

Генетика, 2021, T. 57, № 4, стр. 445-453
Исследование разнообразия гидробионтов Лиственничного залива озера Байкал с использованием ДНК-метабаркодинга
Л. С. Кравцова 1, *, Т. Е. Перетолчина 1, Т. И. Трибой 1, И. А. Небесных 1, А. Б. Купчинский 2, А. Е. Тупикин 3, М. Р. Кабилов 3
1 Лимнологический институт Cибирского отделения Российской академии наук
664033 Иркутск, Россия
2 Байкальский музей Иркутского научного центра Cибирского отделения Российской академии наук
664520 Иркутская область, пос. Листвянка, Россия
3 Институт химической биологии и фундаментальной медицины Cибирского отделения
Российской академии наук
630090 Новосибирск, Россия
* E-mail: lk@lin.irk.ru
Поступила в редакцию 30.04.2020
После доработки 07.09.2020
Принята к публикации 01.10.2020
Аннотация
Исследовано таксономическое разнообразие гидробионтов Лиственничного залива оз. Байкал с помощью ДНК-метабаркодинга. Расшифровка нуклеотидных последовательностей фрагмента гена (313 пн) митохондриальной ДНК, кодирующего первую субъединицу цитохром с оксидазы (СОI), после амплификации с внутренних праймеров (mlCOIintF, jgHCO2198), проведена с использованием Illumina MiSeq. Биоинформатический анализ прочтений СОI позволил выявить 187 ОТЕ (операционных таксономических единиц), среди которых преобладают Arthropoda, Annelida, Mollusca, Plathelminthes, на их долю приходится соответственно 52, 30, 10, 4%. В целом видовое разнообразие фауны залива по показателю Шеннона (3.2 бит) сопоставимо с другими районами Байкала. Наиболее разнообразный состав таксонов (22 ОТЕ) видового уровня приходится на личинок хирономид, что подтверждает эффективность метода для идентификации ювенильных стадий беспозвоночных животных. Результаты исследования служат первым шагом на пути совершенствования системы гидробиологического мониторинга оз. Байкал, основанного на синтезе молекулярно-генетической диагностики беспозвоночных животных и классической морфологической таксономии.
В настоящее время в связи с глобальным изменением климата стал актуален вопрос как будет меняться биоразнообразие природных экосистем в пространстве и времени? Отклик морских и пресноводных водоемов на изменение природно-климатических факторов регистрируют во всем мире, одним из показателей служит таксономический состав гидробионтов [1–3]. В оз. Байкал обитают 2565 видов и подвидов животных, эндемики составляют около 60% [4]. В результате глобальных климатических изменений температура воздуха в Байкальском регионе увеличилась на 1.2°С, а воды в поверхностном слое оз. Байкал (в мае–сентябре) – на 1°С [5]. В связи с нарастающим региональным потеплением климата важны методы экспресс-оценки состояния фауны Байкала для выявления изменений в ее составе, особенно в эндемичном комплексе, адаптированном к обитанию в холодноводном озере. Однако исследование фаунистического разнообразия Байкала сопряжено с трудоемким процессом идентификации видов и необходимостью привлечения большого числа специалистов-морфологов. Хотя в изучении и сохранении биоразнообразия любого водоема по-прежнему базовой остается морфология видов, для оценки современного состояния байкальской фауны мы решили апробировать современный молекулярно-генетический метод – ДНК-метабаркодинг на основе высокопроизводительного секвенирования (next generation sequencing – NGS). Этот метод широко применяется для исследования разнообразия пресноводных и морских экосистем, ДНК-метабаркодинг можно рассматривать как альтернативу традиционной морфологической диагностике видов [6–11].
Преимущество ДНК-метабаркодинга заключается в том, что при анализе большого набора организмов не требуется выделения ДНК из отдельных индивидуумов или клонирования. К тому же одновременно можно получить большое количество нуклеотидных последовательностей для каждого организма, смешанного с другими в одной пробе. При этом мини-баркод фрагментов гена мтДНК, кодирующего первую субъединицу цитохром с оксидазы (СОI), позволяет идентифицировать виды с разрешением более 97–98% [12–14].
Цель работы заключается в исследовании таксономического разнообразия гидробионтов на основе ДНК-метабаркодинга на примере Лиственничного залива оз. Байкал.
