

Генетика, 2021, T. 57, № 10, стр. 1121-1130
Гемофилия в Лейден: литературные и собственные данные
Т. С. Бескоровайная 1, *, В. В. Забненкова 1, Р. А. Зинченко 1, О. А. Щагина 1, А. В. Поляков 1
1 Медико-генетический научный центр им. академика Н.П. Бочкова
115522 Москва, Россия
* E-mail: t-kovalevskaya@yandex.ru
Поступила в редакцию 30.11.2020
После доработки 29.01.2021
Принята к публикации 11.02.2021
Аннотация
Гемофилия В – это моногенное Х-сцепленное рецессивное заболевание, связанное с нарушением свертываемости крови, вызванное мутациями в гене фактора свертывания IX (F9). Описана необычная форма данного заболевания “гемофилия В Лейден”, обусловленная мутациями в промоторной области гена F9, при которой происходит значительное, (до нормальных значений), увеличение фактора IX в плазме после полового созревания больного. Мутации в промоторе группируются в дискретных участках связывания транскрипционных факторов и по-разному изменяют экспрессию гена F9 в процессе развития. Данное заболевание иллюстрирует, как однонуклеотидные патогенные варианты в цис-регуляторных регионах могут оказывать радикальное воздействие на экспрессию генов. В статье представлены современные данные о структуре промотора гена F9 и результаты обследования трех пациентов с гемофилией В Лейден, проходивших генетическое тестирование в лаборатории ДНК-диагностики ФГБНУ “МГНЦ им. академика Н.П. Бочкова”.
Гемофилия В (MIM 306900) – это Х-сцепленное рецессивное заболевание, при котором происходит нарушение свертываемости крови. Его частота составляет 1 : 30 000 мужчин [1]. Различают легкую степень гемофилии В – уровень фактора IX в плазме составляет 5–40%, среднюю степень тяжести – 1–5% и тяжелую – менее 1% (https:// www.ehc.eu/ru/). Заболевание обусловлено мутациями гена F9, которые влияют на экспрессию или функцию фактора свертывания крови IX (фактор Кристмаса). Фактор IX – это витамин К‑зависимая сериновая протеаза, которая совместно с кофактором – фактором VIII участвует во внутреннем пути коагуляции, активируя фактор X. Ген F9 (MIM 300746) локализован на длинном плече хромосомы Х в локусе Xq27. Белок синтезируется в печени [2], после чего подвергается множественным посттрансляционным модификациям [3, 4]. Известно, что в норме у плода до 20-й нед. гестации количество мРНК и белка фактора IX составляет <10% от уровня взрослого человека [5]. В постнатальный период уровень FIX составляет 50%, а в раннем детстве – 75% от показателей у взрослых [6]. Следующее значительное повышение содержания фактора IX в плазме наблюдается у человека после полового созревания, когда у обоих полов зафиксировано увеличение экспрессии на 25% [6, 7].
В настоящее время нумерация нуклеотидов гена F9, согласно рекомендациям HGVS (https:// varnomen.hgvs.org/), начинается со стартового кодона трансляции, однако ранее нумерация велась от основного стартового сайта транскрипции (major transcription start site, TSS), который находится на 29 нуклеотидов ранее стартового кодона ATG. Соответственно данные 29 нуклеотидов теперь включены в промоторную некодирующую область, что необходимо учитывать при изучении ранней литературы, посвященной гену F9.
В 1970 г. в городе Лейден (Нидерланды) была описана необычная форма гемофилии В [8]. Этот подтип заболевания, получивший название “гемофилия В Лейден”, имеет некоторую особенность клинического течения. У больных мальчиков в возрасте до 15 лет отмечается значительное снижение уровня фактора IX:C в плазме (≤1–10%), а после пубертатного периода уровень данного фактора восстанавливается со скоростью около 5% в год практически до нормальных значений (максимально до 40–80%) [9–11]. Клиническое течение заболевания в детстве может быть как мягким, умеренным, так и тяжелым, приводящим к частым кровотечениям, гемартрозам, и может требовать заместительной терапии. На сегодняшний день зарегистрировано более 100 случаев гемофилии В Лейден [12].
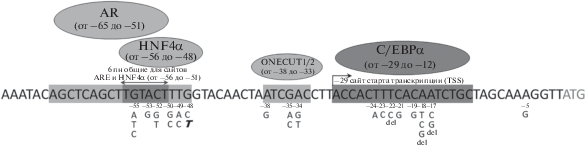
При секвенировании кодирующей последовательности гена F9, сайтов сплайсинга и регуляторных областей у больных из семей с Лейден-подобными симптомами гемофилии В были выявлены точковые мутации в области промотора данного гена [9, 13–21]. В базах “The Human Gene Mutation Database”, “Factor IX Gene Variant Database” и “CHBMP” всего зарегистрировано 30 патогенных вариантов промотора гена F9. Данные варианты сгруппированы в три кластера: один в области с.–55–48 пн, второй – с.–38–34 пн и третий – с.–24–17 пн выше стартового кодона трансляции (рис. 1). Эти участки были признаны потенциальными регуляторными элементами. Известно, что помимо основных факторов транскрипции, которые непосредственно связывают РНК-полимеразу с промотором рядом со стартовой точкой транскрипции, существуют транскрипционные факторы, которые называются активаторами. Они узнают определенные короткие консенсусные нуклеотидные последовательности промотора (элементы ответа) или энхансера и повышают эффективность связывания основного аппарата транскрипции с промотором, т.е. определяют частоту событий инициации транскрипции, поддерживая ее на адекватном уровне. Было выдвинуто предположение, что каждый кластер мутаций разрушает сайт связывания белка-активатора транскрипции и тем самым нарушает экспрессию гена F9, по крайней мере до наступления половой зрелости. Таким образом, результаты генетического тестирования пациентов с гемофилией В Лейден послужили важным материалом для изучения транскрипции в печени человека. Влияние выявленных у данных пациентов нуклеотидных замен в промоторе гена F9 на его экспрессию изучалось с помощью функционального анализа in vitro на клеточных линиях HeLa и HepG2 и in vivo на трансгенных мышах [10, 22–25].
