitless (половое поведение), sluggish-A (двигательная активность), sevenless (дифференцировка фоторецепторных клеток), methuselah (продолжительность жизни), comatose и no action potential (температурно-чувствительный паралич), drop-dead, spongecake, eggroll (морфология мозга и продолжительность жизни).
Бензер оставил после себя десятки воодушевленных учеников, коллег и последователей; продолжал браться за новые задачи, описывал функции разных генов дрозофилы, в последние годы интересовался моделированием нейродегенеративных заболеваний человека у дрозофилы. Его блестящие и элегантные экспериментальные подходы вскрыли “тонкую” структуру гена. Он обосновал роль генов в контроле поведения, показал возможность разложения сложных поведенческих признаков на элементарные, способствовал открытию множества генов дрозофилы, показал их роль в нервной системе и предложил способ выявления участка нервной системы, ответственного за тот или иной фенотип с помощью генетически маркированных мозаиков. Потому Сеймура Бензера называют одним из основоположников современной нейрогенетики. Однако заметим, что параллельно с нейрогенетическими работами Бензера, в России были заложены основы нейроэпигенетики М.Е. Лобашёвым. Им был предложен принципиально иной, эволюционно-физиологический, подход к наследованию поведенческих признаков и их изменчивости [12–14].
Значимый вклад в развитие нейрогенетических исследований внес последователь Бензера из университета Вюрцбурга – Мартин Хайзенберг, желавший сопоставить поведенческие фенотипы с особенностями строения нервной системы. Традиционные подходы к решению такой задачи основывались на хирургическом препарировании нервной системы, что влекло за собой два недостатка. Во-первых, гетерогенность материала, обусловленная невозможностью нанести идентичные повреждения разным особям. Во-вторых, малое количество материала, обусловленное трудоемкостью процедуры. Пытаясь решить указанную проблему, Бензер получал наборы различных мозаиков-гинандроморфов, где мутантная (мужская гемизиготная) часть организма маркировалась внешне отличимыми фенотипическими признаками (yellow, forked, white). Тем самым он сделал первый шаг к определению звена нервной системы, подверженного действию мутации: определял части организма, мутация в которых ведет к характерному поведенческому фенотипу. При этом Бензер замечал, что внутренние ткани могут иметь генотип, отличный от генотипа внешних тканей, что сильно ограничивает выводы, полученные при анализе мозаиков [15]. Хайзенберг выбрал другой путь решения упомянутой проблемы, что привело к разработке нового метода, который помог вести отбор мутантов еще по одному признаку – морфологии головного мозга насекомых. Созданный Хайзенбергом и его коллегами метод массового гистологического скрининга позволил достаточно быстро выявлять новых мутантов дрозофилы, у которых нарушалась морфология структур мозга или его частей [16]. Например, среди обнаруженных в таком исследовании мутантов был вариант, названный Vacuolar medulla, который характеризовался дегенерацией нескольких нейронов в медулле и втором зрительном ганглии, униполярных нейронов L1 и L2 ламины. Поведение мутантных особей оказалось нарушено: они были не способны различать движущиеся объекты, реализовывать реакцию посадки. Электроретинограмма выявила отсутствие возбуждающих и тормозных реакций в ламине, однако фоторецепторный потенциал не отличался от нормального. А это уже означало присутствие нарушений на уровне обработки зрительной информации, но не ее сенсорного восприятия [17]. Так Хайзенберг подошел к решению сложной проблемы расшифровки центрального звена в анализе сенсорной информации. Другие полученные Хайзенбергом мутанты характеризовались изменениями грибовидного тела: vacuolar pedunculi, beta lobes fused, mushroom body defect, mushroom bodies deranged. Рассматривая гипотезы о причинах возникновения того или иного фенотипа у указанных мутантов, Хайзенберг приблизился к выявлению механизмов генетического контроля процессов развития нервной системы, поддержания ее жизнеспособности и функционирования [18]. Таким образом, нейрогенетические исследования вышли на новый уровень. Схема эксперимента оставалась классической: индукция генетических нарушений, селекция мутанта по фенотипу, картирование гена, исследование его молекулярных и клеточных функций. Но это оставалось бы генетикой поведения, если бы свойства “гена поведения” рассматривали вне контекста его роли в нервной системе. Метод Хайзенберга разрешил ограничение метода Бензера – гистологический анализ наглядно показывал изменения структур нервной системы. Благодаря работам Бензера, Хайзенберга и их коллег было открыто множество генов, играющих роль в нервной системе.
Один из них – swiss cheese (sws, от англ. swiss cheese – швейцарский сыр), названный по фенотипу мутанта, полученного в 1979 г. в ходе гистологического скрининга [16, 19]. На срезах мозга мутантных особей было обнаружено множество полостей; нейродегенерация прогрессировала с возрастом. Нейроны мозга погибали путем апоптоза, также наблюдалась гибель глиальных клеток. Заметнее всего последствия нейродегенерации были видны в ламине. Мозг молодых мутантных мух не отличался от нормального, однако на пятый–шестой день их жизни обнаруживались первые полости. Кроме того, было показано, что глиальные клетки, оборачивающие тела и отростки нейронов, образуют аберрантные мембранные структуры и множественные слои, покрывающие нейроны [19]. При этом оказалось, что sws является аллельным вариантом гена olfE, обнаруженного в 1990 г. в скрининге индуцированных этилметансульфонатом мутантов. Мухи с мутацией olfE характеризовались нарушенным поведением в ответ на запах бензальдегида [20]. Вскоре ген olfE был клонирован [21], а уже в 1997 г. в комплементационном тесте доказан аллелизм sws и olfE [19].
Таким образом, был открыт ген, ответственный за поддержание жизнеспособности нейронов и глии, контролирующий обонятельную функцию. А значит, открылись новые возможности для исследования особенностей жизнедеятельности нервной ткани, в том числе и связанных с регуляцией поведения. Исследованиям свойств гена swiss cheese и его ортологов посвящено множество работ. В настоящем обзоре мы попытались обобщить имеющуюся на сегодняшний день информацию об этом гене и его ортологах, их роли в нервной системе.
ОБЩАЯ ХАРАКТЕРИСТИКА ГЕНА sws И ЕГО ОРТОЛОГОВ
Ген sws D. melanogaster картирован на X-хромосоме в положении 7D1. Его длина составляет 11 417 п.н. Ген кодирует три транскрипта, два из которых образуются вследствие альтернативного сплайсинга: содержат либо 8-й, либо 9-й экзон. Длина обоих транскриптов, названных sws-RA и sws-RC, составляет 5571 нуклеотид (размер соответствующих белков – 1425 аминокислот). Третий транскрипт sws-RB образуется вследствие наличия альтернативной точки старта транскрипции, не содержит 1–8-й экзоны, имеет длину 1928 нуклеотидов, кодирует белок длиной в 341 аминокислоту (рис. 1,а, б). Показано, что ген sws является консервативным, его ортологи найдены у различных видов от бактерий до человека. Сходство последовательностей продуктов этого гена при сравнении между мышью и человеком составляет 96%, между мышью и дрозофилой – 36% [22]. Белок SWS (обозначение заимствовано из литературы и удобно, хоть оно и отходит от классической традиции обозначения белков дрозофилы) имеет консервативный пататин-подобный лизофосфолипазный (эстеразный) домен, поэтому одно из названий белка-ортолога SWS у человека и некоторых других животных – PNPLA6 (patatin-like lysophospholipase domain containing 6). Сходство эстеразных доменов SWS дрозофилы и PNPLA6 человека составляет 61% [23]. Эстеразный домен SWS образован аминокислотным участком 952–1118, имеет два активных центра: нуклеофильный Ser985 и протон-акцепторный Asp1105, а также регуляторный мотив GXGXXG (положение 956–961, рис. 1,в).
Рис. 1.
Белок SWS и его ортологи. а – общая информация о цитогенетическом положении гена sws, длине известных транскриптов и изоформ белка; б – структура известных изоформ белка SWS у дрозофилы. Указаны названия, основные структурные элементы и аминокислотная длина изоформ; в – названия генов-ортологов sws у разных видов, структура канонических изоформ соответствующих белков и их длина в аминокислотах. Под трансмембранными, нуклеотид-связывающими и эстеразными участками указано их аминокислотное положение в белке.
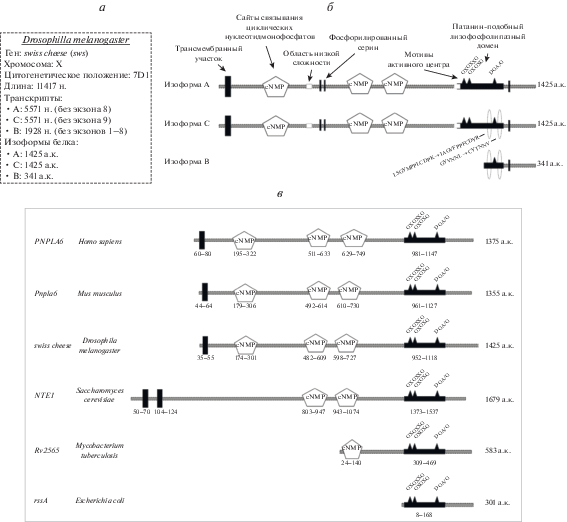
Кроме эволюционно-консервативного эстеразного домена в структуре SWS есть сайты, связывающие циклические нуклеотидмонофосфаты. В соответствующих белках у дрозофилы, человека, мыши и большинства других Metazoa по три таких сайта, у одноклеточных грибов Candida albicans, Schizosaccharomyces pombe, Saccharomyces cerevisiae присутствует два сайта, у белощекого хохлатого гиббона Nomascus leucogenys и бактерии Mycobacterium tuberculosis только один, а у Escherichia coli они вовсе отсутствуют. В структуре SWS дрозофилы нуклеотид-связывающие сайты занимают следующие аминокислотные позиции: 174–301; 482–609; 598–727 (рис. 1,в).