МАТЕРИАЛЫ И МЕТОДЫ
Пробы зообентоса были собраны в 2019 г. в заливе Лиственничный. На глубинах 0.3–0.5 м донные отложения (крупная и мелкая галька с отдельными валунами) отбирали вручную с использованием сачка. На глубинах 2–5 м валуны, неокатанные обломки пород с щебнем, а также заиленный песок отбирали водолазы. Беспозвоночных животных с поверхности камней счищали щеткой в кювету с водой, из заиленного песка их извлекали путем флотации в насыщенном растворе сахара плотностью 1.12. Пробы промывали через сачок из мельничного сита № 23 и фиксировали 100%-ным этанолом. Всего было собрано 15 количественных проб зообентоса.
В лабораторных условиях пробы разбирали под микроскопом МБС-10 при увеличении 2 × 10, беспозвоночных животных очищали от остатков терригенного материала (детрита, песка), водорослей. Для выделения ДНК все организмы предварительно вымачивали (один час) в дистиллированной воде, причем от крупных особей (размерами более 5 мм) брали кусочки ткани по 2–3 мм. Беспозвоночных животных с однотипных донных отложений помещали в фарфоровую чашку и растирали пестиком с добавлением 2%-ного раствора CTAB (в пропорции: на 1 мм3 ткани 300 мкл CTAB). ДНК экстрагировали по модифицированной методике Дойла и Диксон [15]. Всего для метабаркодинга было подготовлено пять проб геномной ДНК (не менее 20 нг в каждой) беспозвоночных животных, собранных на разных типах донных отложений.
Амплификацию проводили с набором реактивов ПЦР с HS-Taq (“Биолабмикс”, г. Новосибирск www.biolabmix.ru) в 25 мкл реакционной смеси в термоциклере Bio-Rad T100 (Bio-Rad, США). Фрагмент гена COI амплифицировали с использованием праймеров mlCO1int F: ggw-acw-ggw-tga-acw-gtw-tay-ccy-cc [14] и igHCO2198: ta(I)- acy-tc(I)-ggr-tg(I)-ccr-aar-aay-ca [16].
Условия ПЦР при 30 циклах амплификации были следующими: денатурация 94°C – 1 мин (преденатурация – 5 мин), отжиг 50°C – 1 мин 30 с, элонгация 72°C – 1 мин 20 с (постэлонгация – 5 мин). Продукты реакции анализировали электрофоретически в 1%-ном агарозном геле. Фрагменты ожидаемого размера вырезали и очищали с помощью набора для элюции ДНК из агарозного геля (Biosilica, Новосибирск).
К ампликонам лигировали адаптеры с помощью набора NebNext Ultra II DNA Library Prep Kit (NEB). Полученные ДНК-библиотеки секвенировали в ЦКП “Геномика” СО РАН (ИХБФМ СО РАН) на секвенаторе MiSeq (Illumina), используя набор Reagent Kit v3 (2 × 300, Illumina).
Полученные парные последовательности анализировали с помощью UPARSE-скриптов [17], используя Usearch v11.0.667 [18]. Биоинформатическая обработка включала перекрывание парных прочтений, фильтрацию по качеству и длине, учет одинаковых последовательностей, отбрасывание синглетонов, удаление химер и получение ОТЕ (операционной таксономической единицы) с помощью алгоритма кластеризации UNOISE2 [19]. Таксономическую принадлежность последовательностей ОТЕ определяли с использованием алгоритмов SINTAX [20] и референсной базы MIDORI_UNIQUE_20180221_COI_SINTAX [21], а также BLAST. Анализ репрезентативности выборок ОТЕ проводили c помощью iNEXT 2.0.15 [22]. Все нуклеотидные последовательности выявленных ОТЕ были протестированы на наличие стоп-кодонов с использованием программы Emboss Transeq, размещенной на сервере: www.ebi.ac.uk.
Зависимость между численностью представителей высших таксонов (экз.) в пробах (до экстракции ДНК) и количеством прочтений идентифицированных ОТЕ оценивали по коэффициенту корреляции Спирмена. Видовое β-разнообразие фауны в Лиственничном заливе характеризовали по показателю Шеннона, а степень сходства населения разных биотопов – с помощью кластерного анализа (UPGMA), в качестве меры расстояния использовали коэффициент Жаккара. ОТЕ, представленные менее 10 прочтениями, в расчетах не использовали.
