

Генетика, 2021, T. 57, № 4, стр. 371-383
Толерантность к повреждениям ДНК у дрожжей Saccharomyces cerevisiae
Е. А. Алексеева 1, *, В. Г. Королев 1
1 Петербургский институт ядерной физики им. Б.П. Константинова Национального исследовательского центра “Курчатовский институт”
188300 Ленинградская область, Гатчина, Россия
* E-mail: alekseeva_ea@pnpi.nrcki.ru
Поступила в редакцию 23.12.2019
После доработки 27.10.2020
Принята к публикации 03.11.2020
Аннотация
У эукариот толерантность к повреждениям ДНК (ТПД) обеспечивается двумя механизмами. Первый опосредован белками гомологичной рекомбинационной репарации. Второй находится под контролем RAD6-эпистатической группы генов и разделяется еще на два пути: безошибочный и склонный к ошибкам. Склонный к ошибкам механизм, который называют синтез ДНК в обход повреждений (TLS – Translesion Synthesis), осуществляется при участии специализированных TLS ДНК-полимераз. TLS является существенным источником мутационных изменений в ДНК. Напротив, при реализации RAD6-зависимого безошибочного механизма ТПД относительно более высокая точность синтеза ДНК обеспечивается благодаря использованию неповрежденной сестринской хроматиды или гомологичной хромосомы в качестве матрицы для продолжения репликации. При этом после остановки вилки репликации на повреждении 3'-конец синтезируемой нити переносится на неповрежденную гомологичную молекулу ДНК, синтез продолжается на некотором протяжении на новой матрице и затем удлиненная нить переносится обратно на исходную хроматиду. Инактивация большинства генов, контролирующих безошибочный механизм ТПД, либо не влияет на уровень УФ-индуцированного мутагенеза, либо его понижает. К исключениям можно отнести гены, входящие в эпистатическую группу HSM3. Мутации в генах этой группы приводят к существенному повышению частоты УФ-индуцированного мутагенеза. В данном обзоре авторы рассматривают безошибочную ветвь ТПД и делают попытку обосновать роль генов HSM3-эпистатической группы в ряду молекулярных событий, которые ведут к безошибочному обходу повреждений, блокирующих репликацию у почкующихся дрожжей.
В норме репликация ДНК завершается точным копированием генетического материала. Однако репликативная вилка часто сталкивается с множеством структурных препятствий, таких как повреждения ДНК, сшивки белок–ДНК и пр. Повреждения часто генерируются при нормальных условиях роста клетки [1], а также индуцируются экзогенными генотоксическими агентами химической или физической природы. В течение эволюции клетки приобрели такие механизмы, как эксцизионная репарация нуклеотидов и эксцизионная репарация поврежденных оснований, которые устраняют большинство повреждений ДНК [2]. Тем не менее некоторые повреждения сохраняются в ДНК и во время репликации, что приводит к остановке репликативного комплекса. Репликативный комплекс не может подставлять нуклеотиды напротив поврежденных нуклеотидов, и если повреждение не обойти или не устранить, то это может привести к гибели клетки. Для решения этой проблемы клетки выработали механизм, позволяющий преодолевать возникшие препятствия благодаря обходу повреждений специализированными TLS ДНК-полимеразами (от англ. Translesion Synthesis – синтез ДНК в обход повреждений) или посредством смены поврежденной матрицы на гомологичную неповрежденную и продолжать процесс синтеза ДНК.
Данный механизм, позволяющий обходить повреждения ДНК, возникшие во время репликации, первоначально называли пострепликативной репарацией (ПРР). Это связано с тем, что УФ-облучение клеток почкующихся дрожжей приводило к образованию однонитевых брешей (ОНБ) в реплицирующейся ДНК [3]. Также было отмечено, что УФ-индуцированные димеры пиримидина, вызывающие ОНБ, часто сохраняются после завершения клеточного цикла [4–6]. Репраймирование и пострепликативное заполнение брешей позже было подтверждено экспериментами, показавшими, что RAD6-зависимый путь обхода повреждений у дрожжей активен в G2/M фазах клеточного цикла [7, 8]. Так как в то время считали, что процесс репарации происходит после процесса репликации, то данный механизм был назван ПРР. Но в отличие от других механизмов репарации ПРР фактически не устраняет повреждения ДНК, а обеспечивает временную устойчивость к ним. Исходя из этого, в настоящее время данный механизм называют толерантностью к повреждениям ДНК (ТПД) [9].
ТПД обеспечивается двумя механизмами: первый осуществляется за счет белков гомологичной рекомбинационной репарации (ГРР); второй контролируется RAD6-эпистатической группой генов. В свою очередь второй механизм подразделяется на два пути: безошибочный и склонный к ошибкам.
Склонный к ошибкам путь (TLS-путь) осуществляется за счет работы специализированных TLS ДНК-полимераз. К TLS ДНК-полимеразам у почкующихся дрожжей относятся полимеразы Rev1 и Polη, принадлежащие к Y-семейству, а также полимераза Polζ, принадлежащая к В-семейству полимераз [10]. TLS-путь является источником большого количества мутационных изменений в ДНК, вносимых в ходе матричного синтеза ДНК TLS ДНК-полимеразами, обладающими относительно низкой точностью.
Безошибочный путь ТПД, контролируемый RAD6-эпистатической группой генов, в свою очередь осуществляется благодаря использованию неповрежденной сестринской хроматиды или гомологичной хромосомы в качестве матрицы для продолжения репликации и поэтому является более точным по сравнению с TLS. Кроме того, относительно высокая точность этого процесса достигается за счет привлечения к синтезу ДНК репликативных ДНК-полимераз, обладающих большей точностью по сравнению с TLS ДНК-полимеразами. К репликативным ДНК-полимеразам почкующихся дрожжей относятся: Polα, Polδ и Polε. В ТПД у почкующихся дрожжей большую роль в безошибочном обходе повреждений ДНК играет Polδ.
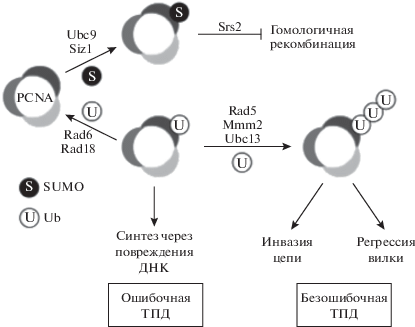
Выбор пути, по которому будет осуществляться обход повреждения ДНК, в основном зависит от типа посттрансляционной модификации ядерного антигена пролифелирующих клеток (PCNA) (рис. 1) [11]. PCNA (субъединицы которого кодируются геном POL30) является важным компонентом реплисомы, а также участвует в таких процессах как репарация ДНК, метилирование ДНК, ремоделирование хроматина и регуляция клеточного цикла [12]. Моноубиквитинирование PCNA комплексом Rad6/Rad18 активирует склонный к ошибкам путь TLS. А полиубиквитинирование PCNA стимулирует переход к безошибочному обходу повреждений ДНК [13].
Рис. 1.
Роль посттрансляционной модификации PCNA в обеспечении толерантности к повреждению ДНК у S. cerevisiae. После присоединения одного Ub PCNA может быть дополнительно последовательно модифицирована путем добавления Lys63-связанной цепи полиубиквитина. Моноубиквитинирование PCNA привлекает подверженные ошибкам и безошибочные TLS-полимеразы для прямого обхода повреждения ДНК. Полиубиквитинирование PCNA является сигналом для активации безошибочного механизма обхода повреждения ДНК с использованием переключения матрицы. Сумоилирование PCNA рекрутирует геликазу Srs2 в вилку репликации, которая предотвращает образование филаментов Rad51-онДНК и тем самым предотвращает нежелательную гомологичную рекомбинацию [11].