Белок SWS является интегральным, имеет трансмембранный участок с 35-й по 55-ю аминокислоты, заякоривающий SWS в клеточной мембране или в мембране эндоплазматического ретикулума (ЭР) [24–26]. Длинный С-концевой фрагмент белка обращен в цитоплазму. Белок может быть фосфорилирован по остаткам Ser в позициях 444, 453, 1160 [27].
Функциональное сходство SWS дрозофилы и PNPLA6 человека было продемонстрировано при изучении мутанта sws5. Индуцированная с помощью пищевой добавки RU486 экспрессия гетерологичного PNPLA6 в нейронах взрослых генетически модифицированных особей sws5;GS-elav-GAL4>UAS-PNPLA6 препятствовала развитию характерных для мутантов sws5 нейропатологических и поведенческих изменений. Более того, заметного улучшения удавалось добиться даже при индукции экспрессии PNPLA6 на 5-й день жизни особей, что предотвращало развитие дальнейшей нейродегенерации [28]. Еще один эксперимент показал функциональное сходство генов человека и Danio rerio. Подавление экспрессии гена pnpla6 рыбы с помощью введения морфолиновых олигонуклеотидов ведет к нарушению ее эмбриогенеза, которое удается предотвратить инъекцией мРНК PNPLA6 человека [29]. Эти данные свидетельствуют о функциональной консервативности ортологов гена sws.
ИЗУЧЕНИЕ ФУНКЦИЙ ОРТОЛОГОВ sws С ПОМОЩЬЮ ФОСФОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Ортолог sws человека – ген PNPLA6 – имеет еще одно название, историческое, а именно NTE (от английских названий соответствующего белка: neurotoxic esterase, neuropathy target esterase). Ген NTE был предсказан в середине XX в. при исследованиях причин возникновения отсроченного синдрома, вызванного отравлением токсичными органическими соединениями фосфора (OPIDN, organophosphorus compound induced delayed neurotoxicity). Оказалось, что продукт этого гена является мишенью для действия ряда веществ, содержащих пятивалентный фосфор: фосфатов, фосфонатов, фосфорамидатов, фосфинатов. Эти соединения ковалентно связываются с активным центром белка NTE (Ser966 для изоформы-2) через атом фосфора, обратимо ингибируя фермент. Некоторые из этих веществ способны участвовать во второй реакции, называемой “старением”, в ходе которой от атома фосфора отщепляется один из боковых радикалов, в результате чего электронная плотность перераспределяется между атомом фосфора и двумя связанными с ним атомами кислорода. Такой интермедиат приобретает отрицательный заряд и не может быть реактивирован даже сильными нуклеофилами. Реакция “старения” необратима, продолжается она несколько минут.
Исследованные фосфорорганические соединения являются по своей природе эфирами. Было предсказано, что ингибируемый ими фермент взаимодействует с последними из-за их подобия естественному субстрату (предположительно тоже эфиру) [30–32]. Из этого следует, что сам фермент является эстеразой.
Возможность взаимодействия белка NTE с некоторыми фосфорорганическими эфирами позволила разработать метод определения активности NTE (и его ортологов). Для этого используют специальный субстрат (фенилвалерат), красящие агенты (аминоантипирин-4 и гексацианоферрат(III) калия) и колориметрически определяют долю расщепившегося субстрата. Однако фениловый эфир пентановой кислоты (фенилвалерат) может быть расщеплен не только NTE, но и ацетилхолинэстеразой и бутирилхолинэстеразой. Поэтому для определения активности NTE в биологическом образце параллельно проводят два эксперимента с добавлением в образец фосфорорганических ингибиторов эстераз: только параоксона (ингибирует эстеразы кроме NTE) или мипафокса (ингибирует NTE) вместе с параоксоном. Чувствительное к мипафоксу, но устойчивое к параоксону расщепление субстрата в таком дифференциальном тесте и вызвано активностью NTE [33, 34].
Накопленные токсикологические данные позволяют предположить, что для развития синдрома OPIDN необходимы два явления: во-первых, ингибирование ферментативной активности NTE более чем на 70% и, во-вторых, “старение” белка. Считается, что “постаревший” интермедиат обладает собственным токсическим эффектом. Некоторые органофосфаты (фосфинаты), а также сульфонаты и карбаматы (эти вещества относят к типу ингибиторов “класса Б”) ингибируют NTE, но не вызывают его “старения”. Поэтому даже если падение активности фермента превышает 70%, синдром OPIDN у модельных животных не развивается. Более того, такие ингибиторы предотвращают последующее “старение” белка при добавлении так называемых ингибиторов “класса А” (фосфатов, фосфонатов, фосфорамидатов, вызывающих “старение”). Например, один из органофосфатов, способных индуцировать OPIDN, триортокрезилфоcфат (англ. TOCP) известен как соединение, вызвавшее отравление около 70 000 человек в XX в. Еще одним распространенным ингибитором NTE является диизопропилфторфосфат (DFP), а самым сильным ингибитором NTE является этилоктилфторфосфонат (EOPF) [33, 34].
Изучение свойств белка с помощью фосфорорганических соединений привело к идентификации функций, которые белок выполняет в клетке. Оказалось, что подавление эстеразной активности PNPLA6 мыши (здесь и далее в тексте мы будем использовать современное название белка и соответствующего гена у человека и мыши – PNPLA6) с помощью органофосфатов как in vivo, так и in vitro (в лизатах мозга) коррелирует со снижением активности лизофосфолипазы (расщепляющей лизолецитин), из чего был сделан вывод, что 1‑пальмитоиллизофосфатидилхолин (лизолецитин) является субстратом PNPLA6. Подтверждением этой гипотезы стал и факт того, что в мозге гетерозиготных мышей с генотипом Pnpla6+/– активность лизофосфолипазы составила 59%, а эстеразная активность PNPLA6 в дифференциальном тесте – 55% (от значений, полученных для мышей дикого типа) [35]. Таким образом была охарактеризована эстеразная функция белков-ортологов SWS.
СВОЙСТВА ЭСТЕРАЗНОГО ДОМЕНА
Как было упомянуто выше, у дрозофилы активным центром является Ser985, ответственный за реализацию эстеразной функции SWS. Результатом экспрессии мутантного sws, полученного методом направленного мутагенеза, приводящего к замене Ser985Asp, стало отсутствие эстеразной активности SWS в лизатах мозга дрозофилы при анализе в дифференциальном тесте. Напротив, гиперэкспрессия нормального sws в нейронах увеличивала активность эстеразы в 2 раза и не вызывала нейродегенерацию. Гиперэкспрессия функционального sws в глии повышала эстеразную активность в 12 раз и приводила к дегенерации глиальных клеток (в особенности эпителиальной астроцитоподобной глии) в ламине и в первой зрительной хиазме [24].
Активность SWS выражается в гидролизе фосфатидилхолина (ФХ) до глицерофосфохолина и двух молекул жирных кислот. Это означает, что SWS выполняет функции фосфолипазы Б, расщепляющей фосфолипиды по сайтам А1 и А2. Мутантная линия sws1, у которой вследствие мутации c.[C1616A] появляется преждевременный нонсенс-кодон Ser375*, характеризуется повышенным содержанием ФХ. Линия с гиперэкспрессией sws дикого типа, напротив, имеет сниженный уровень ФХ по сравнению с линией дикого типа CantonS [24].
Эксперименты in vitro доказали, что эстеразный домен PNPLA6 человека (727–1216 а.к. в изоформе-2) расщепляет фосфолипиды, лизофосфолипиды и моноацилглицериды, высвобождая жирные кислоты. Бóльшая активность проявляется в sn-1 реакциях: в случае расщепления моноацилглицеридов скорость реакций по положению sn-1 в 8–10 раз выше, чем по sn-2. Наибольшая скорость реакции наблюдается при расщеплении лизофосфолипидов, в 10 раз превышающая значения известных специфических лизофосфолипаз. Жирные кислоты высвобождаются еще быстрее из 1‑пальмитоил-лизоФХ – в 200 раз быстрее, чем если бы субстратом был 1-пальмитоил-2-арахидонил-ФХ [36].
СВОЙСТВА НУКЛЕОТИД-СВЯЗЫВАЮЩЕГО ДОМЕНА
Участок SWS, содержащий сайты связывания циклических нуклеотидмонофосфатов, структурно похож на фрагмент регуляторной субъединицы R1α протеинкиназы А (PKA) дрозофилы и человека. Нуклеотид-связывающий домен SWS взаимодействует с каталитической субъединицей C3 протеинкиназы PKA дрозофилы, но не с другими субъединицами (С1 и С2). Любопытно, что то же самое способен делать и белок PNPLA6 человека, что вновь доказывает эволюционную консервативность молекулярных процессов, контролируемых ортологами sws. Нормальный SWS ингибирует активность PKAС3: увеличение экспрессии sws в нейронах у особей с генотипом elav-GAL4;UAS-sws+ ведет к снижению общей активности PKA [37].
Обнаружена колокализация PKAС3 и SWS в нервных клетках мозга дрозофилы на мембранах ЭР и цитоплазматических везикул. В клетках мутантной линии sws1 такого явления не замечено, поэтому предполагают, что нормальный SWS рекрутирует PKAС3 к мембранам компартментов клетки, обращенным в цитоплазму. Функциональные нарушения SWS, наблюдаемые в случае мутации sws1, приводят к невозможности связывания регуляторных субъединиц SWS c каталитическими субъединицами PKAС3, в результате чего последние становятся, как предполагают авторы исследования [25], конститутивно активными. Гиперэкспрессия гена, кодирующего PKAС3, в мутантных особях sws1 приводит к усугублению патологических процессов в мозге дрозофилы. При этом не изменяется эстеразная активность фермента SWS; т.е. к патологическим проявлениям приводит именно измененная активность PKAС3, а не нарушенная эстеразная активность SWS. Удаление одной копии гена, кодирующего PKAС3, не влияет на нейродегенерацию в центральном мозге у мутанта sws1. Более того, в кортексе ламины наблюдается супрессия мутантного фенотипа у таких мух [25]. Однако нокдаун гена PKA-C3 сам по себе тоже способствует нейродегенерации, проявляющейся на 30-й день жизни имаго [37].