Рис. 1.
Патогенные варианты в промоторе гена F9, описанные в базах “The Human Gene Mutation Database”, “Factor IX Gene Variant Database”, “CHBMP”. Курсивом выделена неописанная нуклеотидная замена, выявленная в данном исследовании.
Мутации в промоторе генов интересны тем, что демонстрируют, как изменения последовательности нуклеотидов в данной области могут приводить к глубоким нарушениям экспрессии генов в разные периоды развития организма. В частности, в случае гена F9 экспрессия начинается только после полового созревания (рис. 2).
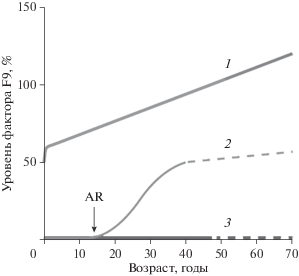
Рис. 2.
Экспрессия гена F9 в норме, у больных гемофилией В Лейден и гемофилией В Бранденбург [26]. У здоровых людей (линия 1) уровень экспрессии гена F9 постепенно растет в течение жизни. У больных гемофилией В Лейден (линия 2) этот рост начинается только после полового созревания согласно одной из гипотез в связи с повышением активности экспрессии гена при связывании с сайтом тестостерон-зависимого фактора андрогеновых рецепторов (AR). У больных гемофилией В Бранденбург (линия 3) такого роста не наблюдается и после полового созревания, так как данный сайт связывания поврежден. Пунктирные линии представляют собой экстраполяции из-за отсутствия доступных клинических данных.
Поиск транскрипционных факторов, которые связываются с тремя функциональными цис-регуляторными элементами в промоторе гена F9, начался в конце 1980-х гг., когда были идентифицированы первые транскрипционные факторы млекопитающих. Ген F9 экспрессируется преимущественно в гепатоцитах. Печень – большой и относительно однородный орган, поэтому она была подходящей моделью для ранних исследований по выделению и очистке транскрипционных факторов. Несколько исследовательских групп успешно идентифицировали и клонировали кДНК специфичных для печени ДНК-связывающих белков [27, 28]. Были определены консенсусные последовательности для этих факторов транскрипции и соответственно были идентифицированы некоторые кандидатные ДНК-связывающие белки, регулирующие экспрессию фактора IX.
Первым открытым белком-активатором гена F9 был CCAAT/энхансер-связывающий белок α (C/EBPα), относящийся к транскрипционным факторам из группы “лейциновых молний”. Данный фактор связывается с сайтом TTGNNCAA, последовательность которого похожа на –24–17 регион промотора гена F9 (рис. 1). После связывания с ДНК данный белок может индуцировать транскрипцию через свой активационный домен, взаимодействуя с компонентами основного аппарата транскрипции. Более поздние исследования на мышах, нокаутированных по C/EBPα, подтвердили, что данный фактор функционально необходим для нормального уровня экспрессии гена [29].
Далее был открыт ядерный фактор гепатоцитов 4α (HNF4α). Ген HNF4A клонирован в начале 1990-х гг. [28]. Было показано, что очищенный белок связывается с областью с.–56–48 промотора гена F9 [22, 30], а известные мутации из данного региона нарушают это связывание и ингибируют трансактивацию посредством HNF4α. В настоящее время данный ядерный рецептор широко известен как главный регулятор дифференцировки печени, так как влияет на экспрессию большого количества специфичных для гепатоцитов генов.
Примерно в то же время исследователи начали изучать причину увеличения экспрессии гена F9 у больных с гемофилией В Лейден после полового созревания. Систематизированный анализ пациентов с гемофилией В позволил выявить еще один подтип заболевания, получивший название “гемофилия В Бранденбург” [30]. Пациенты имели очень низкий уровень фактора IX, и геморрагические симптомы наблюдались на протяжении всей жизни. У данных пациентов были выявлены мутации в положении с.–55 (рис. 1) [12, 30–34], при которых, в отличие от других мутаций в промоторе F9, не происходит восстановления экспрессии гена в период полового созревания (рис. 2). Было отмечено, что данный регион промотора напоминает элемент андрогенного ответа (androgen response element, ARЕ), т.е. сайт связывания для транскрипционного фактора андрогенового рецептора (AR). Белок AR является транскрипционным фактором из семейства рецепторов стероидных гормонов. Он взаимодействует с последовательностями ДНК, соответствующими консенсусу ARЕ, и активирует транскрипцию в присутствии тестостерона. Было показано, что AR может связываться с областью вокруг позиции с.–55 и что мутации при гемофилии B Бранденбург нарушают данное связывание [30]. При этом пациенты с патогенными вариантами промотора гена F9 в позициях с.–48–50, с.–38–34 и с.–24–17 действительно имели увеличение экспрессии фактора после полового созревания, так как ARЕ оставался интактным. Следует отметить, что сайты связывания факторов HNF4α и AR частично перекрываются (на протяжении шести нуклеотидов) (рис. 1). Таким образом, при гемофилии В Бранденбург повреждаются оба сайта связывания – для HNF4α и для AR, обусловливая такую форму гемофилии В, при которой не наблюдается улучшения клинической картины при воздействии тестостерона [33]. Отсутствие в базах данных описанных мутаций в позициях с.–57–65, вероятно, свидетельствует о несущественном влиянии на уровень экспрессии гена F9 нуклеотидных замен в сайте связывания только андрогенового рецептора.