РЕЗУЛЬТАТЫ
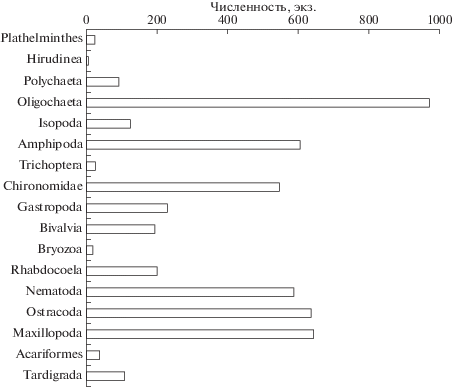
Всего в собранных пробах было найдено 5042 экз., из них 2803 экз. макробеспозвоночных (размеры >2 мм) и 2239 экз. мейобеспозвоночных животных (размеры <2 мм). Наиболее многочисленными были макробеспозвоночные Oligochaeta (972 экз.), Amphipoda (605 экз.), Chironomidae (546 экз.), Mollusca (225 экз.) и мейобеспозвоночные Ostracoda (642 экз.), Nematoda (588 экз.), Rhabdocoela (198 экз.). Зарегистрированы пять кладок и 62 кокона, а также фрагменты колоний губок. Всего в разобранных пробах (до экстракции ДНК) отмечены представители 17 высших таксонов (рис. 1).
Рис. 1.
Численность беспозвоночных животных в бентосных пробах из Лиственничного залива оз. Байкал до экстракции ДНК.
В результате высокопроизводительного секвенирования для каждого образца было получено не менее 75 тыс. парных чтений. Биоинформатический анализ данных позволил выявить 251 ОТЕ на 3% уровне различий между кластерами последовательностей видового ранга. Перед оценкой таксономического разнообразия из полученного массива были удалены: 10 ОТЕ, не соответствующих длине прочтения; 5 ОТЕ, имеющих стоп-кодоны; 49 ОТЕ, не идентифицируемых BLAST из-за неопределенности таксономического статуса. Нижняя граница гомологии ОТЕ высших таксонов с референсными последовательностями GenBank была принята на уровне 80% и выше.
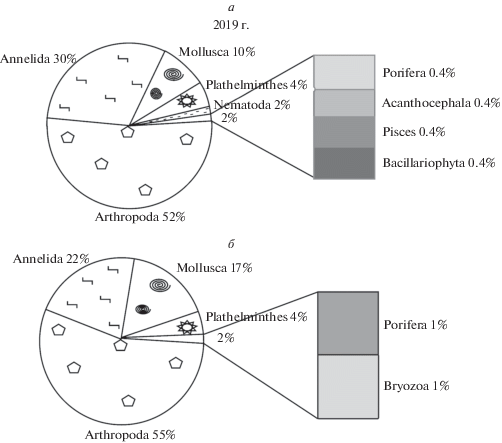
Среди идентифицированных таксонов Arthro-poda составляют 52% от общего количества ОТЕ, Annelida – 30%, Mollusca – 10%, Plathelminthes – 4%, Nematoda – 2%, представители других типов: губки, колючеголовые черви, рыбы и растительные организмы – <1% (рис. 2,а). Tardigrada и Bryozoa, найденные в бентосных пробах (рис. 1), в прочтениях короткого фрагмента СОI геномной ДНК беспозвоночных не отмечены.
Рис. 2.
Процентное соотношение надвидовых таксонов гидробионтов в Лиственничном заливе оз. Байкал. а – доля (%) таксонов в общем разнообразии, установленном в 2019 г. с использованием ДНК-метабаркодинга; б – доля (%) таксонов в общем разнообразии, установленном ранее классическими морфологическими методами [4].
Наблюдается прямая зависимость между численностью представителей высших таксонов в бентосных пробах (до экстракции ДНК) и количеством прочтений, коэффициент корреляции Спирмена составляет 0.49 (р < 0.05).
До видового и родового уровней (при гомологии ≥97 и ≥95% соответственно) идентифицированы 68 ОТЕ (табл. 1), на их долю приходится 55% общего количества (189497) прочтений фрагмента СОI (табл. 2). Причем зарегистрированные по морфологии в бентосных пробах и детектированные по СОI олигохеты рода Nais (Oligochaeta/Naididae), водяные ослики Baicalasellus (Isopoda), Cytherissa (Ostracoda), водяные клещи (Arachnida) не были включены в табл. 1, так как гомология с референсными последовательностями составляла от 80 до 95%.
Таблица 1.