Показано, что PKAС3 мало похожа на другие известные субъединицы (С1 и С2), и отсутствие ее активности у мутантных D. melanogaster не может быть компенсировано, например, дополнительной экспрессией гена PKA-С1. Экспрессия PKA-C3 обнаружена у имаго в промежуточных натализин-положительных нейронах, связывающих нейроны мозга и мотонейроны брюшной нервной цепочки. Оказалось, что мутантные по PKA-C3 самцы не спариваются с самками, хотя их половое поведение остается нормальным: они способны проявлять реакцию “ухаживания” за виргинными самками; их локомоторная активность не отличается от нормы [38].
ВЗАИМОДЕЙСТВИЕ ДОМЕНОВ
Любопытны результаты исследований взаимодействия доменов SWS (или его ортологов). На культурах трансфицированных клеток фибробластов почки зеленой мартышки (COS) была проанализирована эстеразная активность и локализация белка PNPLA6 человека в зависимости от наличия того или иного домена. Оказалось, что отсутствие трансмембранного участка ведет к попаданию белка в цитоплазму и снижению эстеразной активности в 2 раза по сравнению с полноценным белком. Эстеразная активность PNPLA6 не зависит ни от присутствия нуклеотид-связывающего домена, ни от цАМФ. Белок без нуклеотид-связывающего домена колокализуется с маркером ЭР, кальнексином, при этом сам ЭР меняет морфологию, образуя тубулообразные выпячивания. Такая же картина наблюдается при экспрессии полноценного гена, а также мутантного его варианта, имеющего замену S966A. Следует заметить, что эти эксперименты проведены на фоне нормальной активности эндогенного PNPLA6, т.е. имела место “сверхэкспрессия” генетической конструкции. Авторы предположили, что вследствие гидрофобности эстеразного домена белки PNPLA6 агрегируют С-концами, притягивая мембраны ЭР друг к другу [39]. Это явление было обнаружено и у дрожжей Saccharomyces cerevisiae, белок NTE1 которых заякоривается в мембране гладкого ЭР. Гиперэкспрессия NTE1 ведет к образованию тубулообразных выпячиваний ЭР за счет межмолекулярных взаимодействий гидрофобных эстеразных доменов [40].
В другой работе исследователи проанализировали взаимодействие доменов PNPLA6 в клетках нейробластомы человека (SH-SY5Y). Оказалось, что эстеразный домен (в том числе и тот, активность которого отсутствовала вследствие замены S966A) сам по себе способен взаимодействовать с липидными каплями (или их предшественниками) – специализированными органеллами, накапливающими в своем составе липиды, в первую очередь триацилглицериды [41]. Несмотря на обнаруженное сродство эстеразного домена к липидной фазе, реализация эстеразной активности PNPLA6 не нуждается в липидном окружении [36]. Нуклеотид-связывающий домен ингибирует взаимодействие эстеразного домена с липидными каплями. Отсутствие первого ведет к тому, что белок остается на поверхности ЭР, но собирается в небольшие скопления рядом с липидными каплями или их предшественниками, формирующимися на мембране ЭР. Таким образом, полноценный PNPLA6 находится в мембране ЭР и не способен взаимодействовать с липидными каплями. До сих пор неизвестно могут ли какие-либо воздействия изменить конформацию нормального белка таким образом, чтобы он смог связать липидные капли. В работе [41] также было показано, что дополнительный синтез эстеразного домена приводил к увеличению количества липидных капель в клетках SH-SY5Y, снижению уровня ФХ, но не влиял на количество триацилглицеридов. Подавление экспрессии PNPLA6 на 70% с помощью интерференции РНК не влияло ни на уровень ФХ, ни на уровень триацилглицеридов, ни на распределение липидных капель в клетках. Это может означать, что в такой модели остаточный 30%-ный уровень мРНК обеспечивает нормальную функцию PNPLA6. Однако эта гипотеза остается недоказанной [41].
По-видимому, в норме нуклеотид-связывающий участок влияет на эстеразную активность белка. Мутация sws5 у дрозофилы, имеющая вследствие замены c.[G2431A] миссенс-эффект Gly648Arg в нуклеотид-связывающем домене белка, вызывает снижение эстеразной активности SWS. Это приводит к прогрессирующей с возрастом нейродегенерации, выявляемой в мозге в виде образования полостей; нарушению нормальной реакции отрицательного геотаксиса, наблюдаемому как на первой, так и на четвертой неделе жизни имаго; ускорению утомляемости мух, непрерывно скидываемых на дно стаканчика в автоматизированной установке. Например, если особи дикого типа BerlinK перестают подниматься по стенкам стаканчика через 7 ч постоянного стряхивания, то особи sws5 устают уже через 2 ч. Анализ кривых продолжительности жизни показывает сокращение и этого показателя у мутантов по сравнению с контролем [28].
С другой стороны, эстеразный домен, вероятно, влияет на нуклеотид-связывающий участок. Ингибирование эстеразной активности SWS с помощью TOCP приводит к снижению оной до 20% от первоначального уровня у особей дикого типа (после их кормления TOCP в течение 16 ч) и вызывает нейродегенерацию на 14-й день жизни имаго. Заметнее всего последствия нейродегенерации видны в ламине. У самок с генотипом sws1/CantonS снижена эстеразная активность на 50% по сравнению с особями дикого типа. Действие на них TOCP не вызывает нейродегенерацию к 14-му дню жизни имаго. Гиперэкспрессия sws в нейронах, осуществляющаяся у особей с генотипом elav-GAL4;UAS-sws+, увеличивает эстеразную активность до 160% по сравнению с таковой у особей дикого типа. Это не ведет к нейродегенерации. Кормление таких особей в течение 16 ч TOCP вызывает у них гораздо более сильную нейродегенерацию на 14-й день жизни, чем у особей дикого типа, принимавших TOCP. Сходные наблюдения оказались справедливыми и для поведенческого теста на фототаксис у всех перечисленных особей, коррелируя с уровнем нейродегенерации, выражающимся в доле полостей в мозге особей [37]. Такие противоречивые результаты не удается объяснить, рассматривая взаимодействие TOCP только с эстеразным доменом.
Вместе с тем было обнаружено, что кормление особей дикого типа TOCP в течение суток значительно снижает активность протеинкиназы А в лизатах, полученных из голов имаго. Такой же эффект имеет действие TOCP на культуру клеток гиппокампа крысы. Напротив, не нейротоксичный ингибитор эстераз параоксон (не ингибирует SWS/PNPLA6) не вызывает подобных изменений. Как было уже упомянуто, нокдаун PKA-C3 в нейронах дрозофилы способствует нейродегенерации, наблюдаемой на 30-й день жизни имаго. По-видимому, TOCP не только ингибирует эстеразную активность SWS/PNPLA6, связывая активный центр Ser, но и приводит к удержанию PKAC3, инактивируя ее [37]. Однако эту гипотезу еще предстоит проверить.
КЛЕТОЧНЫЕ ПРОЦЕССЫ, ЗАВИСИМЫЕ ОТ SWS И ЕГО ОРТОЛОГОВ
Главной и наиболее изученной функцией SWS и его ортологов является (лизо)фосфолипазная, реализуемая эстеразным доменом. Нарушение этой функции приводит к различным патологическим эффектам вследствие гибели нервных клеток. Но некоторые клетки способны адаптироваться к дисфункции такой липазы, среди них – клетки дрожжей.
Эстеразный домен ортолога SWS у Saccharomyces cerevisiae – NTE1 – на 39% похож на такой же домен PNPLA6 у человека. Известно, что белок дрожжей в среднем представлен 500 копиями на клетку. С помощью метода измерения активности фермента при дифференциальном ингибировании эстераз фосфорорганическими соединениями было показано, что NTE1 способен расщеплять фенилвалерат, а замена в активном центре Ser1406 ведет к отсутствию эстеразной активности. Этот белок чувствителен к действию “нейропатических” фосфорорганических соединений, но в 5–10 раз меньше, чем человеческий PNPLA6. Белок дрожжей способен расщеплять как лизоФХ, так и ФХ и, по-видимому, является мажорной, если не единственной, фосфолипазой Б у дрожжей по отношению к ФХ. Однако нокаут NTE1 не влияет ни на жизнеспособность, ни на рост клеток. Это объясняется следующей динамикой метаболизма холина. Если пометить холин изотопом [14C] и выращивать дрожжи на содержащей его среде, а затем перевести культуру на среду с немеченым холином и выращивать их при температуре 37°С, то у клеток-нокаутов по гену NTE1 [14C]-меченный холин попадает в клетку в 2 раза медленнее, чем у клеток дикого типа. Холин используется для образования промежуточного продукта – CDP-холина, который нужен для синтеза ФХ. В эксперименте у дрожжей-нокаутов концентрация меченого CDP-холина, как и ФХ, была в разы ниже, чем у нормальных клеток. Полученный из CDP-холина ФХ расщепляется в клетках дрожжей: в норме через 1 ч при 37°С концентрация меченого ФХ сокращается вдвое. У нокаутов она остается практически неизменной. Основным (>85%) продуктом расщепления ФХ является глицерофосфохолин. У нокаутов эта молекула отсутствовала в течение 25 ч проведения эксперимента, т.е. они были неспособны расщеплять ФХ с образованием глицерофосфохолина и двух жирных кислот. Оказывается, однако, что общий уровень ФХ одинаков у нормальных и мутантных клеток. Это косвенно доказали в эксперименте, где сперва дрожжи выращивали на [метил-14C]-метионине, который служил источником для метилирования фосфатидилэтаноламина, в результате чего образовывался меченый ФХ: его концентрация не отличалась у нокаута и дикого типа. Как показывают исследования, изменение уровня ФХ токсично для дрожжей. Поэтому при нокауте NTE1 замедляется метаболизм ФХ – дрожжи меньше потребляют холин и медленнее синтезируют ФХ, так как медленнее его расщепляют (активной остается только фосфолипаза Д, отщепляющая холин от ФХ) [40].