Гипотеза о влиянии половых гормонов на экспрессию гена F9 была проверена на мышах [24]. Было выявлено, что у самцов линии Tfm, имеющих укороченный белок AR, не способный связываться с тестостероном, не происходит увеличения экспрессии фактора IX после полового созревания. В дальнейшем у человека важность сайта ARE в промоторе гена F9 также подтвердилась. Было выявлено, что введение тестостерона подростку в пре-пубертатном возрасте с гемофилией В Лейден привело к увеличению уровня FIX:C [35]. Кроме того, в литературе описан случай, когда мужчина с гемофилией B Лейден использовал анаболические стероиды для увеличения мышечной массы, что побочно вызвало еще и повышение уровня FIX:C в плазме [36]. Однако после прекращения использования стероидов количество фактора IX у него снизилось. Данные наблюдения дают весомые аргументы в пользу причастности андрогенов к регуляции экспрессии гена F9.
В позициях c.–53 и с.–52, в которых также перекрываются сайты связывания AR и HNF4α, зарегистрированы еще три миссенс-мутации (рис. 1). К сожалению, у данных пациентов не было проведено лонгитюдного наблюдения, поэтому невозможно сделать вывод о влиянии данных нуклеотидных вариантов на клиническое течение заболевания после полового созревания больного [12, 37]. Вместе с тем описан пациент с делецией c.–52del, которому диагноз гемофилия В был поставлен в возрасте трех лет, и уровень фактора FIX:C в его плазме был <1% [38]. После пубертатного периода качество жизни больного значительно улучшилось, FIX:C варьировал от 4 до 11%. Как ни странно, у его племянника с такой же мутацией, также страдающего гемофилией В (FIX:C < 1%), повышение уровня фактора IX не произошло [39]. Исходя их данного семейного случая, можно предположить, что дополнительные факторы – генетические, эпигенетические, факторы внешней среды – также могут влиять на симптомы и клиническую тяжесть гемофилии B Лейден.
Несколько позже был предложен другой механизм регуляции экспрессии фактора IX, обусловленный возрастом. Он был убедительно продемонстрирован в экспериментах Kurachi et al. на трансгенных мышах [25]. Согласно данной гипотезе, в регуляции задействован элемент возрастной стабильности (ASE, age-related stability element), расположенный в 5'-некодирующей области на расстоянии 831 нуклеотида (802 нуклеотидов согласно исторической номенклатуре) от промотора гена F9. В экспериментах было показано специфическое связывание ядерного белка ETS1 с данной областью. ETS1 является транскрипционным фактором, ранее известным своими протоонкогенными свойствами [40]. В исследованиях Kurachi et al. [25] была показана новая роль ETS1 в физиологической регуляции возрастной экспрессии генов. Связывание данного транскрипционного фактора с возрастным элементом стабильности обусловлено действием гормона роста (соматропином). Данное явление было продемонстрировано на мышах, которым хирургическим путем был резецирован гипофиз. Это позволило проанализировать прямое действие экзогенно вводимых гормонов и исключило возможность нисходящих эффектов, опосредованных другими гормональными путями. Было показано, что при введении дигидротестостерона или эстрадиола уровень FIX восстанавливался только на 10% от показателей до гипофизэктомии, однако при введении гормона роста этот уровень восстанавливался на 80%. Причем содержание фактора в плазме возрастало как у самцов, так и у самок. Таким образом, была установлена важная роль гормона роста в регуляции возраст-зависимой экспрессии гена F9 независимо от пола. Результаты данных экспериментов на модельных животных в дальнейшем нашли свое подтверждение у женщин – носительниц мутаций гемофилии В Лейден. Например, была описана семья, в которой у двух девочек с неравной инактивацией хромосомы Х, имеющих мутацию c.–22T>C в гетерозиготном состоянии, в возрасте 6 и 11 лет уровень фактора IX составлял 11 и 17% соответственно [41]. При этом у первой пациентки этот уровень увеличился к 16 годам до 21%, а у второй пациентки к 14 годам – до 35%. Нормализация уровня FIX:C наблюдалась и у двух носительниц мутации c.–35G>A из Ирландии [42]. Также было выявлено, что при серийном измерении количества фактора IX в группе детей с мутацией c.–35G>A в течение первых трех лет жизни наблюдается постепенное увеличение его содержания в плазме от значений, соответствующих тяжелой/среднетяжелой форме, до значений при мягкой форме гемофилии В [43]. Причем данное увеличение в столь раннем возрасте не может быть обусловлено андрогенами, а, вероятно, связано с постепенным повышением содержания гормона роста от низких значений в раннем детстве до максимальных пиковых значений в период полового созревания [44]. Несмотря на столь убедительные свидетельства в пользу данной гипотезы, на сегодняшний день ни одной мутации из области c.–831 не описано. Кроме того, гипотеза не объясняет отсутствия улучшения состояния у больных с гемофилией В Бранденбург.