ОСТАНОВКА КЛЕТОЧНОГО ЦИКЛА В ОТВЕТ НА ПОВРЕЖДЕНИЯ ДНК
В обеспечении толерантности к повреждениям ДНК ключевую роль играет остановка клеточного цикла в ответ на повреждение ДНК (DDR от англ. DNA Damage Response), так как DDR является отправной точкой начала процесса обхода повреждения ДНК. Ведь именно в момент ареста клеточного цикла, вызванного повреждениями ДНК, клеткой принимается решение продолжить синтез ДНК или уйти в апоптоз, в случае если повреждение не будет устранено и репликация не возобновится.
Основные функции DDR – остановка движения по клеточному циклу и активация процессов репарации поврежденной ДНК. При нормальных условиях роста клеток ответ на повреждения глобально не индуцируется. Однако локальное привлечение белков, специфических для этого ответа, может происходить при столкновении репликативной машины со спонтанными повреждениями ДНК. Индукция глобального ответа на повреждения, приводящего к аресту клеточного цикла, происходит при появлении в ДНК значительного числа повреждений. При большом количестве повреждений ДНК при репликации накапливается большое число ОНБ. У почкующихся дрожжей именно однонитевая ДНК (онДНК) является сигналом для запуска процессов, приводящих к аресту клеточного цикла, которые опосредованы активацией киназы Mec1. Это происходит, когда комплекс Mec1/Ddc2 связывается с онДНК через физическое взаимодействие Ddc2 с RPA. После образования тройного комплекса Mec1/Ddc2/RPA происходит освобождение активной формы киназы Mec1, которая активирует одну из важнейших своих мишеней – эффекторную киназу Rad53 [14]. Rad53 регулирует множество аспектов ответа на повреждения, включая подавление активации поздних ориджинов, контроль синтеза дезоксинуклеотидов, регуляцию транскрипции, индуцированную повреждениями, и задержку клеточного цикла [15].
Таким образом, после активации Rad53 происходят события, способствующие возобновлению репликации и дальнейшему движению по клеточному циклу. Одним из таких событий является инициация ТПД, которая способствует обходу повреждений ДНК и предотвращает гибель клетки.
ИНИЦИАЦИЯ ТПД У ДРОЖЖЕЙ Saccharomyes cerevisiae
Долго полагали, что у эукариот лидирующая нить копируется непрерывно и инициация синтеза лидирующей нити происходит исключительно в ориджинах репликации. Непрерывность синтеза на поврежденной матрице объяснялась процессом, названным синтезом ДНК в обход повреждения (TLS) [16]. Накопление ОНБ и их медленное устранение связывали с низкой скоростью TLS. Но в работе [6] было показано, что у почкующихся дрожжей после облучения их УФ наблюдается прерывистость синтеза на обеих нитях ДНК. В других работах [6, 8] было показано, что репраймирование на лидирующей цепи приводит к образованию длинных ОНБ. Таким образом, ОНБ могут существовать достаточно продолжительное время. ОНБ являются опасными для ДНК, так как эндо- и экзонуклеазы, присутствующие в клетке, могут привести к деградации незащищенных концов ОНБ ДНК и двунитевым разрывам ДНК. В связи с этим оба конца ДНК, ограничивающие ОНБ, нуждаются в защите от деградации. С 3'-стороны бреши защита осуществляется репликативным комплексом, который стабилизируется фосфорилированием киназой Rad53. Однонитевой участок бреши защищает от атаки эндонуклеаз комплекс RPA. Одновременно RPA, связанный с ДНК, активно привлекает погрузчик Rad24 к 5'-стороне бреши, где происходит загрузка на ДНК комплекса 9–1–1 (Rad17/Mec3/Ddc1), являющегося альтернативным фактором процессивности [14, 17], данный комплекс связывается с участком повреждения ДНК и приводит к активации киназы Rad53. Комплекс 9–1–1 служит защитой 5'-стороны ОНБ от экзонуклеазной активности. Организованная таким образом ОНБ служит субстратом для белков, инициирующих ТПД. Ключевую роль при склонном к ошибкам обходе повреждений играют специализированные ДНК-полимеразы Rev1, Polη и Polζ (TLS) [18, 19], а безошибочный (рекомбинационный) обход повреждений находится под контролем комплекса Mms2/Ubc13/Rad5 и ГРР [20, 21].
ОБХОД ПОВРЕЖДЕНИЙ ДНК ПО МЕХАНИЗМУ TLS
Генетические исследования на модели дрожжей Saccharomyces cerevisiae показали критическую роль убиквитин-конъюгирующего ферментативного комплекса Rad6/Rad18 в контроле репликации через повреждения ДНК [7, 22, 23]. Связывание этого комплекса с ДНК в районе ОНБ является первым этапом ТПД. Способность белка Rad18 связываться с однонитевой ДНК служит средством доставки белка Rad6 к заблокированной повреждением репликационной вилке, где Rad6 ковалентно модифицирует PCNA, присоединяя одну молекулу убиквитина (Ub) к Lys164 (рис. 1). Когда PCNA моноубиквитинирован, белки TLS могут быть привлечены для прямого обхода повреждения ДНК [24]. Эта модификация PCNA резко повышает сродство комплекса к связыванию двух TLS ДНК-полимераз Polη и Rev1, но не влияет на эффективность связывания ДНК-полимераз Polδ и Polζ [25].
Polη является уникальной среди эукариотических TLS ДНК-полимераз. Она способна осуществлять эффективный и безошибочный синтез ДНК на матричной цепи, содержащей циклобутановые пиримидиновые димеры, образующиеся в результате воздействия УФ-излучения [18], однако как и другие TLS ДНК-полимеразы Polη с низкой точностью копирует неповрежденную ДНК. Polη не только способна эффективно подставлять два адениновых нуклеотида напротив димера ТТ, но и обеспечивает сохранение рамки считывания. Более того, когда димер выходит из активного сайта полимеразы, стерические препятствия гарантируют, что ДНК освободится от фермента. Диссоциация полимеразы происходит после того, как за повреждением будут встроены три нуклеотида [26]. Это спасает клетки от синтеза протяженных участков ДНК при помощи часто ошибающейся Polη. Поэтому у дрожжей мутанты по гену RAD30, кодирующему Polη, проявляют повышенную чувствительность к летальному, но не мутагенному действию УФ [27]. В отличие от дрожжей инактивация Polη в человеческих клетках приводит к повышенному УФ-индуцированному мутагенезу и определяет предрасположенность к вариантной форме пигментной ксеродермы и раку кожи [28–31].
В отличие от Polη Polζ способна продолжать синтез от нуклеотида, встроенного напротив повреждения другими ДНК-полимеразами, что, как правило, сопровождается мутагенезом [32, 33]. Polζ является мультисубъединичным комплексом, состоящим из четырех белков Rev3/Rev7/Pol31/Pol32 [10]. В нуклеоплазме этот комплекс чаще всего представлен двумя субъединицами – каталитической Rev3 и вспомогательной Rev7. Для сборки остальных субъединиц TLS-полимеразы Polζ, вероятно, служит платформа, состоящая из белка Rev1p, связанного с PCNA и Rad5p. Белок Rev1 является dCMP-трансферазой и дополнительно обладает некаталитическими функциями в TLS. Rev1 имеет повышенное сродство к моноубиквитинированному PCNA и стабильно взаимодействует с Rad5 через С‑терминальный домен Rev1 и N-терминальный домен Rad5 [34]. Комплекс Rev3/Rev7/Pol31/Pol32, возможно, при взаимодействии с репликативной полимеразой Polδ осуществляет склонный к ошибкам обход повреждений ДНК. Так как концентрация белка Rev1 в нуклеоплазме очень низкая [34], можно предположить, что Polη будет выигрывать у Rev1 конкуренцию за связывание с моноубиквитинированным PCNA. В результате первая стадия обхода пиримидиновых димеров будет осуществляться в основном Polη.