Изменения липидома, вызванные дисфункцией SWS/PNPLA6, могут влиять на различные свойства клетки, например на электрическую проводимость мембран. В эксперименте было показано, что подавление эстеразной функции с помощью нейропатических органофосфатов или при замене S966A в активном центре ведет к изменению профиля ионных токов через мембрану липосом, состоящих из диолеилФХ и эстеразного домена PNPLA6 человека [42].
Локализованный в мембране ЭР белок SWS может влиять и на эту органеллу. Было замечено, что у мутантов sws1 с возрастом менялась форма ЭР в нейронах, что выявили методом электронной микроскопии [24]. Позднее выяснилось, что дисфункция гена sws ведет к стрессовому ответу ЭР. Мутация sws1 вызывает четырехкратное увеличение уровня сплайсированного транскрипта, кодирующего транскрипционный фактор XBP1, который отвечает за стресс ЭР посредством активации шаперонов, ферментов синтеза липидов, факторов деградации белков. При этом увеличивается количество шаперона ЭР GRP78/BiP в головах молодых мух (возраст которых составил 4–6 дней жизни). Это сопровождается уменьшением уровня кальциевой АТФазы сарко-ЭР (SERCA) и соответствующим увеличением концентрации Ca2+ в цитоплазме натализин-положительных нейронов, экспрессирующих sws. В результате увеличивается уровень моно- и полиненасыщенных форм ФХ, а также различных форм лизоФХ [43].
Регуляция обнаруженных стрессовых реакций способна уменьшить патологические проявления, наблюдаемые у мутанта sws1. Экспрессия в нейронах дополнительного XBP1 снижала уровень GRP78/BiP к нормальному. На 7-й день жизни такие мухи не имели отклонений в локомоторной активности, анализируемой в тестах на отрицательный геотаксис; замедлялось развитие нейродегенерации: на 14-й день жизни мух полостей в мозге было больше, чем в норме, но меньше, чем у мутантов. При этом снижался уровень лизоФХ (но не достигал нормальных значений), в то время как уровень ФХ не менялся. К подобным фенотипическим эффектам приводила и дополнительная экспрессия SERCA в нейронах, однако она не влияла на уровень GRP78/BiP [43].
Нейродегенеративные и поведенческие изменения, наблюдаемые у мутантов sws1, удалось предотвратить, используя таурурсодеоксихолевую кислоту (TUDCA), производное одной из желчных кислот. TUDCA оказывает противоапоптотическое действие, предотвращая активацию каспазы-3, высвобождение Bax и цитохрома C [43].
Еще одно исследование, посвященное изучению реакции организма на дисфункцию sws, показало, что мутанты sws1 и swsolfE имеют более низкую выживаемость при 96-часовой инкубации на пище с окислителями (метилвиолет и перекись водорода). Мутант swsolfE при этом характеризуется бóльшим уровнем окисленных липидов (измеренным с помощью тиобарбитуровой кислоты) по сравнению с контролем, что свидетельствует о возможном изменении окислительно-восстановительного статуса клеток при нарушении функции гена sws [44].
Другие исследования показали связь sws и митохондрий. У особей с гиперэкспрессией sws в нейронах (elav-GAL4;UAS-sws+) выявлено нарушение формы и распределения митохондрий в нейронах, выявляемое методом электронной микроскопии. И как было отмечено выше, нейродегенерация у таких мух не наблюдается [37]. Напротив, у мутантов sws1 наблюдается нейродегенерация, но не замечено изменение митохондрий в нейронах [24]. Однако у личинок-мутантов sws1, так же как и у личинок с нокдауном sws в нейронах, в пресинаптических окончаниях нейромышечных соединений заметно уменьшалось количество митохондрий [45].
Представленные исследования показывают множественность клеточных функций, зависимых от действия гена sws, и указывают на сложный ответ клетки при изменении работы гена sws.
ВЗАИМОДЕЙСТВИЕ sws С ДРУГИМИ ГЕНАМИ
Существуют данные о взаимодействии sws с некоторыми другими генами. Например, исследования обонятельной функции у мутантов swsolfE выявили взаимодействие sws с генами, кодирующими субъединицы интегринов: myospheroid и inflated. Несмотря на то что использованные мутации в этих генах рецессивны и не влияют на обоняние будучи в гетерозиготном состоянии, в положении транс-аллельного взаимодействия (дигетерозиготное носительство) некоторые мутации ведут к нарушению чувствительности мух как к бензальдегиду, так и к изоамилацетату (восприятие которого нарушено у мутантов по гену myospheroid). Однако мутация, определяющая полную дисфункцию гена интегрина, не вызывает такие эффекты. Подобные результаты получены и для гена α2-субъединицы интегрина (inflated): транс-дигетерозиготы хуже чувствовали бензальдегид, чем моногетерозиготы [46].
Еще одна работа посвящена изучению двух генов, открытых при участии Сеймура Бензера. Было показано, что нейродегенерация у мутанта sws1 усиливается при потере функции гена циркадных ритмов per: увеличивались как количество, так и площадь полостей в нейропиле дейтоцеребрума. Это свидетельствует о взаимодействии указанных генов по типу кумулятивной полимерии при рассмотрении нейродегенеративного фенотипа, а значит, существует независимый контроль ими клеточных функций [47]. В свою очередь это говорит о сложных взаимодействиях генов, определяющих тот или иной фенотип, и предлагает рассматривать более сложные модели в нейрогенетике.
РЕГУЛЯЦИЯ ЭКСПРЕССИИ sws И ЕГО ОРТОЛОГОВ
Вопрос регуляции экспрессии sws пока остается неизученным. На сегодняшний день нам известна только одна работа, посвященная исследованию контроля экспрессии ортолога sws, а именно гена PNPLA6 человека. Добавление в питательную среду к клеткам нейробластомы человека (SK-N-SH) активатора протеинкиназы С – форбол-12-миристат-13-ацетата – вело к снижению уровня мРНК гена PNPLA6 на 60–70%, что обусловило остаточный уровень белка и соответственно остаточную эстеразную активность на уровне около 1/3 от нормы. Ингибитор протеинкиназы C – стауроспорин – не влиял на исследуемые показатели, как и введение в клетки вектора для гиперэкспрессии PNPLA6 под собственным промотором. Исходя из полученных данных и информации о времени жизни мРНК гена PNPLA6, авторы заключили, что активация протеинкиназы С влияет на экспрессию эндогенного PNPLA6, но не на сам белок или его мРНК [48].
ОСОБЕННОСТИ ЭКСПРЕССИИ sws У ДРОЗОФИЛЫ
Длинные транскрипты гена sws (sws-RA и sws-RC) появляются на первой–третьей стадиях эмбриогенеза в большом количестве, затем к стадии личинки третьего возраста и куколки уровень длинного транскрипта падает. У имаго длинные транскрипты обнаружены как в теле, так и в голове. Короткий транскрипт (sws-RB) появляется только у имаго в голове [21].
Исследование экспрессии sws с помощью транскриптомных методов выявило, что транскрипты sws можно обнаружить на всех стадиях онтогенеза особей. Наибольший уровень мРНК sws найден в первые часы развития эмбриона и у взрослых особей. Низкая экспрессия sws характерна для слюнных желез личинок, умеренный уровень экспрессии наблюдается в пищеварительной системе личинок и имаго, высокая экспрессия происходит в головах особей вне зависимости от их возраста, и очень высокий уровень транскриптов sws обнаружен в семенниках и жировом теле имаго [49, 50].
У личинок третьего возраста sws экспрессируется в нейронах и в глиальных клетках мозга и брюшной нервной цепочки. С помощью иммуногистохимического окрашивания белок SWS был обнаружен в периферических нервах: аксонах нейронов и в глиальных клетках, их окружающих, а также в нейромышечных окончаниях [45].
Дисфункция sws влияет на развитие нейромышечных контактов личинок. Так, у самцов sws1 и особей с нокдауном sws в нейронах было обнаружено увеличение числа сателлитных бутонов и нарушение цитоскелета. Вместе с тем количество активных зон в одном бутоне, содержащих активные синапсы, оказалось уменьшенным у личинок с нарушенной функцией sws. Хотя сам SWS располагается пресинаптически и отсутствует в постсинапсе, было замечено снижение уровня белка DLG – постсинаптического маркера – ортолога PSD-95 млекопитающих [45].
Экспрессия sws в мозге имаго D. melanogaster в первые дни жизни происходит повсеместно, но с возрастом ограничивается крупными клетками [24]. Ген sws экспрессируется в фоторецепторных клетках; его дисфункция ведет к прогрессирующей с возрастом дегенерации фасеток [26].