Белок-активатор для третьего локуса, в котором располагается кластер мутаций (с.–38–34), искали в течение 20 лет. Мотивация для поиска этого белка была особенно сильной, потому что на долю этого кластера приходится около половины всех случаев гемофилии В Лейден [12, 31]. Кроме того, разные нуклеотидные замены на этом участке приводят к различной степени тяжести течения заболевания [10, 12, 14, 16]. Понимание данного механизма регуляции представляло большой интерес. Указанная область содержит динуклеотид CpG, который, как известно, является горячей мутационной точкой как в гене F9, так и в других локусах [45]. Загадка данного регуляторного участка промотора была решена одновременно двумя исследовательскими группами, которые совместно опубликовали свои результаты [46]. Данные метода ChIP-Seq для транскрипционного фактора ONECUT1 (также известного как ядерный фактор гепатоцитов 6, HNF6) позволили предположить его в качестве кандидатного белка для связывания с элементом с.–38–34 в промоторе F9 [47]. Исследования показали, что ONECUT1 специфически связывается с областью с.–38–34 промотора F9 в гепатоцитах человека [46] (рис. 1). Он также занимает проксимальный промотор гена F9 в гепатоцитах мышей [48], а нокаутированные по ONECUT1 эмбрионы мышей имеют пониженный уровень FIX [46]. Родственный фактор ONECUT2 тоже связывается с сайтом с.–34–38 in vitro, а исследования на эмбрионах мышей с двойным нокаутом Onecut1/Onecut2 подтвердили, что при отсутствии обоих белков ген F9 не транскрибируется [46].
В базе HGMD описан еще один патогенный вариант в позиции c.–5A>G у пациента с мягкой формой гемофилии В Лейден. Данный вариант не попадает ни в один из известных кластеров для транскрипционных факторов [49]. Кроме того, в базе Genome Aggregation Database (gnomAD, https:// gnomad.broadinstitute.org) у одной женщины в промоторе гена F9 в гетерозиготном состоянии встретилась нуклеотидная замена c.–4G>T (частота аллеля 0.0005%), у другой женщины – c.–3G>T (частота аллеля 0.0005%). Вероятно, они также являются патогенными и могут нарушать транскрипцию или процессинг РНК, тем не менее данных, подтверждающих их патогенность, на настоящий момент нет.
Существует предположение, что в природе могут встречаться нуклеотидные варианты в промоторной области гена F9, которые не имеют клинических проявлений [46]. Однако в выборке gnomAD, в которую включены результаты секвенирования 125 748 экзомов и 15 708 геномов людей без тяжелых патологий, в 5'-UTR области изучаемого гена не представлено ни одного частого полиморфизма, кроме упомянутых редких вариантов c.–3G>T и c.–4G>T. В базе dbSNP в промоторе F9 зарегистрированы еще несколько вариантов с крайне низкой частотой альтернативного аллеля: c.–7A>T (rs1569481540), c.–13G>C (rs372382059), c.–15C>G (rs375594466), c.–19C>T (rs761036720), c.–39A>C (rs371045754), c.–41C>A (rs750827465), c.–44C>G (913707665), но для них не установлено клинического значения и не представлены данные о клиническом статусе людей, у которых они были выявлены. В выборке, включающей данные секвенирования 1223 экзомов пациентов, не страдающих коагулопатиями, обследованных в МГНЦ им. академика Н.П. Бочкова, в промоторе гена F9 ни одной нуклеотидной замены не обнаружено. Из этого можно предположить, что изучаемая область гена F9 все же более консервативна, чем предполагалось. Это также подтверждается филогенетической консервативностью данной последовательности у разных видов млекопитающих (рис. 3).
Рис. 3.
Промоторная область гена F9 у млекопитающих (https://genome.ucsc.edu). Пики отображают консервативность данных последовательностей, которые являются кандидатными сайтами связывания транскрипционных факторов.

В некоторых исследованиях было предсказано наличие еще одного сайта для связывания транскрипционных факторов в локусе с.–231–248, с которым в частности взаимодействует альбумин D-сайт-связывающий белок (DBP) и GA-связывающий белок (GABPα/β), которые также могут влиять на регуляцию экспрессии фактора IX после полового созревания [50, 51], хотя на сегодняшний день ни одной мутации из этой области не описано.
МАТЕРИАЛЫ И МЕТОДЫ
В данной работе проведено генетическое тестирование трех пациентов, имевших/имеющих в детстве диагноз “гемофилия В, тяжелая форма”. От пациентов или их законных представителей было получено информированное согласие на проведение исследования.
Выделение ДНК из цельной крови, забранной в пробирку с антикоагулянтом ЭДТА, проводили с помощью набора реактивов Wizard® Genomic DNA Purification Kit (Promega, США) согласно протоколу производителя.
Секвенирование по Сенгеру ПЦР-продуктов, полученных с праймеров, комплементарных последовательности гена F9, фланкирующих экзоны 1–8 и промоторную область, проводили на приборе для капиллярного электрофореза 3130 ABI genetic analyzer (Applied Biosystems, США). Частоту выявленных вариантов гена F9 определяли по базам данных dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/) и Genome Aggregation Database (http:// gnomad.broadinstitute.org/), патогенность – по их описанию в базах “The Human Gene Mutation Database” (https://my.qiagendigitalinsights.com/bbp/) и “Factor IX Gene Variant Database” (http:// www.factorix.org/), “CHBMP” (https://www.cdc.gov/ ncbddd/hemophilia/champs.html).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
При обследовании пациентов с гемофилией В, проходивших генетическое тестирование гена F9 в ФГБНУ “Медико-генетический научный центр им. академика Н.П. Бочкова”, у троих были обнаружены мутации в промоторной области гена.