РЕКОМБИНАЦИОННЫЙ ПУТЬ ТПД
Полиубиквитинирование PCNA является сигналом для инициации безошибочного механизма ТПД [35]. Этот механизм осуществляется при помощи репликативных ДНК-полимераз, у почкующихся дрожжей он в основном осуществляется за счет работы Polδ.
Безошибочный механизм ТПД может осуществляться по двум различным путям: один контролируется генами гомологичной рекомбинации (ГР), второй контролируется генами RAD5-группы.
Полиубиквитинирование PCNA осуществляет комплекс Rad5/Mms2/Ubc13. Механизм ТПД, контролируемый генами RAD5, MMS2 и UBC13, обеспечивает обход повреждения путем временного переключения синтеза с поврежденной нити на сестринскую хроматиду (переключение матрицы). В экспериментах in vitro было показано, что этот процесс может инициироваться благодаря изомеризации репликативной вилки, генерируемой белком Rad5 [36]. Белок Rad5 не только входит в состав комплекса, обеспечивающего моноубиквитинирование PCNA, но и выполняет функцию сборки комплекса для последующей модификации PCNA. Rad5 физически взаимодействует с двумя белками Mms2 и Ubc13, составляющими единый комплекс. Этот комплекс может функционировать in vitro как убиквитин лигаза E2 для синтеза поли-Ub-цепей, связанных с Lys63 PCNA [37, 38]. Mms2 напоминает фермент E2, но в его активном сайте отсутствует остаток Cys [35]. Образование комплекса Mms2/Ubc13 приводит к тому, что образуется уникальный активный сайт для катализа реакции образования только Ub-цепей, связанных с Lys63 [37, 39–41]. Rad5 взаимодействует с Ubc13, обеспечивая роль комплекса Mms2/Ubc13 в качестве фермента E3 [11, 42–46]. В соответствии с этим комплекс Rad5/Mms2/Ubc13 имеет всe необходимое для полиубиквитинирования PCNA [45].
В работе Янга с сотрудниками [11] в определенной мере раскрывается функциональная роль полиубиквитинирования PCNA в сигнальном механизме ТПД. В ней было показано, что моноубиквитинированный PCNA стимулировал TLS напротив апурин-апиримидиновых сайтов с помощью Polη. Однако эффективность включения нуклеотидов напротив повреждения снижалась по мере того, как длина убиквитиновой цепи росла от моно- до тетра-Ub. На основании проведенных экспериментов авторы пришли к заключению, что полиубиквитинирование PCNA подавляет TLS, уменьшая активность полимераз в месте повреждения. Кроме того, полиубиквитинирование PCNA ослабляет связь Polη с PCNA, что способствует их диссоциации и предотвращает образование нового комплекса Polη/PCNA [11]. Таким образом, предполагается, что полиубиквитинирование PCNA служит в качестве сигнала, блокирующего репликацию, посредством предотвращения доступа полимераз семейства B и Y в вилку репликации.
Современные генетические и биохимические данные способствовали созданию двух моделей, объясняющих безошибочный обход повреждений ДНК. Первая модель рассматривает в качестве основного механизма регрессии вилки репликации (рис. 2) [11]. Согласно этой модели вилка репликации останавливается, когда сталкивается с повреждением в матричной цепи ДНК во время синтеза лидирующей нити. Это ведет к нарушению координации синтеза ведущей и отстающей нитей [47]. Синтез отстающей нити продолжается, что ведет к генерации ОНБ, которая может служить субстратом для связывания гетеродимера Rad6/Rad18. Последний будет моноубиквитинировать PCNA. Взаимодействие белка Rad18 с Rad5 приведет к регрессии вилки репликации в результате активации геликазной активности последнего. При этом родительская и вновь синтезированная нити раскручиваются и отжигаются в структуру “куриных лапок”. Благодаря формированию такой структуры вновь синтезированная нить будет служить матрицей для восстановления информации о поврежденной области в родительской нити и с 3'-конца будет инициирован нормальный процесс репликации.
Рис. 2.
Модель молекулярного механизма переключения матрицы и/или регрессии вилки репликации, реализуемого в ходе безошибочного пути ТПД. а – задержка репликации из-за повреждения в матричной цепи ДНК приводит к накоплению онДНК, это является сигналом для Rad6/Rad18-зависимой активации ТПД, опосредованной моноубиквитинированием PCNA; б – комплекс Rad5–Ubc13–Mms2 полиубиквитинирует PCNA, что активирует безошибочную ветвь ТПД. Rad5 способен осуществлять регрессию вилки репликации благодаря своей геликазной активности, что приводит к формированию структуры “куриные лапки”. Факторы гомологичной рекомбинации (ГР) опосредуют инвазию цепи и переключение матрицы на вновь синтезированную сестринскую хроматиду [11].
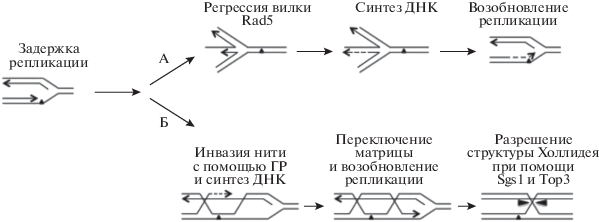
Вторая модель предполагает, что механизм переключения матриц для безошибочного механизма ТПД включает процесс обмена нитями, осуществляемый при участии факторов гомологичной рекомбинации (ГР) (рис. 2) [11]. После диссоциации полимеразы Polη в результате полиубиквитинирования PCNA 3'-конец вновь синтезированной ДНК подвергается деградации под действием экзонуклеазной активности белкового комплекса MRX, который активно привлекается к блокированной вилке [48–50]. При этом степень деградации ДНК находится под контролем белка Rad51. В отсутствие Rad51 ДНК подвергается обширной деградации, которую возможно осуществляет геликаза Pif1 при содействии флэп-эндонуклеазы Fen1 [50]. Предполагается, что Mre11 и Rad51 находятся в динамическом равновесии на онДНК и противодействуют активности друг друга [49]. Роль Pif1 в данном процессе, по-видимому, сводится к процессингу ОНБ и подготовке свободного однонитевого 3'-конца ДНК для последующих этапов [50]. Свободный однонитевой 3'-конец ДНК служит для инициации рекомбинационного процесса путем образования D-петли (Displacement-loop (D-loop)) (рис. 3). Инактивация каждого из трех белков (Mre11, Rad51, Pif1) нарушает безошибочный механизм ТПД, опосредованный рекомбинацией [50].

Физические доказательства участия процессов рекомбинации в механизме безошибочной ТПД получены в результате визуализации специфических структур – Х-ДНК (Х-образной двухцепочечной ДНК) [51] (рис. 4). Обработка клеток любого из мутантов sgs1, top3, ubc9 или mms21 ДНК-повреждающим агентом приводит к накоплению структур X-ДНК, которые можно визуализировать с помощью 2D гель-электрофореза [52, 53]. Поскольку ген RAD51, кодирующий один из основных белков рекомбинации, абсолютно необходим для накопления структур X-ДНК [51], это подтверждает связь между безошибочным механизмом ТПД и гомологичной рекомбинацией.