Реакция нервных и глиальных клеток на дисфункцию sws различается. Тканеспецифичная экспрессия нормального аллеля sws в глии мутантов sws1 препятствовала патологическому множественному обертыванию, однако нейроны продолжали гибнуть. Экспрессия аллеля sws дикого типа только в нейронах мутантных особей sws1, но не в глии, приводила к отсутствию полостей в мозге, однако морфология глиальных клеток имела фенотип как у мутанта. Похожие явления были обнаружены и при тканеспецифичной экспрессии у дрозофилы мышиного Pnpla6, что вновь показывает функциональное сходство соответствующих белков [24]. Позднее выяснилось, что нокдаун sws только в нейронах или только в глии не влияет на образование множественных мембранных слоев, образованных глиальными клетками, из чего следует, что для этого необходима дисфункция sws во всех клетках нервной системы (проявляющаяся у мутантов sws1) [19, 51]. Более того, для поддержания жизнедеятельности клеток требуется конститутивная экспрессия sws. Использование факультативного промотора генов теплового шока для экспрессии нормального sws не супрессировало мутантный фенотип sws1 [24].
Ингибирование эстеразной активности SWS с помощью TOCP имеет отсроченный нейродегенеративный эффект. После однодневного кормления токсичным органофосфатом у только что вылупившихся дрозофил фиксировали снижение эстеразной активности SWS на 80%, и к 14-му дню жизни этих особей образовывались полости в мозге, нарушалась быстрая реакция фототаксиса [37].
Действие TOCP на первичную культуру нейронов дрозофилы вело к заметному сокращению длины отростков, прямо пропорционально зависящему от дозы токсина [37].
Наличие глиального фенотипа у мутанта sws1 подтолкнуло исследователей к выявлению типов глии, где экспрессируется sws. С помощью иммунофлуоресцентного окрашивания выявили белок SWS в пограничной глии между кортексом и нейропилем в протоцеребруме, фенестрированной (периневральной) и сателлитной (кортексной) глии ламины, глии нейропиля медуллы. В глии нейропиля в протоцеребруме белок не был обнаружен таким методом, так же как и в гигантской глии первой зрительной хиазмы, и в эпителиальной (астроцитоподобной) глии ламины [24].
Однако факт экспрессии гена в том или ином типе клеток сам по себе не свидетельствует о функциональной роли исследуемого гена в этих клетках. Поэтому с помощью метода электронной микроскопии был проведен анализ влияния подавления экспрессии гена sws в глии на морфологию этих клеток. Индуцированный интерференцией РНК посттранскрипционный сайленсинг sws во всех глиальных клетках (с помощью драйверов экспрессии repo-GAL4 или loco-GAL4) вызывал образование полостей, наиболее заметных в кортексе ламины, на границе между ретиной и ламиной. Количество полостей увеличивалось с возрастом имаго, однако число глиальных клеток при этом не менялось. Оказалось, что при подавлении экспрессии sws апоптоз глии происходит до второго дня жизни имаго. У двухдневных мух число глиальных клеток было ниже, чем у нормальных особей. Наибольшее снижение – до 58% – выявили для глиальных клеток, окружающих оптические доли. В меньшей степени падало число клеток эпителиальной (астроцитоподобной) глии ламины [51].
К подобным изменениям приводило подавление экспрессии sws в отдельных типах глии, а именно в субпериневральной или оборачивающей глии центральной нервной системы, но не в астроцитоподобной или кортексной. При нокдауне sws в субпериневральной глии образовывались крупные аберрантные мембранные структуры, происходило это преимущественно в ламине. Напротив, подавление экспрессии sws в оборачивающей глии центральной нервной системы вело к дегенерации ее отростков в нейропиле ламины, медуллы, лобулы и лобулярной пластинки, где аксоны нейронов становились в результате оголенными [51].
Авторы предположили, что именно недостаток эстеразной функции SWS ведет к нарушениям жизнедеятельности глиальных клеток, так как синтез дополнительного мутантного по активному центру SWS не влияет на развитие нейродегенерации у особей с нокдауном sws, в то время как мутантный по нуклеотид-связывающему домену белок снижает темпы нейродегенерации. Обнаруженная при дисфункции SWS в глии нейродегенерация влияла на поведение особей – с возрастом ухудшалась реакция фототаксиса. Такие изменения наблюдались как при нокдауне sws во всех типах глии, так и в оборачивающей и субпериневральной в отдельности, и прогрессировали с возрастом. При этом зрительная или моторная функция не нарушалась, а страдало центральное звено обработки информации. Мухи с нокдауном sws во всех типах глии хуже различали специальные ориентиры в “Буридановом” эксперименте, и с возрастом их гигантские интернейроны (giant fibres, обрабатывают входящую зрительную информацию от сенсорных нейронов и передают сигнал на моторные нейроны) хуже отвечали на электрическое возбуждение. Нокдаун не влиял на электроретинограмму или возбудимость моторных нейронов. Не менялся и уровень маркера активных зон нейромышечного синапса – белка Bruchpilot, но увеличивался уровень белка CSP, ответственного за Ca2+-индуцируемый экзоцитоз [51].
РОЛЬ ГЕНА sws В КОНТРОЛЕ ПОВЕДЕНИЯ
Выше мы уже отмечали, что связанная с дисфункцией sws нейродегенерация может влиять на нарушение поведения у D. melanogaster: подавлять реакции положительного фототаксиса [37, 51] или отрицательного геотаксиса [28]. Мутация swsolfE была обнаружена у мух с нарушенным поведением в ответ на запах бензальдегида [20]. Мутант olfE был изучен среди прочих (из группы мутантов olfactory) в исследовании группового поведения мух, в ходе которого было показано, что в группах более 50 особей есть мухи, которые ищут пищу, а затем оставшиеся особи собираются вокруг “первопроходцев”. Однако при описании результатов работы авторы не указали данные, полученные при исследовании мутанта olfE. Утверждалось, что мутанты группы olf имеют более низкие разработанные количественные показатели для оценки такого поведения, чем контрольные мухи Canton-Special [52].
ОСОБЕННОСТИ ЭКСПРЕССИИ Pnpla6 У МЫШИ
У мыши экспрессия Pnpla6 начинается на 7-й день жизни эмбриона и важна для его выживания [53]. Экспрессия гена у эмбрионов обнаружена в мезонефросе, метанефросе, обонятельном, кишечном, трахеальном эпителии, в языкоглоточном и тройничном ганглиях. Постнатальная экспрессия повсеместна в центральной нервной системе, однако с возрастом сокращается до структур моста, нижних бугров четверохолмия, гипоталамуса, зубчатого ядра мозжечка, продолговатого мозга и коры. С возрастом экспрессия Pnpla6 отчетливо выявляется в крупных нейронах (например, в пирамидных клетках гиппокампа и в клетках Пуркинье мозжечка) [22]. Кроме того, экспрессия Pnpla6 обнаружена в гормон-продуцирующих клетках семенников – клетках Лейдига [53].
Нокаут Pnpla6 имеет летальный эффект – особи гибнут на 9-й день эмбриогенеза из-за дефектов развития плаценты матери. Особи, несущие лишь один нормальный аллель Pnpla6, жизнеспособны и развиваются нормально, хотя и имеют сниженную на 50% активность PNPLA6 [53, 54].
Гетерозиготные мыши Pnpla6+/– не имеют нейропатологических изменений, характеризуются нормальными обучаемостью и памятью. Введение таким мышам малой дозы нейропатического органофосфата EOPF вызывает увеличение локомоторной активности, при этом ни в головном, ни в спинном мозге не наблюдается патологических изменений. Введение токсичных доз EOPF вызывает снижение двигательной активности и судороги [53].
Ингибирование PNPLA6 с помощью фосфорорганического соединения MOCDPP при ежедневном (в течение пяти дней) добавлении токсина в пищу мышам вело к увеличению уровня ФХ до 120% только через неделю после окончания отравления, затем уровень восстанавливался до нормального. Вместе с тем развивалась дегенерация дистальных отделов сенсорных и моторных нейронов в спинном мозге, но это не влияло на двигательную активность особей [55].
Инактивация экспрессии Pnpla6 в нейронах мыши также ведет к нейродегенерации – гибнут клетки гиппокампа, таламуса, клетки Пуркинье мозжечка [56]. У мышей с нокаутом Pnpla6 в нейронах (nestin-cre:NTEfl/fl) происходит дегенерация дистальных участков сенсорных и моторных нейронов в спинном мозге уже у молодых особей (возраст 1–3 мес.), эффект сохраняется и у взрослых (возраст 6–12 мес.). При этом, как заметили авторы работы [56], мыши страдают от двигательных нарушений в конечностях. Взятые у 7-дневных мышей клетки гранулярных нейронов мозжечка характеризовались сниженным образованием конститутивно секретируемых sAPPα и рилина (до 80% от нормального уровня). В мозге таких особей уровень ФХ увеличивался до 120% от нормальных значений [55].
В периферической нервной системе белок PNPLA6 обнаружен в аксонах, а также в зрелых немиелинизирующих и миелинизирующих шванновских клетках. Белок локализован вокруг ядра. Нормальный Pnpla6 не влияет на образование миелиновой оболочки вокруг отростков нервных клеток в периферической нервной системе, но важен для образования шванновской оболочки (Remak bundles) вокруг немиелинизированных нервных волокон (Remak fibers). Дефицит PNPLA6 в немиелинизирующих шванновских клетках способствует неполному обертыванию нервных волокон и индуцирует их дегенерацию. При повреждении нервного волокна значительно усиливается экспрессия Pnpla6 в шванновских клетках, что может играть роль в демиелинизации и защитных функциях периферической глии [57].
ОСОБЕННОСТИ ЭКСПРЕССИИ PNPLA6 У ЧЕЛОВЕКА
Согласно транскриптомным данным мРНК гена PNPLA6 найдена практически во всех тканях человека [58]. Эстеразная активность PNPLA6 обнаружена помимо нервной ткани в лимфоцитах [59] и тромбоцитах [60], плаценте [61], фибробластах [62].