Пациент ГВ_382.1 2002 г. рождения. На момент обследования пациенту 14 лет. Наблюдается у гематолога с 5 лет c диагнозом “гемофилия В” (болезнь Кристмаса). При осмотре: нормальное телосложение, умственное и физическое развитие соответствует возрасту, выявлены коленные гемартрозы. Из семейного анамнеза известно, что в детстве у дедушки по материнской линии наблюдались частые кровотечения, однако обследования он не проходил. К сожалению, более подробный анамнез собрать не удалось из-за проживания больного в труднодоступной местности. При проведении генетического тестирования в гене F9 выявлена описанная ранее Reitsma et al. мутация c.–17А>G в гемизиготном состоянии [9]. Родословная больного из семьи 382 схожа с родословной американской семьи. У представленного в статье [9] пробанда в детстве уровень фактора IX в плазме был <1%, наблюдались частые кровотечения, но после 14 лет кровотечения стали более редкими, уровень FIX:C постепенно увеличивался и к 18 годам достиг 24%, оставшись в дальнейшем на постоянном уровне. Известно также о дяде больного по материнской линии с такими же клиническими проявлениями, которому впервые обследование было проведено в 39 лет и FIX:C у него составил 42%. У матери больного, которая являлась носительницей данной мутации, FIX:C был 79%. Интересно, что у мужчин из одной семьи, описанной в статье [9], после полового созревания уровень фактора IX вырос в разной степени – у пробанда остался диагноз “мягкая форма гемофилии В”, а у дяди пробанда уровень фактора достиг нормальных значений. На сегодняшний день известно, что данная мутация находится в сайте связывания транскрипционного фактора C/EBPα. Исходя из этого, а также из данных о семейном анамнезе больного 382.1, можно предположить, что в ближайшее время состояние данного пациента улучшится.
Пациент ГВ_266.1 1985 г. рождения. На момент обследования пациенту 29 лет. Обследование проводилось с целью дальнейшего уточнения статуса сестры пробанда на предмет носительства гемофилии В. Из анамнеза известно, что пациент с 5 лет наблюдался в НМИЦ гематологии г. Москвы с клиническим диагнозом “тяжелая коагулопатия, гемофилия В”. У больного в детстве наблюдались кровотечения из слизистых, геморрагии на коже, гематомы мягких тканей, гемартрозы крупных суставов. Больной получал гемостатическую терапию концентратом фактора IX, несколько раз проходил лечение в стационаре. В 16 лет пациенту поменяли диагноз на “гемофилия В, легкая форма”, FIX:C в плазме составлял 13%, в 18 лет – 26%. Далее больной перестал наблюдаться у гематолога в связи с исчезновением клинических проявлений заболевания. При проведении секвенирования гена F9 у данного пациента был обнаружен ранее не описанный вариант с.–48G>T. В 1995 г. Ketterling в данной позиции описал вариант с.–48G>С у 12‑летнего мальчика с гемофилией В средней тяжести (FIX:C 3%) [37]. Также известны патогенные варианты в соседних нуклеотидах (c.–49T>A, c.–49T>C, c.–50T>G, c.–50T>C) у больных со схожим фенотипом [13, 21, 52, 53]. У всех больных произошло значительное смягчение клинических симптомов гемофилии В после полового созревания, как и у пробанда 266.1. Известно, что в позициях с.–56–48 находится сайт связывания для транскрипционного фактора HNF4α, при этом только в позициях с.–50–48 он не перекрывается с сайтом для андрогенового рецептора, что обеспечивает увеличение концентрации фактора IX после полового созревания. Вариант с.–48G>T также выявлен у сестры пробанда в гетерозиготном состоянии.
Пациент ГВ_401.1 2014 г. рождения. На момент обследования пациенту 4 года, наблюдается у гематолога с клиническим диагнозом “гемофилия В, тяжелая форма”. Из анамнеза известно, что в 2016 г. он проходил стационарное лечение по причине образовавшейся гематомы в области лба после травмы и повторявшихся кровотечений из раны после проведенного вскрытия гематомы. Также были отмечены множественные небольшие подкожные кровоизлияния по всему телу и умеренное увеличение селезенки, суставы были не изменены. Выявлены отклонения в коагулограмме: АЧТВ – 102.2 с, фактор IX – 0.9%. На сегодняшний день ребенок получает заместительную терапию плазматическим концентратом фактора IX. При генетическом тестировании у пациента был обнаружен ранее описанный патогенный вариант в промоторе гена F9 – c.–52C>T в гемизиготном состоянии. Мать пробанда является гетерозиготной носительницей данного патогенного варианта. Согласно описанным в литературе случаям при наличии у больных мутации в этой области, выздоровления после полового созревания не происходит в связи с тем, что данная нуклеотидная замена попадает на сайты связывания как фактора HNF4α, так и тестостерон-зависимого фактора AR. Однако дает надежду разное течение заболевания у двух больных из одной семьи с мутацией c.–52del, при которой у одного из больных наступило улучшение состояния [38].
Гемофилия В Лейден является примером генетического заболевания, обусловленного мутациями в промоторной области гена F9, при котором в большинстве случаев происходит выздоровление или облегчение состояния пациента после полового созревания. Степень ингибирования транскрипции и соответственно тяжесть течения гемофилии В зависят от мутации, в той или иной мере нарушающей связывание белка-активатора с промотором. При этом следует подчеркнуть, что на фенотип также могут влиять дополнительные факторы (генетические, эпигенетические и др.), поэтому тяжесть течения заболевания не может быть достоверно предсказана только по выявленной мутации.
Были предложены две гипотезы, объясняющие улучшение состояния больных в период полового созревания. Обе они нашли подтверждение в ряде проведенных экспериментов. Возможно, что оба механизма – регуляция экспрессии гена F9 половыми гормонами и гормоном роста – являются составными частями еще более сложного биологического процесса. Поэтому причины выздоровления при гемофилии В Лейден являются предметом дальнейших исследований.