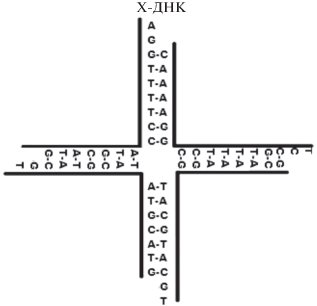
Участие генов ГРР в безошибочной ТПД также было подтверждено в недавнем исследовании роли гена RAD5 в завершении репликации после повреждения ДНК [53]. В данном исследовании использовался ДНК-алкилирующий агент адозелезин. Использование адозелезина позволило авторам изучить остановленную репликацию в определенных местах хромосомы. У мутанта rad5 обработка клеток адозелезином приводила к замедлению репликации и образованию структур X-ДНК в результате остановки репликативной вилки [54]. X-ДНК по сути представляли собой структуры Холлидея, для образования которых требовались функциональные продукты генов RAD51, RAD52, RAD54 и RAD55. Полученные данные подтверждают связь между безошибочным механизмом ТПД, инициируемым Rad5, и ГРР. Наконец, выявление эпистатического взаимодействия между генами, участвующими в ГРР и ТПД, также указывает на наличие связи между ними [55].
Решающую роль в выборе между Rad5-зависимым ТПД и ГРР определяют события, происходящие в D-петле, промежуточном продукте ГР (рис. 3). В ходе Rad5-зависимого пути ТПД происходит синтез относительно коротких фрагментов ДНК, способных перекрыть повреждение на материнской молекуле. При реализации RAD52-зависимого механизма осуществляется синтез более протяженных участков ДНК. На первых порах после возникновения D-петли представляют собой неустойчивые структуры, которые нуждаются в стабилизации при помощи специфических белков.
Одним из таких белков является Hmo1. Было показано, что он контролирует образование промежуточных продуктов рекомбинации, в том числе D‑петли, при инициации безошибочного механизма ТПД, так как Hmo1 демонстрирует высокое сродство к крестообразным структурам ДНК и измененным конформациям онДНК [56, 57]. Делеция гена HMO1 дестабилизирует D-петли, что приводит к снижению чувствительности мутантов rad5Δ, mms2Δ и ubc13Δ к MMS. Снижение чувствительности данных мутантов происходит в результате разрушения продуктов ГР (RAD52-зависимый путь), образование которых в большом количестве оказывает цитотоксическое действие на клетку [56].
Белковый комплекс, известный как SHU-комплекс, также играет определенную роль в ГРР, стабилизируя D-петли [58, 59]. Гены CSM2, PSY3, SHU1 и SHU2, кодирующие все четыре субъединицы данного комплекса, были выделены в полногеномном синтетическом скрининге генетического массива и могут играть роль в безошибочном механизме ТПД [55]. Субъединица Csm2 комплекса SHU стимулирует ГРР путем физического взаимодействия с гетеродимером Rad55/Rad57. Последний привлекает комплекс SHU в остановленную репликативную вилку, таким образом облегчая образование филамента Rad51 на онДНК [60, 61]. Противоположную роль у почкующихся дрожжей играет геликаза Srs2, которая дестабилизирует D‑петли и является ингибитором ГР [62]. Геликаза Srs2 разрушает рекомбинационные интермедиаты, удаляя белок Rad51 из нуклеопротеинового комплекса. Ген SRS2 был открыт как супрессор УФ чувствительности мутантов rad6Δ и rad18Δ и в независимом скрининге мутантов с сверхвысоким уровнем рекомбинации [18, 22]. Мутации в гене SRS2 в большой степени снижают УФ-индуцированный мутагенез в клетках дрожжей [20]. Продукт еще одного гена связан с дестабилизацией D-петель. Это АТФ-зависимая ДНК-геликаза с 3'–5'-полярностью, которая кодируется геном MPH1 [62, 63]. Продукт этого гена вовлечен в диссоциацию D-петель независимо от геликазы Srs2 для того, чтобы подавлять избыточное образование кроссоверных событий [64]. Таким образом, в отличие от белков комплекса SHU и Hmo1, которые стабилизируют D-петли, геликазы Mph1 и Srs2 разрушают вновь образованные рекомбинационные интермедиаты.
Ген HMO1 (HSM2) входит в HSM3-группу генов, которая принимает участие в контроле безошибочного механизма ТПД [65].
РОЛЬ HSM-ГРУППЫ ГЕНОВ В БЕЗОШИБОЧНОЙ ВЕТВИ ТПД
Ранее были выделены мутанты почкующихся дрожжей hsm, которые отличаются высокой частотой индуцированного мутагенеза и практически неизмененной устойчивостью к летальному действию мутагенов [66, 67], а также характеризуются повышенной частотой спонтанных мутаций устойчивости к канаванину. Выраженность мутаторного фенотипа варьировала от двукратного повышения частоты спонтанных мутаций для hsm1 до десятикратного для hsm2 по сравнению с диким типом [68, 69]. Исследование этих мутантов выявило шесть неаллельных мутаций (hsm1–hsm6), которые приводят к повышению спонтанного и УФ-индуцированного мутагенеза и не изменяют чувствительности клеток к УФ- и гамма-лучам, а также метилметансульфонату. Кроме того, были выделены мутанты him по признаку повышенного уровня мутагенеза, индуцированного азотистой кислотой, которые оказались еще и спонтанными мутаторами. Эти мутанты обладали повышенным уровнем УФ-индуцированного мутагенеза [66]. Генетический анализ позволил разделить обе коллекции мутантов на четыре эпистатические группы. В первую группу попали четыре мутанта hsm2Δ, hsm3Δ, hsm6Δ и him1Δ [69]. Наиболее важным для данного обзора свойством мутантов по генам HSM2, HSM3, HSM6 и HIM1 является их влияние на частоту индуцированной митотической рекомбинации. Показано, что частота кроссинговера у всех мутантов значительно уменьшена, а частота генной конверсии наоборот увеличена [68, 70, 71]. Это доказывает, что изучаемые мутации влияют негативно на длину гибридной ДНК, возникающей в течение репарационного синтеза в D-петлях [72]. Таким образом, все четыре гена имеют отношение к контролю рекомбинационного процесса. Мутанты по этим генам обладали выраженным мутаторным фенотипом, который зависел от гена REV3, что позволило отнести их к пострепликативному типу репарации и более определенно к генам, контролирующим безошибочный механизм ТПД [65]. Этот результат оказался неожиданным, так как практически все известные гены этого пути отличаются либо неизмененным, либо пониженным УФ-индуцированным мутагенезом. Более того, биохимические функции четырех изучаемых генов, выявленные или предполагаемые, предполагают участие соответствующих белков в различных процессах метаболизма хроматина [67]. Эти данные позволяют выдвинуть предположение, что причиной повышенного индуцированного мутагенеза у рассматриваемых мутантов является нарушение сборки хроматина в ходе репарационного процесса.
Среди обсуждаемых мутантов наибольшим влиянием на скорость спонтанного мутагенеза обладали мутанты по гену HSM2 [68]. Было показано, что мутация hsm2-1 является аллелем гена HMO1. Продукт этого гена относится к семейству HMG-белков, ассоциированных с хроматином и участвующих в поддержании стабильности генома [73].
Белок Hsm3 физически взаимодействует с белками Hat1, Hat2 и Hif1 – субъединицами гистонацетилтрансферазного комплекса NuB4, принимающего участие в модификации и сборке хроматина [74, 75]. С другой стороны, в ряде работ показано, что белок Hsm3 участвует в сборке протеасомного комплекса S26 [76, 77]. Для нахождения ответа на вопрос о связи процессов сборки протеасомы и мутагенеза были проведены исследования доменной структуры белка Hsm3 и показано, что за регуляцию мутационного процесса отвечает С-терминальный домен белка, а за сборку протеасом – центральный домен [78].