Состав ОТЕ видового уровня, имеющих сходство с референсными последовательностями GenBank
| Таксон | Вид | № референсной последовательности из GenBank |
Сходство с референсной последовательносью из GenBank, % |
|---|---|---|---|
| Porifera | Baikalospongia intermedia Dyb. | KF835270 | 99.7 |
| Acanthocephala | Echinorhynchus cinctulus Porta | KP261014 | 98.6 |
| Polychaeta/Fabriciidae | Manayunkia baicalensis (Nusb.) | KF289881 | 99.6 |
| Manayunkia godlewskii (Nusb.) | KF289841 | 100 | |
| Manayunkia sp. | MK393734 | 95.9 | |
| Manayunkia zenkewitschii Sit., Shcherb. et Kharch. | KF289867 | 98.8 | |
| Oligochaeta | Lamprodrilus sp. | GU328642 | 95 |
| Lamprodrilus nigrescens Mich. | GU328643 | 91.6 | |
| Rhynchelmis alyonae Mart., Ferrag. et Kayg. | GU328669 | 98.3 | |
| Rhynchelmis sp. | AJ577632 | 95 | |
| Styloscolex chorioidalis Isoss. | AJ577636 | 96.2 | |
| Baikalodrilus sp. | MK984619 | 95.6 | |
| Rhyacodriloides sp. | FN796445 | 95 | |
| Teleuscolex baicalensis Grube | AJ577630 | 99 | |
| Hirudinea/Piscicolidae | Baicalobdella torquata Grube | AY336018 | 99.3 |
| Copepoda/Cyclopidae | Diacyclops arenosus (Mazep.) | KT075074 | 99.6 |
| Diacyclops incolotaenia (Mazep.) | KT075079 | 98.3 | |
| Amphipoda | Eulimnogammarus vittatus (Dyb.) | MK887750 | 99.7 |
| Baikalogammarus pullus (Dyb.) | FJ756303 | 99.7 | |
| Gmelinoides fasciatus (Stebb.) | KF478573 | 99.7 | |
| Linevichella vortex (Dyb.) | MN148355 | 99.7 | |
| Linevichella sp. | MN148355 | 95.4 | |
| Micruropus wahli (Dyb.) | AY926683 | 99.7 | |
| Pallaseopsis kessleri (Dyb.) | GQ919207 | 100 | |
| Pallaseopsis sp. | MG936047 | 95.7 | |
| Brandtia latissima lata (Dyb.) | FJ756301 | 97.2 | |
| Diptera/Chironomidae | Chironomus nipponensis Tokun. | LC096177 | 99.3 |
| Cricotopus bicinctus (Meig.) | JN275346 | 99.7 | |
| Cricotopus conf. fuscus | MN673037 | 99 | |
| Cricotopus rufiventris (Meig.) | HQ941597 | 99.3 | |
| Cricotopus sp. | JF870941 | 99.3 | |
| Micropsectra radialis Goetgh. | MT048255 | 99.3 | |
| Microtendipes sp. | LC329125 | 95 | |
| Orthocladius conf. ashei Sopon. | HQ941639 | 98.6 | |
| Orthocladius conf. glabripennis Goetgh. | LC462332 | 97.9 | |
| Orthocladius gregarius Linev. | KC879234 | 100 | |
| Orthocladius nitidoscutellatus Lundstr. | MT048224 | 98.6 | |
| Orthocladius sp. | HM421332 | 95.5 | |
| Orthocladius sp. 1 | KC894910 | 100 | |
| Orthocladius sp. 2 | KT248920 | 100 | |
| Orthocladius sp. 3 | KC894908 | 99.3 | |
| Orthocladius sp. 4 | KC894911 | 99.3 | |
| Diptera/Chironomidae | Orthocladius sp. 5 | KC894907 | 100 |
| Pagastia orientalis (Tshern.) | KY640356 | 97.6 | |
| Paratanytarsus baicalensis (Tshern.) | MT020734 | 99.3 | |
| Polypedilum albicorne (Meig.) | MG949931 | 99.4 | |
| Prodiamesa olivacea (Meig.) | KT248930 | 99 | |
| Sergentia baicalensis Tshern. | AF116586 | 99.3 | |
| Trichoptera | Baicalina bellicosa Mart. | KR153132 | 99.7 |
| Baicalina reducta Mart. | KR153134 | 99.7 | |
| Baicalinella foliata (Mart.) | KR153101 | 99.7 | |
| Baicaloides ovalis (Mart.) | KR153105 | 99.7 | |
| Thamastes dipterus Hag. | KR153094 | 99.7 | |
| Mollusca/Bivalvia | Pisidium casertanum (Poli) | KF483386 | 99.7 |
| Pisidium henslowanum (Shepp.) | MN913398 | 98.9 | |
| Pisidium sp. | KF000182 | 99.4 | |
| Mollusca/Gastropoda | Baicalancylus sp. | KR822545 | 95 |
| Pseudancylastrum sibiricum (Gerstf.) | KR822557 | 100 | |
| Maackia herderiana (Lindh.) | KY697388 | 99.7 | |
| Parabaikalia dubiosa (Kozh.) | KT885121 | 99.7 | |
| Pseudobaikalia contabulata (W. Dyb.) | Z92987 | 99.7 | |
| Teratobaicalia duthiersii (W. Dyb) | KT885132 | 97.7 | |
| Benedictia baicalensis (Gerstf.) | Z92983 | 99 | |
| Choanomphalus amauronius Bourg. | Y14721 | 100 | |
| Choanomphalus aorus Bourg. | Y14718 | 97.5 | |
| Choanomphalus maacki Gerstf. | EF012168 | 99.7 | |
| Choanomphalus sp. | Y14722 | 95.8 | |
| Pisces/Cottidae | Batrachocottus baicalensis (Dyb.) | LC125703 | 99.7 |
Таблица 2.