В настоящее время известно около 1600 факторов транскрипции человека [54, 55]. Основополагающим для этого было клонирование практически всех ДНК-связывающих белков и появление ChIP-Seq метода. Современные методы позволяют определять функциональное значение для все большего количества SNPs и мутаций в регуляторных областях генов. Несомненно существуют многочисленные сложности, которые затрудняют этот процесс. Например, исследования ChIP-Seq показали, что транскрипционные факторы обычно связываются с тысячами сайтов по всему геному, и многие такие события связывания считаются функционально несущественными [56]. Кроме того, эухроматические цис-регуляторные области часто совместно заняты множеством транскрипционных факторов [56]. Таким образом, отличить функционально значимые белки от тех, которые связываются просто вследствие доступности хроматина, может оказаться затруднительным. Кроме того, стандартные анализы ChIP-Seq обычно определяют относительно широкую область, которая может содержать несколько предполагаемых сайтов связывания для данного фактора транскрипции. В таких случаях более точно сайты связывания могут быть идентифицированы только путем последующего функционального анализа или с помощью методов высокого разрешения, таких как ChIP-exo [57]. Наконец, хотя число установленных факторов транскрипции человека увеличивается, определить их функцию в каждой ткани и влияние на регуляцию генов in vivo остается сложной задачей. Тем не менее значительный технический прогресс позволяет надеяться, что постепенно будет достигнуто понимание всех процессов генной регуляции.
Работа выполнена в рамках Государственного задания Минобрнауки России.
Настоящая статья не содержит каких-либо исследований с использованием в качестве объекта животных.
От пациентов или их законных представителей было получено информированное согласие на проведение исследования и публикацию результатов на условии анонимности.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Mannucci P.M., Tuddenham E.G. The hemophilias–from royal genes to gene therapy // N. Engl. J. Med. 2001. V. 344. № 23. P. 1773–1779. https://doi.org/10.1056/nejm200106073442307
Yoshitake S., Schach B.G., Foster D.C. et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B) // Biochemistry. 1985. V. 24. № 14. P. 3736–3750. https://doi.org/10.1021/bi00335a049
Larson P.J., Stanfield-Oakley S.A., VanDusen W.J. et al. Structural integrity of the gamma-carboxyglutamic acid domain of human blood coagulation factor IXa is required for its binding to cofactor VIIIa // J. Biol. Chem. 1996. V. 271. № 7. P. 3869–3876. https://doi.org/10.1074/jbc.271.7.3869
Derian C.K., VanDusen W., Przysiecki C.T. et al. Inhibitors of 2-ketoglutarate-dependent dioxygenases block aspartyl beta-hydroxylation of recombinant human factor IX in several mammalian expression systems // J. Biol. Chem. 1989. V. 264. № 12. P. 6615–6618.
Hassan H.J., Leonardi A., Chelucci C. et al. Blood coagulation factors in human embryonic-fetal development: Preferential expression of the FVII/tissue factor pathway // Blood. 1990. V. 76. № 6. P. 1158–1164.
Andrew M., Vegh P., Johnston M. et al. Maturation of the hemostatic system during childhood // Blood. 1992. V. 80. № 8. P. 1998–2005.
Sweeney J.D., Hoernig L.A. Age-dependent effect on the level of factor IX // Am. J. Clin. Pathol. 1993. V. 99. № 6. P. 687–688. https://doi.org/10.1093/ajcp/99.6.687
Veltkamp J.J., Meilof J., Remmelts H.G. et al. Another genetic variant of haemophilia B: haemophilia B Leyden // Scand. J. Haematol. 1970. V. 7. № 2. P. 82–90. https://doi.org/10.1111/j.1600-0609.1970.tb01873.x
Reitsma P.H., Mandalaki T., Kasper C.K. et al. Two novel point mutations correlate with an altered developmental expression of blood coagulation factor IX (hemophilia B Leyden phenotype) // Blood. 1989. V. 73. № 3. P. 743–746.
Crossley M., Brownlee G.G. Disruption of a C/EBP binding site in the factor IX promoter is associated with haemophilia B // Nature. 1990. V. 345. № 6274. P. 444–446. https://doi.org/10.1038/345444a0
Briët E., Bertina R.M., van Tilburg N.H., Veltkamp J.J. Hemophilia B Leyden: A sex-linked hereditary disorder that improves after puberty // N. Engl. J. Med. 1982. V. 306. № 13. P. 788–790. https://doi.org/10.1056/nejm198204013061306
Rallapalli P.M., Kemball-Cook G., Tuddenham E.G. et al. An interactive mutation database for human coagulation factor IX provides novel insights into the phenotypes and genetics of hemophilia B // J. Thromb. Haemost. 2013. V. 11. № 7. P. 1329–1340. https://doi.org/10.1111/jth.12276
Reitsma P.H., Bertina R.M., Ploos van Amstel J.K. et al. The putative factor IX gene promoter in hemophilia B Leyden // Blood. 1988. V. 72. № 3. P. 1074–1076.