Ген HSM6 был картирован на левом плече хромосомы II в районе локализации гена PSY4. Функциональный тест на аллелизм показал, что мутация hsm6-1 является аллелем гена PSY4 [79]. Белок Psy4 является третьей субъединицей фосфатазного комплекса PPH3, который дефосфорилирует гистон γH2A [79].
Ген HIM1 был картирован на правом плече хромосомы IV; биохимическая функция белка, кодируемого данным геном, в настоящее время неизвестна. Известно, что делеция гена HIM1 приводит к дестабилизации D-петли [80].
Изучаемые мутации негативно влияют на длину гибридной ДНК, возникающей в ходе удлинения D-петли ДНК-полимеразами. Это подтверждается данными генетического анализа о взаимодействии мутаций him1Δ, hsm3Δ и hsm2Δ с мутацией srs2Δ, которая влияет на процессинг D-петли. У двойных мутантов him1Δ srs2Δ, hsm3Δ srs2Δ и hsm2Δ srs2Δ уровень мутагенеза оказался ниже, чем у всех одиночных мутантов [69, 70, 80]. Подобный результат был получен в экспериментах с двойными мутантами him1Δ mph1Δ и hsm3Δ mph1Δ [80, 81]. Основываясь на этих результатах, было сделано предположение, что, по крайней мере, мутации him1Δ, hsm3Δ и hsm2Δ дестабилизируют D-петли, что и приводит к повышенному мутагенезу. Это предположение было доказано прямыми биохимическими экспериментами с мутантами по гену HMO1 (HSM2) [56, 57]. Hmo1 может способствовать рекомбинационному процессу, помогая образованию сестринских хроматидных соединений посредством изгиба ДНК [82]. Известно, что белковый комплекс SHU, в состав которого входит субъединица Shu1, также стабилизирует вновь образованные D-петли, препятствуя работе антирекомбинационной геликазы Srs2 [83]. Как показали эксперименты, мутация shu1Δ приводит к резкому увеличению уровня УФ-индуцированного мутагенеза по сравнению с клетками дикого типа, что подтверждает высказанное выше предположение о роли стабилизации D-петли в предотвращении УФ-индуцированного мутагенеза [81].
Удивительные данные были получены при изучении взаимодействия мутаций генов HSM3-группы между собой. Двойной мутант him1Δ hsm2Δ показал уровень УФ-индуцированного мутагенеза, равный аналогичному показателю у штамма дикого типа [70]. Примерно такой же эффект наблюдался у двойного мутанта hsm2Δ hsm3Δ [84]. С другой стороны, у двойного мутанта him1Δ hsm3Δ уровень мутагенеза соответствовал уровню одиночного мутанта hsm3Δ, что указывает на эпистатическое взаимодействие между соответствующими генами.
Ранее было показано, что блок репарации неспаренных оснований у мутантов группы HSM3 приводит к синергическому увеличению частоты УФ-индуцированного мутагенеза, в частности у двойных мутантов pms1Δ him1Δ, pms1Δ hsm2Δ и pms1Δ hsm3Δ [65, 69, 70]. Это кажется странным, так как пиримидиновые димеры, возникающие при действии УФ-лучей, не являются субстратом белков репарации неспаренных оснований, следовательно мутации HSM3-группы генов инициируют образование ошибочно спаренных оснований на неповрежденной матрице, которые и являются субстратом Pms1-зависимой системы репарации. Интересный результат был получен в эксперименте с тройным мутантом pms1Δ him1Δ hsm2Δ. УФ-индуцированный мутагенез у тройного мутанта оказался таким же, как у одиночного hsm2Δ. Эти результаты позволяют сделать предположение, что причиной появления ошибочно спаренных оснований является работа склонных к ошибкам полимераз и что мутации в генах HSM3-группы приводят к переключению высокоточных ДНК-полимераз на TLS ДНК-полимеразы в ходе репарационного синтеза ДНК. Это скорее всего и влияет на фенотип мутантов по генам HSM3-группы. Пока неизвестно какие именно полимеразы участвуют в данном процессе.
На какой стадии процесса ТПД принимают участие гены HSM3-группы? В безошибочной ветви ТПД можно рассматривать два механизма: ГРР и Rad5-зависимый механизм. Для установления на какой из этих механизмов оказывают влияние HSM-мутанты были использованы мутанты по генам SRS2 и MPH1. Известно, что на Rad5-зависимом пути активно работает геликаза Mph1, которая способна разрушать D-петли, возвращая внедрившийся в сестринскую хроматиду однонитевой “хвост” на материнскую нить [84, 85]. У двойных мутантов him1Δ srs2Δ, mph1Δ him1Δ и hsm3Δ srs2Δ, mph1Δ hsm3Δ практически все возникшие D-петли будут процессироваться по механизму ГРР.
Для того чтобы разрешить гибридную структуру D-петли, необходимо вмешательство резолваз. Было сделано предположение, что на эту роль вполне может претендовать эндонуклеаза Mus81/Mms4. Эта структурно-специфическая эндонуклеаза требуется для подавления накопления летальных рекомбинационных интермедиатов, возникающих в процессе репликации поврежденной ДНК [4, 6–8, 18]. Mus81/Mms4 используется клеткой в процессинге рекомбинационных интермедиатов, которые возникают при репарации поврежденных репликативных вилок.
При исследовании УФ-индуцированного мутагенеза у двойного мутанта hsm3Δ mms4Δ было показано, что двойной мутант не отличался от штамма дикого типа по уровню индуцированного мутагенеза [81]. Следовательно, можно предположить, что основным ферментом, осуществляющим завершение процесса обмена матриц, является эндонуклеаза Mus81/Mms4. Таким образом, скоординированное действие геликазы Mph1 и эндонуклеазы Mus81/Mms4 может обеспечить необходимую длину вновь синтезированной ДНК в D-петле, чтобы перекрыть брешь, образованную вокруг повреждения.
Суммируя представленные результаты, можно сделать заключение, что причиной высокого индуцированного мутагенеза у мутантов по генам HSM2, HSM3 и HIM1 является преждевременное вытеснение онДНК из D-петли и заполнение оставшейся бреши полимеразой, склонной к ошибкам.
Большое количество ОНБ, оставшихся в ДНК после завершения репликации, приводит к индукции ареста клеточного цикла и активации киназы Rad53. Одной из основных биохимических функций Rad53 является контроль за эффективностью индукции комплекса RNR и, как следствие, концентрации дезоксинуклеотидов (дНТФ) [86]. При активации чекпойнта концентрация дНТФ в нуклеоплазме возрастает в 8 раз по сравнению с уровнем G0-фазы [87]. До настоящего времени остается нерешенным вопрос: для чего необходимо такое увеличение концентрации дНТФ? Единственное, что удалось показать, это то, что высокие концентрации дезоксинуклеотидов значительно повышают скорость синтеза ДНК. В клетках дикого типа максимальная активность Rad53 и высокий уровень дНТФ достигаются перед началом активной работы механизма ТПД [87]. Однако при этом мы не видим высокого уровня мутагенеза, характерного для мутантов him1Δ и hsm3Δ. Такой же результат мы наблюдаем в клетках дикого типа при быстром выходе из ареста клеточного цикла и быстром снижении уровня дНТФ. Отсюда следует, что, по-видимому, промежуточные концентрации дНТФ могут быть причиной высокого мутагенеза у мутантов по генам HSM3 эпистатической группы.