Ингибирование продукта гена PNPLA6, наблюдающееся у отравившихся TOCP, приводит к развитию синдрома OPIDN, характеризующегося дегенерацией длинных аксонов моторных нейронов в пирамидном тракте спинного мозга. В меньшей степени страдают сенсорные нейроны. Это вызывает спастичность, поражающую нижние конечности сильнее, чем верхние.
Рецессивные мутации в гене PNPLA6 в состоянии гомозиготы или транс-дигетерозиготного компаунда приводят к различным нейродегенеративным заболеваниям. При этом могут погибать нейроны спинного мозга, мозжечка, гипофиза, фоторецепторные клетки. Это, в свою очередь, вызывает спастичность, атаксию, гипогонадотропный гипогонадизм, пигментный ретинит, хориоретинальную дистрофию. Более подробно роль гена PNPLA6 в нейропатологиях человека описана в нашем обзоре [63].
ФУНКЦИОНАЛЬНОЕ ЗНАЧЕНИЕ МУТАЦИЙ В ГЕНЕ PNPLA6
Обнаружение заболеваний, ассоциированных с мутациями в гене PNPLA6, инициировало ряд исследований по изучению функций “мутантных” белков. Например, короткий белок PNPLA6D376GfsX18, образующийся вследствие инсерции c2494_2495InsTGTGGGCCTGGGG у пациентов с синдромом Gordon Holmes [23], не является функциональным. Экспрессия соответствующего гена у мух-мутантов sws1 не улучшала ни один из показателей: долю полостей в мозге мух, параметры реакции быстрого фототаксиса, уровень ФХ или лизоФХ [64].
Эксперименты по восстановлению эмбриогенеза Danio rerio с нокдауном гена pnpla6, индуцированным с помощью введения морфолиновых олигонуклеотидов, показали, что человеческая мРНК PNPLA6 дикого типа почти полностью восстанавливает морфологию личинок, в отличие от мутантных мРНК, ведущих к заменам в белке, встречающимся у пациентов с синдромами Oliver–McFarlane/Laurence–Moon. Наблюдаемые явления подтверждают негативное влияние указанных мутаций на функциональную состоятельность белка PNPLA6 [29].
Оказалось, что мутации, обнаруженные у пациентов с различными PNPLA6-ассоциированными заболеваниями, по-разному влияют на функции белка. Для проведения исследования анализировали гены, приводящие к заменам в нуклеотид-связывающих доменах (L524P, G578W, T629R) или в эстеразном домене (A1029T, R1099Q). Чтобы изучить свойства дефектных вследствие мутаций белков, экспрессировали соответствующие генетические конструкции у мух, имеющих мутацию sws1. Чтобы получить одинаковый уровень экспрессии конструкций, трансгенез проводили с помощью интегразы PhiC31, а затем подтверждали равенство уровней белка в тканях головы с помощью Вестерн-блоттинга. Оказалось, что белки с заменами L524P и G578W, по-видимому, менее стабильны, так как экспрессия соответствующих генов не приводила к образованию того же уровня белка, что и в других вариантах [64].
Для изучения биологических функций различных вариантов PNPLA6 использовали систему UAS-GAL4: в нейронах (с помощью драйвера Appl‑GAL4) у мутантных мух (sws1) экспрессировали варианты гена человека (UAS-PNPLA6). Расщепляющий ФХ SWS не способен эффективно делать это в случае мутации sws1. При этом экспрессия ни нормального аллеля PNPLA6, ни какого-либо из исследованных мутантных аллелей не ведет к нормализации уровня ФХ у мух, возраст которых составил 3–5 дней. Однако иная картина обнаруживается с другим субстратом SWS – лизоФХ. Если нормальный аллель PNPLA6 возвращает уровень лизоФХ к норме у мух того же возраста, то ни один из изученных мутантных аллелей делать это не способен. При этом 7-дневные мухи с экспрессией какого-либо из мутантных вариантов PNPLA6 имеют нормальную или близкую к нормальной локомоторную активность в тесте на фототаксис. Но у 14-дневных мух ни аллель дикого типа, ни мутантные аллели PNPLA6 не способны восстановить поведение мух до нормального. Тем не менее экспрессия гена дикого типа или дефектных по фосфолипазному домену вариантов “спасает” мух от нейродегенерации, характерной для мутанта sws1. Кроме того, если экспрессировать “мутантные” конструкции в глиальных клетках (с помощью драйвера loco-GAL4), то поведение таких мух в тесте на фототаксис не отличается от поведения контрольных особей [64].
Результаты этих экспериментов показывают, что нормальная функция SWS зависит как от эстеразного, так и от нуклеотид-связывающего участка и в первую очередь связана с расщеплением лизоФХ. Нормальный аллель PNPLA6 способствует восстановлению к норме как поведения, так и морфологии мозга. Мутантные аллели PNPLA6 не способны восстанавливать ни уровень лизоФХ на фоне дисфункции sws, ни поведение у 14-дневных особей; они по-нейродегенерации в мозге. Известно уже множество мутаций, связанных с различными PNPLA6-ассоциированными синдромами у человека, и не замечено корреляции клинической симптоматики и локализации мутации. Авторы обсуждаемой статьи предполагают, что отличия в клинических проявлениях мутаций PNPLA6 вообще связаны с влиянием генетического фона, а не с вариабельностью функций белка в зависимости от поражения нуклеотид-связывающего или эстеразного домена [64].
Результаты другого исследования, в котором анализировали эстеразную активность в фибробластах, полученных от пациентов с различными PNPLA6-ассоциированными заболеваниями, предлагают к рассмотрению альтернативную гипотезу. Полученные значения активности PNPLA6 в фибробластах разделили на четыре группы, сделав вывод, что разные мутации по-разному влияют на функцию белка [29]. Возможно, из-за разной чувствительности к дисфункции PNPLA6 у пациентов страдают разные клетки (чем сильнее снижение эстеразной функции, тем больше клеток погибает). А значит, развиваются разные синдромы. Однако эта любопытная гипотеза еще нуждается в тщательной экспериментальной проверке.
Так, в одной из работ изучали влияние мутаций PNPLA6 на эстеразную активность фермента в первичных культурах фибробластов, полученных от пациентов с наследственной спастической параплегией (тип SPG39) или их родственников [62]. Рецессивные гомозиготы, имеющие замену M1012V, и гетерозиготные компаунды с заменой R890H и нарушенной структурой белка из-за инсерции c2946_2947InsCAGC имели сниженную эстеразную активность на 59–65% по сравнению с клетками, имеющими хотя бы один нормальный аллель PNPLA6. Однако снижения эстеразной активности, как оказалось, недостаточно для развития заболевания. Так, еще большее снижение эстеразной активности обнаружено в фибробластах двух здоровых взрослых родственников пациентов, гетерозиготных носителей аллеля c2946_2947InsCAGC [62]. Это свидетельствует о неоднозначности связи клинических проявлений, эстеразной активности, локализации мутации в гене PNPLA6. А значит, несмотря на многочисленные исследования, мы еще далеки от понимания механизмов развития PNPLA6-ассоциированных патологий, роли этого гена (и его ортологов) в функционировании разных структур нервной системы и поддержании жизнеспособности нервных и глиальных клеток.
ЗАКЛЮЧЕНИЕ
За прошедшие 100 лет, с рождения Сеймура Бензера, генетика человека и животных чрезвычайно преобразилась. Казавшаяся когда-то невероятной расшифровка геномов многих видов значительно упростила и конкретизировала общее представление о наследственной информации, определяющей различные, в том числе и поведенческие, свойства организма. Исследования конкретных генов, обнаруженных вследствие изучения мутантов, позволили описать многообразные функции, находящиеся под контролем изучаемых генов, на клеточном и молекулярном уровнях, и связать эти дискретные единицы наследственности с поведенческими признаками.
На частном примере исследований гена sws (открытого благодаря новаторским подходам С. Бензера и М. Хайзенберга), а также его ортологов, выявляются успехи и проблемы современной нейрогенетики, а также прослеживается история ее развития. Ортолог гена sws впервые был открыт у курицы [65], а затем у человека [66–68] в ходе токсикологических исследований третьей четверти XX в. [34]. Позднее, к концу XX в., описан ген sws у дрозофил, полученных в скрининге нейродегенеративных мутантов [19]. И вскоре обнаружены несколько ортологов у кишечной палочки, микобактерии, дрожжей, нематоды; показана их эволюционная консервативность [69]; а затем исследован ортолог у мыши [22]. Оказалось, что эти гены контролируют функцию фосфолипазы Б и именно ее дефицит ответствен за наблюдаемые нейродегенеративные фенотипы. Однако более подробное исследование структуры белка выявило еще и необычную роль нуклеотид-связывающего домена как в реализации эстеразной активности, так и в развитии нейродегенерации. Таким образом, соотнесение структуры белка с его функциями выявило, как минимум, роль метаболизма фосфатидилхолина в поддержании жизнеспособности нервных клеток. В 2008 г. открыто первое наследственное заболевание, ассоциированное с ортологом sws у человека [70]. А в последующее десятилетие, благодаря внедрению геномных исследований в медицину, обнаружено еще несколько подобных синдромов [63]. Это, во-первых, способствовало более тонкому изучению функциональной структуры соответствующего белка и, во-вторых, открыло его роль в нескольких отделах нервной системы человека. Исследования дрозофилы и мыши помогли открыть типы клеток нервной системы, нуждающиеся в нормальной работе sws/Pnpla6: как нервных, так и глиальных [51, 57]. Так, анализ морфологии чувствительных к дисфункции гена клеток привел к установлению связи различных клеточных популяций с контролем некоторых особенностей поведения. Наконец, проведены исследования, касающиеся ответа клетки на дисфункцию гена sws, и предложен механизм как генетического, так и фармакологического вмешательства для подавления характерных патогенных процессов [43]. Значит, открываются перспективы для лечения редких наследственных заболеваний человека, связанных с геном PNPLA6. Это становится особенно актуально в связи с тем, что функции sws и его ортологов позволили открыть нейропатические свойства целого ряда токсичных фосфорорганических соединений, используемых в качестве пестицидов или рассматриваемых как потенциальное химическое оружие [33, 34, 66].