Hirosawa S., Fahner J.B., Salier J.P. et al. Structural and functional basis of the developmental regulation of human coagulation factor IX gene: factor IX Leyden // Proc. Natl Acad. Sci. USA. 1990. V. 87. № 12. P. 4421–4425. https://doi.org/10.1073/pnas.87.12.4421
Royle G., Van de Water N.S., Berry E. et al. Haemophilia B Leyden arising de novo by point mutation in the putative factor IX promoter region // Br. J. Haematol. 1991. V. 77. № 2. P. 191–194. https://doi.org/10.1111/j.1365-2141.1991.tb07976.x
Vidaud D., Tartary M., Costa J.M. et al. Nucleotide substitutions at the -6 position in the promoter region of the factor IX gene result in different severity of hemophilia B Leyden: consequences for genetic counseling // Hum. Genet. 1993. V. 91. № 3. P. 241–244. https://doi.org/10.1007/bf00218264
Hall A.J., Chuansumrit A., Peake I.R., Winship P.R. A single base pair deletion in the promoter region of the factor IX gene is associated with haemophilia B // Thromb. Haemost. 1994. V. 72. № 6. P. 799–803.
Nielsen L.R., Scheibel E., Ingerslev J., Schwartz M. Detection of ten new mutations by screening the gene encoding factor IX of Danish hemophilia B patients // Thromb Haemost. 1995. V. 73. № 5. P. 774–778.
Crossley P.M., Winship P.R., Black A. et al. Unusual case of haemophilia B // Lancet. 1989. V. 1. № 8644. P. 960. https://doi.org/10.1016/s0140-6736(89)92540-3
Picketts D.J., D’Souza C., Bridge P.J., Lillicrap D. An A to T transversion at position -5 of the factor IX promoter results in hemophilia B // Genomics. 1992. V. 12. № 1. P. 161–163. https://doi.org/10.1016/0888-7543(92)90421-n
Reijnen M.J., Peerlinck K., Maasdam D. et al. Hemophilia B Leyden: Substitution of thymine for guanine at position –21 results in a disruption of a hepatocyte nuclear factor 4 binding site in the factor IX promoter // Blood. 1993. V. 82. № 1. P. 151–158.
Reijnen M.J., Sladek F.M., Bertina R.M., Reitsma P.H. Disruption of a binding site for hepatocyte nuclear factor 4 results in hemophilia B Leyden // Proc. Natl Acad. Sci. USA. 1992. V. 89. № 14. P. 6300–6303. https://doi.org/10.1073/pnas.89.14.6300
Yao S.N., DeSilva A.H., Kurachi S. et al. Characterization of a mouse factor IX cDNA and developmental regulation of the factor IX gene expression in liver // Thromb. Haemost. 1991. V. 65. № 1. P. 52–58.
Brady J.N., Notley C., Cameron C., Lillicrap D. Androgen effects on factor IX expression: in vitro and in vivo studies in mice // Br. J. Haematol. 1998. V. 101. № 2. P. 273–279. https://doi.org/10.1046/j.1365-2141.1998.00694.x
Kurachi S., Huo J.S., Ameri A. et al. An age-related homeostasis mechanism is essential for spontaneous amelioration of hemophilia B Leyden // Proc. Natl Acad. Sci. USA. 2009. V. 106. № 19. P. 7921–7926. https://doi.org/10.1073/pnas.0902191106
Funnell A.P., Crossley M. Hemophilia B Leyden and once mysterious cis-regulatory mutations // Trends Genet. 2014. V. 30. № 1. P. 18–23. https://doi.org/10.1016/j.tig.2013.09.007
Landschulz W.H., Johnson P.F., Adashi E.Y. et al. Isolation of a recombinant copy of the gene encoding C/EBP // Genes Dev. 1988. V. 2. № 7. P. 786–800. https://doi.org/10.1101/gad.2.7.786
Sladek F.M., Zhong W.M., Lai E., Darnell J.E., Jr. Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily // Genes Dev. 1990. V. 4. № 12b. P. 2353–2365. https://doi.org/10.1101/gad.4.12b.2353
Davies N., Austen D.E., Wilde M.D. et al. Clotting factor IX levels in C/EBP alpha knockout mice // Br. J. Haematol. 1997. V. 99. № 3. P. 578–579. https://doi.org/10.1046/j.1365-2141.1997.4603263.x
Crossley M., Ludwig M., Stowell K.M. et al. Recovery from hemophilia B Leyden: An androgen-responsive element in the factor IX promoter // Science. 1992. V. 257. № 5068. P. 377–379. https://doi.org/10.1126/science.1631558
Giannelli F., Green P.M., Sommer S.S. et al. Haemophilia B: Database of point mutations and short additions and deletions–eighth edition // Nucl. Acids Res. 1998. V. 26. № 1. P. 265–268. https://doi.org/10.1093/nar/26.1.265
Belvini D., Salviato R., Radossi P. et al. Molecular genotyping of the Italian cohort of patients with hemophilia B // Haematologica. 2005. V. 90. № 5. P. 635–642.