Гены HSM3 эпистатической группы еще не полностью изучены, и поэтому мы не можем достаточно подробно описать механизм их работы в безошибочной ветви ТПД.
Однако за последнее десятилетие произошел большой прогресс в понимании молекулярных механизмов толерантности клеток к повреждению генетического материала. Теперь стало известно большинство ферментативных стадий в процессах безошибочного и склонного к ошибкам механизмов ТПД.
Авторы надеются, что в дальнейшем роль генов HSM3-группы будет более подробно изучена и приведенная в данном обзоре информация поможет в изучении механизмов ТПД.
Работа поддержана грантом РФФИ № 18-34-00540 мол_а, а также работа выполнена при финансовой поддержке “Курчатовского геномного центра – ПИЯФ” программой развития центров генетических исследований мирового уровня, Соглашение № 075-15-2019-1663.
Настоящая статья не содержит каких-либо исследований с использованием в качестве объекта животных.
Настоящая статья не содержит каких-либо исследований с участием в качестве объекта людей.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Lindahl T. Instability and decay of the primary structure of DNA // Nature.1993. V. 362. P. 709–715. https://doi.org/10.1038/362709a0
Freidberg E.C., Walker G.C., Siede W. et al. DNA Repair and Mutagenesis. 2nd ed. Washington: ASM Press, 2006. 1161 p.
Prakash L. Characterization of postreplication repair in Saccharomyces cerevisiae and effects of rad6, rad18, rev3 and rad52 mutations // Mol. Gen. Genet. 1981. V. 184. № 3. P. 471–478. https://doi.org/10.1007/bf00352525
Bridges B.A., Munson R.J. Mutagenesis in Escherichia coli: evidence for the mechanism of base change mutation by ultraviolet radiation in a strain deficient in excision-repair // Proc. R. Soc. Lond. B: Biol. Sci. 1968. V. 171. P. 213–226. https://doi.org/10.1098/rspb.1968.0065
Ganesan A.K. Persistence of pyrimidine dimers during post-replication repair in ultraviolet light-irradiated Escherichia coli K12 // J. Mol. Biol. 1974. V. 87. P. 103–119. https://doi.org/10.1016/0022-2836(74)90563-4
Lopes M., Foiani M., Sogo J.M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions // Mol. Cell. 2006. V. 21. P. 15–27. https://doi.org/10.1016/j.molcel.2005.11.015
Daigaku Y., Davies A.A., Ulrich H.D. Ubiquitin-dependent DNA damage bypass is eparable from genome replication // Nature. 2010. V. 465. P. 951–955. https://doi.org/10.1038/nature09097
Karras G.I., Jentsch S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase // Cell. 2010. V. 141. № 2. P. 255–267. https://doi.org/10.1016/j.cell.2010.02.028
Andersen P., Xu F., Xiao W. Eykaryotic DNA damage tolerance and translesion synthesis through covalent modifications of PCNA // Cell Res. 2008. V. 18. P. 162–173. https://doi.org/10.1038/cr.2007.114
Pages V., Fuchs R.P. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo // Science. 2003. V. 300. P. 1300–1303. https://doi.org/10.1126/science.1083964
Yang K., Gong P., Gokhale P., Zhuang Z. Chemical protein polyubiquitination reveals the role of a noncanonical polyubiquin chain in DNA damage tolerance // ACS Chem. Biol. 2014. V. 9. № 8. P. 1685–1691. https://doi.org/10.1021/cb500133k
Choe K.N., Moldovan G.-L. Forging ahead through darkness: PCNA, still the principal conductor at the replication fork // Mol. Cell. 2017. V. 65. № 3. P. 380–392. https://doi.org/10.1016/j.molcel.2016.12.020
Amara F., Colombo R., Cazzaniga P. et al. In vivo and in silico analysis of PCNA ubiquitylation in the activation of the post replication repair pathway in S. cerevisiae // BMC Syst. Biol. 2013. 7:24. https://doi.org/10.1186/1752-0509-7-24
Majka J., Binz S.K., Wold M.S., Burgers P.M.J. Replication protein A directs loading of the DNA damage checkpoint clamp to 5'-DNA janctions // JBC. 2006. V. 281. P. 27855–27861. https://doi.org/10.1074/jbc.M605176200
Pardo B., Crabbe L., Pasero P. Signaling pathways of replication stressing yeast // FEMS Yeast Res. 2017. V. 17. № 2. https://doi.org/10.1093/femsyr/fow101
Goodman M.F., Woodgate R. Translesion DNA polymerases // Cold Spring Harb Perspect. Biol. 2013. V. 5. № 10. https://doi.org/10.1101/cshperspect.a010363
Zou L., Liu D., Elledge S.J. Replication protein A-mediated recruitment and activation of Rad17 complexes // PNAS. 2003. V. 100. P. 13827–13832. https://doi.org/10.1073/pnas.2336100100
Johnson R.E., Prakash S., Prakash L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase Polη // Science. 1999. V. 283. P. 1001–1004. https://doi.org/10.1126/science.283.5404.1001
Nelson J.R., Lawrence C.W., Hinkle D.C. Thymine-thymine dimer bypass by yeast DNA polymerase zeta // Science. 1996. V. 272. P. 1646–1649. https://doi.org/10.1126/science.272.5268.1646
Torres-Ramos C., Prakash S., Prakash L. Requirement of RAD5 and MMS2 for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae // Mol. Cell. Biol. 2002. V. 22. P. 2419–2426. https://doi.org/10.1128/MCB.22.7.2419-2426.2002
Gangavarapu V., Prakash S., Prakash L. Requirement of RAD52 group genes for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae // Mol. Cell. Biol. 2007. V. 27. P. 7758–7764. https://doi.org/10.1128/MCB.01331-07
Bailly V., Lamb J., Sung P. et al. Specific complex formation between yeast RAD6 and RAD18 proteins: a potential mechanism for targeting RAD6 ubiquitin-conjugating activity to DNA damage sites // Genes Dev. 1994. V. 8. P. 811–820. https://doi.org/10.1101/gad.8.7.811
Broomfield S., Hryciw T., Xiao W. DNA postreplication repair and mutagenesis in Saccharomyces cerevisiae // Mutat. Res. 2001. V. 486. P. 167–184. https://doi.org/10.1016/s0921-8777(01)00091-x
Bienko M., Green C.M., Crosetto N. et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis // Science. 2005. V. 310. P. 1821–1824. https://doi.org/10.1126/science.1120615
Garg P., Burgers P.M. Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases η and REV1 // PNAS. 2005. V. 102. P. 18361–18366. https://doi.org/10.1073/pnas.0505949102
Biertümpfel C., Zhao Y., Kondo Y. et al. Structure and mechanism of human DNA polymerase eta // Nature. 2010. V. 465. № 7301. P. 1044–1048. https://doi.org/10.1038/nature09196
McDonald J.P., Levine A.S., Woodgate R. The Saccharomyces cerevisiae RAD30 gene, a homologue of Escherichia coli dinB and umuC, is DNA damage inducible and functions in a novel error-free postreplication repair mechanism // Genetics. 1997. V. 147. P. 1557–1568.