Количество прочтений фрагмента гена СОI мтДНК для ОТЕ надвидовых таксонов гидробионтов, собранных в заливе Лиственничный (оз. Байкал, 2019 г.)
| Таксон | Глубина, м | |||
|---|---|---|---|---|
| 0.3–0.5 | 2–5 | |||
| Донные отложения | ||||
| галька, отдельные валуны | валуны | неокатанные обломки пород, щебень | заиленный песок | |
| Porifera/Lubomirskiidae | 0 | 104 | 2 | 0 |
| Plathelminthes/Tricladida | 0 | 26 | 101 | 0 |
| Plathelminthes/Rhabdocoela | 0 | 4 | 23 | 119 |
| Acanthocephala | 3 | 0 | 0 | 611 |
| Nematoda | 0 | 0 | 36 | 88 |
| Annelida | 12 | 2016 | 1185 | 83 |
| Annelida/Polychaeta/Fabriciidae | 14 | 4308 | 1968 | 943 |
| Annelida/Clitellata/Hirudinea | 0 | 37 | 0 | 0 |
| Annelida/Clitellata/Oligochaeta | 12 673 | 19 517 | 10 966 | 17 958 |
| Arthropoda | 26 | 18 | 21 | 255 |
| Arthropoda/Copepoda | 1 | 292 | 0 | 0 |
| Arthropoda/Cyclopidae | 47 | 659 | 0 | 3 |
| Arthropoda/Ostracoda | 164 | 274 | 0 | 1858 |
| Arthropoda/Isopoda | 3 | 384 | 10 | 0 |
| Arthropoda/Amphipoda | 837 | 8166 | 6335 | 720 |
| Arthropoda/Arachnida/Acariformes | 90 | 6 | 0 | 323 |
| Arthropoda/Insecta | 38 | 149 | 292 | 2057 |
| Arthropoda/Trichoptera/Apataniidae | 27 | 2131 | 1 | 0 |
| Arthropoda/Diptera/Chironomidae | 26 795 | 5170 | 8941 | 1860 |
| Mollusca/Gastropoda | 59 | 26 244 | 13 122 | 28 |
| Mollusca/Bivalvia | 29 | 795 | 7709 | 725 |
| Chordata/Cottidae | 0 | 24 | 0 | 0 |
| Bacillariophyta | 0 | 9 | 3 | 0 |
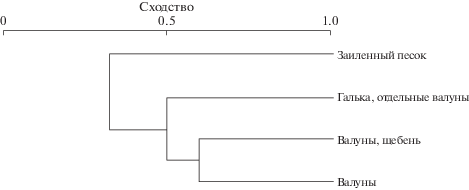
Судя по коэффициенту Жаккара, состав фауны (по идентифицированным ОТЕ) на валунах и неокатанных обломках пород с щебнем в зоне глубин 2–5 м сходен и значительно отличается от такового на заиленных песках (рис. 3). Это соответствует общим закономерностям пространственного распределения гидробионтов в прибрежной зоне оз. Байкал.
Рис. 3.
Фаунистическое сходство донного населения разных биотопов Лиственничного залива оз. Байкал в летний период (2019 г.). Кластерный анализ (на основе идентифицированных ОТЕ) проведен методом UPGMA с использованием коэффициента Жаккара в качестве меры расстояния.