Morgan G.E., Rowley G., Green P.M. et al. Further evidence for the importance of an androgen response element in the factor IX promoter // Br. J. Haematol. 1997. V. 98. № 1. P. 79–85. https://doi.org/10.1046/j.1365-2141.1997.1712991.x
Heit J.A., Ketterling R.P., Zapata R.E. et al. Haemophilia B Brandenberg-type promoter mutation // Haemophilia. 1999. V. 5. № 1. P. 73–75. https://doi.org/10.1046/j.1365-2516.1999.00193.x
Briët E., Wijnands M.C., Veltkamp J.J. The prophylactic treatment of hemophilia B Leyden with anabolic steroids // Ann. Intern. Med. 1985. V. 103. № 2. P. 225–226. https://doi.org/10.7326/0003-4819-103-2-225
Rimmer E.K., Seftel M.D., Israels S.J., Houston D.S. Unintended benefit of anabolic steroid use in hemophilia B Leiden // Am. J. Hematol. 2012. V. 87. № 1. P. 122–123. https://doi.org/10.1002/ajh.22190
Ketterling R.P., Liu J.Z., Liao D. et al. Two novel factor IX promoter mutations: Incremental progress towards ‘saturation in vivo mutagenesis’ of a human promoter region // Hum. Mol. Genet. 1995. V. 4. № 4. P. 769–770. https://doi.org/10.1093/hmg/4.4.769
Lannoy N., Lambert C., Farrugia A. et al. Usual and unusual mutations in a cohort of Belgian patients with hemophilia B // Thromb. Res. 2017. V. 149. P. 25–28. https://doi.org/10.1016/j.thromres.2016.11.006
Nathwani A.C., Reiss U.M., Tuddenham E.G. et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B // N. Engl. J. Med. 2014. V. 371. № 21. P. 1994–2004. https://doi.org/10.1056/NEJMoa1407309
Wasylyk B., Hahn S.L., Giovane A. The Ets family of transcription factors // Eur. J. Biochem. 1993. V. 211. № 1–2. P. 7–18. https://doi.org/10.1007/978-3-642-78757-7_2
Hildyard C., Keeling D. Effect of age on factor IX levels in symptomatic carriers of Haemophila B Leyden // Br. J. Haematol. 2015. V. 169. № 3. P. 448–449. https://doi.org/10.1111/bjh.13223
Lavin M., Jenkins P.V., Healy M.L. et al. Age-related factor IX correction in symptomatic female carriers with haemophilia B Leyden // Haemophilia. 2015. V. 21. № 6. P. e498–e500. https://doi.org/10.1111/hae.12761
Ahmed S.Z., O’Rourke M., Jenkins V. et al. Progressive increase in FIX level in males with haemophilia B Leyden and c.35G>A mutation in early childhood not related to androgen effect // Br. J. Haematol. 2020. V. 189. № 6. P. e262–e265. https://doi.org/10.1111/bjh.16688
Steyn F.J., Tolle V., Chen C., Epelbaum J. Neuroendocrine regulation of growth hormone secretion // Compr. Physiol. 2016. V. 6. № 2. P. 687–735. https://doi.org/10.1002/cphy.c150002
Green P.M., Montandon A.J., Bentley D.R. et al. The incidence and distribution of CpG→TpG transitions in the coagulation factor IX gene. A fresh look at CpG mutational hotspots // Nucl. Acids Res. 1990. V. 18. № 11. P. 3227–3231. https://doi.org/10.1093/nar/18.11.3227
Funnell A.P., Wilson M.D., Ballester B. et al. A CpG mutational hotspot in a ONECUT binding site accounts for the prevalent variant of hemophilia B Leyden // Am. J. Hum. Genet. 2013. V. 92. № 3. P. 460–467. https://doi.org/10.1016/j.ajhg.2013.02.003
Laudadio I., Manfroid I., Achouri Y. et al. A feedback loop between the liver-enriched transcription factor network and miR-122 controls hepatocyte differentiation // Gastroenterology. 2012. V. 142. № 1. P. 119–129. https://doi.org/10.1053/j.gastro.2011.09.001
Odom D.T., Zizlsperger N., Gordon D.B. et al. Control of pancreas and liver gene expression by HNF transcription factors // Science. 2004. V. 303. № 5662. P. 1378–1381. https://doi.org/10.1126/science.1089769
Thompson A.R., Bajaj S.P., Chen S.H., MacGillivray R.T. “Founder” effect in different families with haemophilia B mutation // Lancet. 1990. V. 335. № 8686. P. 418. https://doi.org/10.1016/0140-6736(90)90259-8
Picketts D.J., Lillicrap D.P., Mueller C.R. Synergy between transcription factors DBP and C/EBP compensates for a haemophilia B Leyden factor IX mutation // Nat. Genet. 1993. V. 3. № 2. P. 175–179. https://doi.org/10.1038/ng0293-175
Boccia L.M., Lillicrap D., Newcombe K., Mueller C.R. Binding of the Ets factor GA-binding protein to an upstream site in the factor IX promoter is a critical event in transactivation // Mol. Cell Biol. 1996. V. 16. № 5. P. 1929–1935. https://doi.org/10.1128/mcb.16.5.1929
Ghanem N., Costes B., Martin J. et al. Twenty-four novel hemophilia B mutations revealed by rapid scanning of the whole factor IX gene in a French population sample // Eur. J. Hum. Genet. 1993. V. 1. № 2. P. 144–155. https://doi.org/10.1159/000472401
Сурин В.Л., Демидова Е.Ю., Селиванова Д.С., Лучинина Ю.А., Саломашкина В.В., Пшеничникова О.С., Лихачева Е.А. Мутационный анализ гемофилии В в России с использованием молекулярно-генетических методов // Генетика. 1916. Т. 52. № 4. С. 466–473.
Vaquerizas J.M., Kummerfeld S.K., Teichmann S.A., Luscombe N.M. A census of human transcription factors: function, expression and evolution // Nat. Rev. Genet. 2009. V. 10. № 4. P. 252–263. https://doi.org/10.1038/nrg2538
Babu M.M., Luscombe N.M., Aravind L. et al. Structure and evolution of transcriptional regulatory networks // Curr. Opin. Struct. Biol. 2004. V. 14. № 3. P. 283–291. https://doi.org/10.1016/j.sbi.2004.05.004
Biggin M.D. Animal transcription networks as highly connected, quantitative continua // Dev. Cell. 2011. V. 21. № 4. P. 611–626. https://doi.org/10.1016/j.devcel.2011.09.008
Rhee H.S., Pugh B.F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution // Cell. 2011. V. 147. № 6. P. 1408–1419. https://doi.org/10.1016/j.cell.2011.11.013
Дополнительные материалы отсутствуют.