Stary A., Kannouche P., Lehmann A.R., Sarasin A. Role of DNA polymerase η in the UV mutation spectrum in human cells // J. Biol. Chem. 2003. V. 278. P. 18767–18775. https://doi.org/10.1074/jbc.M211838200
Johnson R.E., Kondratick C.M., Prakash S., Prakash L. hRAD30 mutations in the variant form of xeroderma pigmentosum // Science. 1999. V. 285. P. 263–265. https://doi.org/10.1126/science.285.5425.263
Masutani C., Kusumoto R., Yamada A. et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η // Nature. 1999. V. 399. P. 700–704. https://doi.org/10.1038/21447
Johnson R.E., Haracska L., Prakash S., Prakash L. Role of DNA polymerase η in the bypass of a (6-4) TT photoproduct // Mol. Cell. Biol. 2001. V. 21. P. 3558–3563. https://doi.org/10.1128/MCB.21.10.3558-3563.2001
Johnson R.E., Washington M.T., Haracska L. et al. Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions // Nature. 2000. V. 406. P. 1015–1019. https://doi.org/10.1038/35023030
Kuang L., Kou H., Xie Z. et al. A non-catalytic function of Rev1 in translesion DNA synthesis and mutagenesis is mediated by its stable interaction whith Rad5 // DNA Repair. 2013. V. 12. P. 27–37. https://doi.org/10.1016/j.dnarep.2012.10.003
Blastyák A., Pintér L., Unk I. et al. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression // Mol. Cell. 2007. V. 28. P. 167–175. https://doi.org/10.1016/j.molcel.2007.07.030
Eddins M.J., Carlile C.M., Gomez K.M. et al. Mms2-Ubc13 covalently bound to ubiquitin reveals the structural basis of linkage-specific polyubiquitin chain formation // Nat. Struct. Mol. Biol. 2006. V. 13. P. 915–920. https://doi.org/10.1038/nsmb1148
Hofmann R.M., Pickart C.M. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair // Cell. 1999. V. 96. P. 645–653. https://doi.org/10.1016/s0092-8674(00)80575-9
VanDemark A.P., Hofmann R.M., Tsui C. et al. Molecular insights into polyubiquitin chain assembly: crystal structure of the Mms2/Ubc13 heterodimer // Cell. 2001. V. 105. P. 711–720. https://doi.org/10.1016/s0092-8674(01)00387-7
Broomfield S., Chow B.L., Xiao W. MMS2, encoding a ubiquitin-conjugating-enzyme-like protein, is a member of the yeast error-free postreplication repair pathway // Proc. Natl Acad. Sci. USA. 1998. V. 95. P. 5678–5683. https://doi.org/10.1073/pnas.95.10.5678
McKenna S., Spyracopoulos L., Moraes T. et al. Noncovalent interaction between ubiquitin and the human DNA repair protein Mms2 is required for Ubc13-mediated polyubiquitination // J. Biol. Chem. 2001. V. 276. P. 40120–40126. https://doi.org/10.1074/jbc.M102858200
Moraes T.F., Edwards R.A., McKenna S. et al. Crystal structure of the human ubiquitin conjugating enzyme complex, hMms2-hUbc13 // Nat. Struct. Biol. 2001. V. 8. P. 669–673. https://doi.org/10.1038/90373
Ulrich H.D., Jentsch S. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair // EMBO J. 2000. V. 19. P. 3388–3397. https://doi.org/10.1093/emboj/19.13.3388
Hoege C., Pfander B., Moldovan G.L. et al. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO // Nature. 2002. V. 419. P. 135–141. https://doi.org/10.1038/nature00991
Ulrich H.D. Protein–protein interactions within an E2–RING finger complex. Implications for ubiquitin-dependent DNA damage repair // J. Biol. Chem. 2003. V. 278. P. 7051–7058. https://doi.org/10.1074/jbc.M212195200
Carlile C.M., Pickart C.M., Matunis M.J., Cohen R.E. Synthesis of free and proliferating cell nuclear antigen-bound polyubiquitin chains by the RING E3 ubiquitin ligase Rad5 // J. Biol. Chem. 2009. V. 284. P. 29326–29334. https://doi.org/10.1074/jbc.M109.043885
Parker J.L., Ulrich H.D. Mechanistic analysis of PCNA poly-ubiquitylation by the ubiquitin protein ligases Rad18 and Rad5 // EMBO J. 2009. V. 28. P. 3657–3666. https://doi.org/10.1038/emboj.2009.303
Zhang W., Qin Z., Zhang X., Xiao W. Roles of sequential ubiquitination of PCNA in DNA-damage tolerance // FEBS Lett. 2011. V. 585. P. 2786–2794. https://doi.org/10.1016/j.febslet.2011.04.044
Trenz K., Smith E., Smith S., Costanzo V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks // EMBO J. 2006. V. 25. P. 1764–1774. https://doi.org/10.1038/sj.emboj.7601045
Bentsen I.B., Nielsen I., Lisby M. et al. MRX protects fork integrity at protein-DNA barriers, and its absence causes checkpoint activation dependent on chromatin context // Nucl. Acids Res. 2013. V. 41. P. 3173–3189. https://doi.org/10.1093/nar/gkt051
Hashimoto Y., Chaudhuri A.R., Lopez M., Costanzo V. Rad51 protects nascent DNA from Mre11 dependent degradation and promotes continuous DNA synthesis // Nat. Struct. Mol. Biol. 2010. V. 17. P. 1305–1311. https://doi.org/10.1038/nsmb.1927
Garcia-Rodrigues N., Wong R.P., Ulrich H.D. The helicase Pif1 functions in the template switching pathway of DNA damage bypass // Nucl. Acids Res. 2018. V. 46. № 16. https://doi.org/10.1093/nar/gky648
Liberi G., Maffioletti G., Lucca C. et al. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase // Genes Dev. 2005. V. 19. P. 339–350. https://doi.org/10.1101/gad.322605
Branzei D., Sollier J., Liberi G. et al. Ubc9- and mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks // Cell. 2006. V. 127. P. 509–522. https://doi.org/10.1016/j.cell.2006.08.050
Minca E.C., Kowalski D. Multiple Rad5 activities mediate sister chromatid recombination to bypass DNA damage at stalled replication forks // Mol. Cell. 2010. V. 38. P. 649–661. https://doi.org/10.1016/j.molcel.2010.03.020
Ball L.G., Zhang K., Cobb J.A. et al. The yeast Shu complex couples error-free post-replication repair to homologous recombination // Mol. Microbiol. 2009. V. 73. P. 89–102. https://doi.org/10.1111/j.1365-2958.2009.06748.x
Gonzalez-Huici V., Szakal B., Urulangodi M. et al. DNA bending facilitates the error-free DNA damage tolerance pathway and upholds genome integrity // EMBO J. 2014. V. 33. P. 327–340. https://doi.org/10.1002/embj.201387425
Kamau E., Bauerle K.T., Grove A. The Saccharomyces cerevisiae high mobility group box protein HMO1 contains two functional DNA binding domains // J. Biol. Chem. 2004. V. 279. P. 55234-55240. https://doi.org/10.1074/jbc.M409459200
Shor E., Weinstein J., Rothstein R. A genetic screen for top3 suppressors in Saccharomyces cerevisiae identifies SHU1SHU2PSY3 and CSM2: four genes involved in error-free DNA repair // Genetics. 2005. V. 169. P. 1275–1289. https://doi.org/10.1534/genetics.104.036764
Mankouri H.W., Ngo H.P., Hickson I.D. Shu proteins promote the formation of homologous recombination intermediates that are processed by Sgs1–Rmi1–Top3 // Mol. Biol. Cell. 2007. V. 18. P. 4062–4073. https://doi.org/10.1091/mbc.e07-05-0490
Xu X., Ball L., Chen W. et al. The yeast Shu complex utilizes homologous recombination machinery for error-free lesion bypass via physical interaction with a Rad51 paralogue // PLoS One. 2013. V. 8. № 12. https://doi.org/10.1371/journal.pone.0081371
Godin S., Wier A., Kabbinavar F. et al. The Shu complex interacts with Rad51 through the Rad51 paralogues Rad55–Rad57 to mediate error-free recombination // Nucl. Acids Res. 2013. V. 41. P. 4525–4534. https://doi.org/10.1093/nar/gkt138
Scheller J., Schurer A., Rudolph C. et al. MPH1, a yeast gene encoding a DEAH protein, plays a role in protection of the genome from spontaneous and chemically induced damage // Genetics. 2000. V. 155. P. 1069–1081.