Как мы видим, редукционистский подход к изучению нейрогенетики оказался чрезвычайно плодотворным. Многие вопросы, однако, остаются по-прежнему открытыми и возникают новые проблемы, требующие решения. В частности, не расшифрованы механизмы регуляции экспрессии гена sws (и его ортологов); не выявлены все молекулярные события, происходящие при дисфункции этих генов; неизвестна реакция всех типов клеток организма на нарушение функции генов; неизвестны причины гибели клеток; не ясно, какие именно нарушения в работе гена достаточны для развития патологии. Эти и другие вопросы ожидают дальнейших исследований.
Но еще более важно понимание работы генома как целого функционального комплекса. Ведь показано, что дисфункция гена sws плейотропно влияет на различные процессы в клетке, а также действует на проявление работы других генов. Кроме того, разные клетки организма по-разному чувствительны к дисфункции sws, вероятно, в силу отличий на уровне реализации генетической информации из-за дифференцированности клеточных популяций. Есть и тысячи других генов, экспрессия которых необходима для работы нервной системы. Это означает, что нейрогенетику ждут в будущем новые принципиальные изменения. Требуется применение холистического подхода для интеграции полученных частных знаний о функциях конкретных генов, роли конкретных клеток в реализации той или иной формы поведения. Методы биоинформатики, системной биологии и компьютерного моделирования, возможно, смогут помочь в решении таких задач.
Работа выполнена при финансовой поддержке РФФИ в рамках научного проекта № 20-34-90148.
Настоящая статья не содержит каких-либо исследований с использованием в качестве объекта животных.
Настоящая статья не содержит каких-либо исследований с участием в качестве объекта людей.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Harris W.A. Seymour Benzer 1921–2007 the man who took us from genes to behaviour // PLoS Biol. 2008. V. 6. № 2. P. e41. https://doi.org/10.1371/journal.pbio.0060041
Jan Y.N., Jan L. Seymour Benzer (1921–2007) // Science. 2008. V. 319. № 5859. P. 45. https://doi.org/10.1126/science.1154050
Dudai Y. Seymour Benzer (1921–2007) // Neuron. 2008. V. 57. № 1. P. 24–26. https://doi.org/10.1016/j.neuron.2007.12.01
Tanouye M.A. Seymour Benzer 1921–2007 // Nat. Genet. 2008. V. 40. № 2. P. 121. https://doi.org/10.1038/ng0208-121
Anderson D., Brenner S. Seymour Benzer (1921–2007) // Nature. 2008. V. 451. № 7175. P. 139. https://doi.org/10.1038/451139a
Greenspan R.J. The origins of behavioral genetics // Curr. Biol. 2008. V. 18. № 5. P. R192–R198. https://doi.org/10.1016/j.cub.2008.01.015
Hirsch J., Boudreau J.C. Studies in experimental behavior genetics: I. The heritability of phototaxis in a population of Drosophila melanogaster // J. Comparative and Physiol. Psychology. 1958. V. 51. № 6. P. 647–651. https://doi.org/10.1037/h0039498
Scott J.P. Effects of single genes on the behavior of Drosophila // The Am. Naturalist. 1943. V. 77. № 769. P. 184–190.
Benzer S. Behavioral mutants of Drosophila isolated by countercurrent distribution // Proc. Natl Acad. Sci. USA. 1967. V. 58. № 3. P. 1112. https://doi.org/10.1073/pnas.58.3.1112
Alderson T. Chemically induced delayed germinal mutation in Drosophila // Nature. 1965. V. 207. № 4993. P. 164–167. https://doi.org/10.1038/207164a0
Lewis E.B., Bacher F. Method of feeding ethyl methane sulfonate (EMS) to Drosophila males // Dros. Inf. Serv. 1968. V. 43. № 193. P. 193.
Лобашев М.Е. Сигнальная наследственность // Сб. исследований по генетике. Вып. 1. Л.: Изд-во ЛГУ, 1961. С. 3–11.
Лопатина Н.Г. М.Е. Лобашев и новые главы в науке – биология, физиология и генетика условного рефлекса // Экол. генетика. 2007. Т. 5. № 4. С. 5–10. https://doi.org/10.17816/ecogen545-10
Савватеева-Попова Е.В., Никитина Е.А., Медведева А.В. От нейрогенетики к нейроэпигенетике // Генетика. 2015. Т. 51. № 5. С. 613–624. https://doi.org/10.1134/S1022795415050075
Hotta Y., Benzer S. Genetic dissection of the Drosophila nervous system by means of mosaics // Proc. Natl Acad. Sci. USA. 1970. V. 67. № 3. P. 1156–1163. https://doi.org/10.1073/pnas.67.3.1156
Heisenberg M.A., Böhl K. Isolation of anatomical brain mutants of Drosophila by histological means // Zeitschrift für Naturforschung C. 1979. V. 34. № 1–2. P. 143–147.
Coombe P.E., Heisenberg M. The structural brain mutant Vacuolar medulla of Drosophila melanogaster with specific behavioral defects and cell degeneration in the adult // J. Neurogenetics. 1986. V. 3. № 3. P. 135–158. https://doi.org/10.3109/01677068609106845
Heisenberg M. Mutants of brain structure and function: what is the significance of the mushroom bodies for behavior? // Development and Neurobiol. Drosophila. Boston, MA: Springer, 1980. P. 373–390.
Kretzschmar D., Hasan G., Sharma S. et al. The swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila // J. Neuroscience. 1997. V. 17. № 19. P. 7425–7432. https://doi.org/10.1523/JNEUROSCI.17-19-07425.1997
Ayyub C., Paranjape J., Rodrigues V., Siddiqi O. Genetics of olfactory behavior in Drosophila melanogaster // J. Neurogenetics. 1990. V. 6. № 4. P. 243–262. https://doi.org/10.3109/01677069009107114
Hasan G. Molecular cloning of an olfactory gene from Drosophila melanogaster // Proc. Natl Acad. Sci. USA. 1990. V. 87. № 22. P. 9037–9041. https://doi.org/10.1073/pnas.87.22.9037
Moser M., Stempfl T., Li Y. et al. Cloning and expression of the murine sws/NTE gene // Mechanisms of Development. 2000. V. 90. № 2. P. 279–282. https://doi.org/10.1016/s0925-4773(99)00239-7
Topaloglu A.K., Lomniczi A., Kretzschmar D. et al. Loss-of-function mutations in PNPLA6 encoding neuropathy target esterase underlie pubertal failure and neurological deficits in Gordon Holmes syndrome // The J. Clin. Endocrinology & Metabolism. 2014. V. 99. № 10. P. E2067–Е2075. https://doi.org/10.1210/jc.2014-1836
Mühlig-Versen M., Da Cruz A.B., Tschäpe J.A. et al. Loss of Swiss cheese/neuropathy target esterase activity causes disruption of phosphatidylcholine homeostasis and neuronal and glial death in adult Drosophila // J. Neuroscience. 2005. V. 25. № 11. P. 2865–2873. https://doi.org/10.1523/JNEUROSCI.5097-04.2005
Da Cruz A.B., Wentzell J., Kretzschmar D. Swiss Cheese, a protein involved in progressive neurodegeneration, acts as a noncanonical regulatory subunit for PKA-C3 // J. Neuroscience. 2008. V. 28. № 43. P. 10885–10892. https://doi.org/10.1523/JNEUROSCI.3015-08.2008
Kmoch S., Majewski J., Ramamurthy V. et al. Mutations in PNPLA6 are linked to photoreceptor degeneration and various forms of childhood blindness // Nat. Communications. 2015. V. 6. № 1. P. 1–10. https://doi.org/10.1038/ncomms6614
https://www.uniprot.org/uniprot/Q9U969
Sujkowski A., Rainier S., Fink J.K., Wessells R.J. Delayed induction of human NTE (PNPLA6) rescues neurodegeneration and mobility defects of Drosophila swiss cheese (sws) mutants // PLoS One. 2015. V. 10. № 12. P. e0145356. https://doi.org/10.1371/journal.pone.0145356
Hufnagel R.B., Arno G., Hein N.D. et al. Neuropathy target esterase impairments cause Oliver–McFarlane and Laurence–Moon syndromes // J. Med. Genet. 2015. V. 52. № 2. P. 85–94. https://doi.org/10.1136/jmedgenet-2014-102856
Greenfield J.G., Aring C.D. The Spino-Cerebellar Degenerations // Ed. Charles D. Aring. Blackwell Sci. Publ., 1954. 112 p.
Johnson M.K. The delayed neurotoxic effect of some organophosphorus compounds. Identification of the phosphorylation site as an esterase // Biochemical J. 1969. V. 114. № 4. P. 711–717. https://doi.org/10.1042/bj1140711
Behan W.M.H., Maia M. Strümpell’s familial spastic paraplegia: genetics and neuropathology // J. Neurology, Neurosurgery & Psychiatry. 1974. V. 37. № 1. P. 8–20. https://doi.org/10.1136/jnnp.37.1.8
Richardson R.J., Fink J.K., Worden R.M. et al. Neuropathy target esterase as a biomarker and biosensor of delayed neuropathic agents // Handbook of Toxicology of Chem. Warfare Agents. Acad. Press. 2020. P. 1005–1025.
Richardson R.J., Fink J.K., Glynn P. et al. Neuropathy target esterase (NTE/PNPLA6) and organophosphorus compound-induced delayed neurotoxicity (OPIDN) // Adv. Neurotoxicology. 2020. V. 4. P. 1. https://doi.org/10.1016/bs.ant.2020.01.001
Quistad G.B., Barlow C., Winrow C.J. et al. Evidence that mouse brain neuropathy target esterase is a lysophospholipase // Proc. Natl Acad. Sci. USA. 2003. V. 100. № 13. P. 7983–7987. https://doi.org/10.1073/pnas.1232473100
van Tienhoven M., Atkins J., Li Y., Glynn P. Human neuropathy target esterase catalyzes hydrolysis of membrane lipids // J. Biol. Chemistry. 2002. V. 277. № 23. P. 20942–20948. https://doi.org/10.1074/jbc.M200330200
Wentzell J.S., Cassar M., Kretzschmar D. Organophosphate-induced changes in the PKA regulatory function of Swiss Cheese/NTE lead to behavioral deficits and neurodegeneration // PLoS One. 2014. V. 9. № 2. P. e87526. https://doi.org/10.1371/journal.pone.0087526
Cassar M., Sunderhaus E., Wentzell J.S. et al. The PKA-C3 catalytic subunit is required in two pairs of interneurons for successful mating of Drosophila // Sci. Reports. 2018. V. 8. № 1. P. 1–8. https://doi.org/10.1038/s41598-018-20697-3
Li Y., Dinsdale D., Glynn P. Protein domains, catalytic activity, and subcellular distribution of neuropathy target esterase in mammalian cells // J. Biol. Chemistry. 2003. V. 278. № 10. P. 8820–8825. https://doi.org/10.1074/jbc.M210743200
Zaccheo O., Dinsdale D., Meacock P.A., Glynn P. Neuropathy target esterase and its yeast homologue degrade phosphatidylcholine to glycerophosphocholine in living cells // J. Biol. Chemistry. 2004. V. 279. № 23. P. 24024–24033. https://doi.org/10.1074/jbc.M400830200
Chang P., He L., Wang Y. et al. Characterization of the interaction of neuropathy target esterase with the endoplasmic reticulum and lipid droplets // Biomolecules. 2019. V. 9. № 12. P. 848. https://doi.org/10.3390/biom9120848
Forshaw P.J., Atkins J., Ray D.E., Glynn P. The catalytic domain of human neuropathy target esterase mediates an organophosphate-sensitive ionic conductance across liposome membranes // J. Neurochemistry. 2001. V. 79. № 2. P. 400–406. https://doi.org/10.1046/j.1471-4159.2001.00562.x
Sunderhaus E.R., Law A.D., Kretzschmar D. ER responses play a key role in Swiss-Cheese/Neuropathy Target Esterase-associated neurodegeneration // Neurobiol. Disease. 2019. V. 130. P. 104520. https://doi.org/10.1016/j.nbd.2019.104520
Mohylyak I.I., Matiytsiv N.P., Hrunyk N.I., Chernyk Y.I. Sensitivity of neurodegenerative mutants of Drosophila melanogaster from Swiss cheese group to the oxidative stress conditions // Biopolymers & Cell. 2011. V. 27. № 6.
Ryabova E., Matiytsiv N., Trush O. et al. Swiss cheese, Drosophila ortholog of hereditary spastic paraplegia gene NTE, maintains neuromuscular junction development and microtubule Network // Drosophila melanogaster – Model for Recent Advances in Genetics and Therapeutics. In Tech. 2018. P. 209–225. https://doi.org/10.5772/intechopen.73077
Ayyub C., Paranjape J. Genetic interactions provide evidence for the role of integrins in specifying normal olfactory behavior in Drosophila melanogaster // J. Neurogenetics. 2002. V. 16. № 3. P. 165–174. https://doi.org/10.1080/01677060215308
Krishnan N., Rakshit K., Chow E.S. et al. Loss of circadian clock accelerates aging in neurodegeneration-prone mutants // Neurobiol. Disease. 2012. V. 45. № 3. P. 1129–1135. https://doi.org/10.1016/j.nbd.2011.12.034
Chen R., Chang P.A., Long D.X. et al. Down-regulation of neuropathy target esterase by protein kinase C activation with PMA stimulation // Mol. Cell. Biochemistry. 2007. V. 302. № 1–2. P. 179–185. https://doi.org/10.1007/s11010-007-9439-0
http://flybase.org/reports/FBgn0003656
http://flyatlas.gla.ac.uk/FlyAtlas2
Dutta S., Rieche F., Eckl N. et al. Glial expression of Swiss cheese (SWS), the Drosophila orthologue of neuropathy target esterase (NTE), is required for neuronal ensheathment and function // Disease Models & Mechanisms. 2016. V. 9. № 3. P. 283–294. https://doi.org/10.1242/dmm.022236
Tinette S., Zhang L., Robichon A. Cooperation between Drosophila flies in searching behavior // Genes, Brain and Behavior. 2004. V. 3. № 1. P. 39–50. https://doi.org/10.1046/j.1601-183x.2003.0046.x
Winrow C.J., Hemming M.L., Allen D.M. et al. Loss of neuropathy target esterase in mice links organophosphate exposure to hyperactivity // Nat. Genet. 2003. V. 33. № 4. P. 477–485. https://doi.org/10.1038/ng1131
Moser M., Li Y., Vaupel K. et al. Placental failure and impaired vasculogenesis result in embryonic lethality for neuropathy target esterase-deficient mice // Mol. Cell. Biology. 2004. V. 24. № 4. P. 1667–1679. https://doi.org/10.1128/mcb.24.4.1667-1679.2004
Read D.J., Li Y., Chao M.V. et al. Neuropathy target esterase is required for adult vertebrate axon maintenance // J. Neuroscience. 2009. V. 29. № 37. P. 11594–11600. https://doi.org/10.1523/JNEUROSCI.3007-09.2009
Akassoglou K., Malester B., Xu J. et al. Brain-specific deletion of neuropathy target esterase/swiss cheese results in neurodegeneration // Proc. Natl Acad. Sci. USA. 2004. V. 101. № 14. P. 5075–5080. https://doi.org/10.1073/pnas.0401030101
McFerrin J., Patton B.L., Sunderhaus E.R., Kretzschmar D. NTE/PNPLA6 is expressed in mature Schwann cells and is required for glial ensheathment of Remak fibers // Glia. 2017. V. 65. № 5. P. 804–816. https://doi.org/10.1002/glia.23127
https://www.ncbi.nlm.nih.gov/gene/10908.
Bertoncin D., Russolo A., Caroldi S., Lotti M. Neuropathy target esterase in human lymphocytes // Archiv. Environmental Health: An Intern. J. 1985. V. 40. № 3. P. 139–144. https://doi.org/10.1080/00039896.1985.10545905
Maroni M., Bleecker M.L. Neuropathy target esterase in human lymphocytes and platelets // J. Applied Toxicol. 1986. V. 6. № 1. P. 1–7. https://doi.org/10.1002/jat.2550060102
Williams D.G. Intramolecular group transfer is a characteristic of neurotoxic esterase and is independent of the tissue source of the enzyme. A comparison of the aging behaviour of di-isopropyl phosphorofluoridate-labelled proteins in brain, spinal cord, liver, kidney and spleen from hen and in human placenta // Biochemical J. 1983. V. 209. № 3. P. 817–829.
Hein N.D., Rainier S.R., Richardson R.J., Fink J.K. Motor neuron disease due to neuropathy target esterase mutation: Enzyme analysis of fibroblasts from human subjects yields insights into pathogenesis // Toxicol. Letters. 2010. V. 199. № 1. P. 1–5. https://doi.org/10.1016/j.toxlet.2010.06.020
Melentev P.A., Agranovich O.E., Sarantseva S.V. Human diseases associated with NTE gene // Ecol. Genet. 2020. V. 18. № 2. P. 229–242. https://doi.org/10.17816/ecogen16327
Sunderhaus E.R., Law A.D., Kretzschmar D. Disease-associated PNPLA6 mutations maintain partial functions when analyzed in Drosophila // Frontiers in Neurosci. 2019. V. 13. P. 1207. https://doi.org/10.3389/fnins.2019.01207
Johnson M.K. Organophosphorus and other inhibitors of brain ‘neurotoxic esterase’ and the development of delayed neurotoxicity in hens // Biochemical J. 1970. V. 120. № 3. P. 523–531. https://doi.org/10.1042/bj1200523
Johnson M.K. Organophosphorus esters causing delayed neurotoxic effects: mechanism of action and structure activity studies // Archiv. Toxicology. 1975. V. 34. № 4. P. 259–288. https://doi.org/10.1007/BF00353848
Lotti M., Johnson M.K. Neurotoxicity of organophosphorus pesticides: Predictions can be based on in vitro studies with hen and human enzymes // Archiv. Toxicology. 1978. V. 41. № 3. P. 215–221. https://doi.org/10.1007/BF00354093
Hierons R., Johnson M.K. Clinical and toxicological investigations of a case of delayed neuropathy in man after acute poisoning by an organophosphorus pesticide // Archiv. Toxicology. 1978. V. 40. № 4. P. 279–284. https://doi.org/10.1007/BF00310333
Lush M.J., Li Y., Read D.J. et al. Neuropathy target esterase and a homologous Drosophila neurodegeneration-associated mutant protein contain a novel domain conserved from bacteria to man // Biochemical J. 1998. V. 332. № 1. P. 1–4. https://doi.org/10.1042/bj3320001
Rainier S., Bui M., Mark E. et al. Neuropathy target esterase gene mutations cause motor neuron disease // The Am. J. Hum. Genet. 2008. V. 82. № 3. P. 780–785. https://doi.org/10.1016/j.ajhg.2007.12.018
Дополнительные материалы отсутствуют.