Prakash R., Krejci L., Van Komen S. et al. Saccharomyces cerevisiae MPH1 gene, required for homologous recombination-mediated mutation avoidance, encodes a 3' to 5' DNA helicase // J. Biol. Chem. 2005. V. 280. № 9. P. 7854–7860. https://doi.org/10.1074/jbc.M413898200
Prakash R., Satory D., Dray E. et al. Yeast Mph1 helicase dissociates Rad-51made D-loop: implications for crossingover control in mitotic recombination // Genes Dev. 2009. V. 23. P. 67–79. https://doi.org/10.1101/gad.1737809
Ivanov E.L., Kovaltzova S.V., Korolev V.G. Saccharomyces cerevisiae mutants with enhanced induced mutation and altered mitotic gene conversion // Mutat. Res. 1989. V. 213. P. 105–115. https://doi.org/10.1016/0027-5107(89)90141-3
Blackwell Jr., Wilkinson S.T., Mosammaparast N., Pemberton L.F. Mutational analysis of H3 and H4 N termini reveals distinct roles in nuclear import // J. Biol. Chem. 2007. V. 282. P. 20142–20150.
Иванов Е.Л., Федорова И.В., Ковальцова С.В. Выделение и характеристика новых мутантов дрожжей Saccharomyces cerevisiae с повышенной спонтанной мутабильностью // Генетика. 1992. Т. 28. № 1. С. 47–55.
Федорова И.В., Ковальцова С.В., Иванов Е.Л. Влияние мутаций hsm, повышающих спонтанную мутабильность, на индуцированный мутагенез и митотическую рекомбинацию у дрожжей Saccharomyces cerevisiae // Генетика. 1992. Т. 28. № 1. С. 54–65.
Alekseev S.Yu., Kovaltzova S.V., Fedorova I.V. et al. HSM2 (HMO1) gene participates in mutagenesis control in yeast Saccharomyces cerevisiae // DNA Repair. 2002. V. 1. P. 287–297.
Kelberg E.P., Kovaltsova S.V., Alekseev S.Y. et al. HIM1, a new yeast Saccharomyces cerevisiae gene playing a role in control of spontaneous and induced mutagenesis // Mutat. Res. 2005. V. 578. P. 64–78. https://doi.org/10.1016/j.mrfmmm.2005.03.003
Ковальцова С.В., Грачева Л.М., Евстюхина Т.А. и др. Гены-мутаторы дрожжей Saccharomyces cerevisiae. Взаимодействие him и hsm с мутациями, блокирующими три основные пути репарации индуцированных повреждений ДНК // Генетика. 1996. Т. 32. № 7. С. 927–932.
Fedorova I.V., Gracheva L.M., Kovaltzova S.V. et al. The yeast HSM3 gene acts in one of the mismatch repair pathways // Genetics. 1998. V. 148. P. 963–973.
Inbar O., Liefshitz B., Bitan G., Kupiec M. The relationship between homology length and crossing over during the repair of a broken chromosome // J. Biol. Chem. 2000. V. 275. P. 30833–30838. https://doi.org/10.1074/jbc.C000133200
Ge Z., Wang H., Parthun M.R. Nuclear Hat1p complex (NuB4) components participate in DNA repair-linked chromatin reassembly // J. Biol. Chem. 2011. V. 286. P. 16790–16799.
Le Tallec B., Barrault M.-B., Guerois R. et al. Hsm3/S5b participates in the assembly pathway of the 19S regulatory particle of the proteasome // Mol. Cell. 2009. V. 33. P. 389–399.
Funakoshi M., Tomko R.J., Jr., Kobayashi H., Hochstrasser M. Multiple assembly chaperones govern biogenesis of the proteasome regulatory particle base // Cell. 2009. V. 137. P. 887–899.
Черненков А.Ю., Иванова С.В., Ковальцова С.В. и др. Генетический анализ доменной структуры белка Hsm3 дрожжей Saccharomyces cerevisiae // Генетика. 2010. Т. 46. № 6. С. 742–749.
Gadal C., Labarre S., Djschiero C., Thuriaux P. Hmo1, an HMG-box protein, belongs to the yeast ribosomal DNA transcription system // EMBO J. 2002. V. 21. P. 5498–5507.
Черненков А.Ю., Федоров Д.В., Грачева Л.М. и др. Взаимодействие гена HSM3 с генами, инициирующими гомологичную рекомбинационную репарацию у дрожжей Saccharomyces cerevisiae // Генетика. 2012. Т. 48. № 3. С. 333–339.
Федоров Д.В., Ковальцова С.В, Евстюхина Т.А. и др. Ген HSM6 идентичен гену PSY4 дрожжей Saccharomyces cerevisiae // Генетика. 2013. Т. 49. № 3. C. 328–336.
Алексеева Е.А., Евстюхина Т.А., Пешехонов В.Т., Королев В.Г. Взаимодействие продукта гена HIM1 с геликазами SRS2 (RADH) и MPH1 дрожжей Saccharomyces cerevisiae // Цитология. 2018. Т. 60. № 7. С. 555–557. https://doi.org/10.31116/tsitol.2018.07.13
Xiao L., Williams A.M., Grove A. The C-terminal domain of yeast high mobility group protein HMO1 mediates lateral protein accretion and in-phase DNA bending // Biochemistry. 2010. V. 49. P. 4051–4059. https://doi.org/10.1021/bi1003603
Bernstein K.A., Reid R.J.D., Sunjevaric I. et al. The Shu complex, which contains Rad51 paralogues, promotes DNA repair through inhibition of the Srs2 anti-recombinase // Mol. Biol.Cell. 2011. V. 22. P. 1599–1607.
Ковальцова С.В., Грачева Л.М., Евстюхина Т.А. и др. Гены-мутаторы дрожжей Saccharomyces cerevisiae. Взаимодействие между him и hsm генами // Генетика. 1996. Т. 32. № 7. С. 927–932.
Stafa A., Donnianni R.A., Timashev L.A. et al. Template switching during break-induced replication is promoted by the Mph1 helicase in Saccharomyces cerevisiae // Genetics. 2014. V. 196. P. 1017–1028. https://doi.org/10.1534/genetics.114.162297
Zheng X.-F., Prakash R., Saro D. et al. Processing of DNA structures via DNA unwinding and branch migration by the S. cerevisiae Mph1 protein // DNA Repair (Amst.). 2011. V. 10. № 10. P. 1034–1043. https://doi.org/10.1016/j.dnarep.2011.08.002
Dohrmann P.R., Sclafani R.A. Novel role for checkpoint Rad53 protein kinase in the initiation of chromosomal DNA replication in Saccharomyces cerevisiae // J. Gen. Soc. of America. 2006. № 174. P. 87–99. https://doi.org/0.1534/genetics.106.060236
Chabes A., Georgieva B., Domkin V. et al. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase // Cell. 2003. V. 112. P. 391–401. https://doi.org/10.1016/s0092-8674(03)00075-8
Дополнительные материалы отсутствуют.