Видовое β-разнообразие беспозвоночных животных в Лиственничном заливе по показателю Шеннона составляет 3.2 бит.
ОБСУЖДЕНИЕ
Митохондриальный ген СОI в начале 2000 гг. был принят за стандарт таксономического баркода большинства групп животных организмов [23]. Несмотря на универсальность праймеров (LCO1490 и HCO2198) фолмеровского фрагмента митохондриального гена СОI [24] при реконструкции филогении возникали проблемы с таксономическим статусом некоторых видов беспозвоночных животных, в частности хирономид [25], моллюсков Gastropoda: Cypraeidae [26] и др. Для получения лучшего разрешения на дереве или филогенетического сигнала многие исследователи стали использовать разные, наиболее вариабельные участки гена СОI. Например, для морских нематод в качестве ДНК-баркода использовали районы M1–M6 и I3–M11 [27]. В последнее время, с появлением NGS-технологии, для оценки разнообразия Metazoa стал популярен мини-баркод с внутренних праймеров (mlCOIintF в комбинации с jgHCO2198) фолмеровского фрагмента СОI длиной 313 пн [11, 14, 16]. Проведенные нами в 2019 г. исследования также показали эффективность применения данных праймеров для беспозвоночных, населяющих Лиственничный залив (табл. 1, 2; рис. 2,а). Выявленное в заливе разнообразие фауны по показателю Шеннона (3.2 бит) сравнимо с таковым в других районах оз. Байкал (в бухте Большие Коты, у восточного побережья в районе р. Утулик–р. Хара-Мурина), установленным с помощью классического морфологического анализа [28, 29].
В составе донного населения Лиственничного залива преобладают, как и ранее [4], представители Arthropoda, Annelida, Mollusca, Plathelminthes (рис. 2,a, б). Наиболее разнообразен состав ОТЕ членистоногих, особенно хирономид (табл. 1), несмотря на то что среди них были многочисленны личинки младших возрастов, не поддающиеся видовой диагностике по морфологии. Размеры их составляли до 2 мм, а численность более 300 экз. Тем не менее по мини-баркоду фрагментов СОI выявлены 22 ОТЕ видового уровня, при гомологии прочитанных последовательностей с представленными в GenBank от 97 до 100%. Аналогичная картина наблюдается у личинок ручейников, среди которых по ДНК-метабаркодингу идентифицированы пять видов с гомологией 99.7% с последовательностями из GenBank. Это свидетельствует о преимуществе ДНК-метабаркодинга по сравнению с классической таксономией. Большинство видов амфипод, олигохет, полихет и других беспозвоночных, приведенных в списке (табл. 1), встречались в Лиственничном заливе в предыдущие годы исследований [4]. Таким образом, ДНК-метабаркодинг с внутренних праймеров СОI (mlCOIintF, jgHCO2198) позволяет оценить видовое разнообразие гидробионтов как половозрелых, так и ювенильных стадий, идентификация которых морфологическими методами часто не представляется возможной. Особенно это касается личинок амфибиотических насекомых, а также кладок беспозвоночных животных. О чувствительности метода ДНК-метабаркодинга свидетельствует наличие в массиве данных не только последовательностей рыб Batrachocottus baicalensis (Dyb.), типичного обитателя литорали Байкала [4], но и заражающих их червей Echinorhynchus cinctulus Porta, часто встречающихся в пищеварительном тракте большеголовой широколобки. Непосредственно рыбы B. baicalensis не были обнаружены, но присутствие их в смеси геномной ДНК беспозвоночных обусловлено попаданием в пробы отдельных икринок вместе с коконами.
Несмотря на перечисленные выше преимущества ДНК-метабаркодинга, для Tardigrada из Лиственничного залива (рис. 1) ОТЕ не идентифицированы. Вероятно, для них приемлем другой маркер (18S), поскольку они также не были детектированы по COI при исследовании мейофауны фьордов западного побережья Швеции на основе NGS [30]. Что касается Bryozoa, в работе [30] показано, что они могут быть детектированы по короткому фрагменту COI, однако мы не получили ни одного прочтения. Для выяснения этого обстоятельства необходимы дополнительные исследования, поскольку для большинства байкальских мейобеспозвоночных референсные последовательности СОI отсутствуют в GenBank. К тому же Bryozoa, возможно, не были идентифицированы нами из-за плохой экстракции ДНК, так как в пробах встречались лишь остатки колоний (18 экз.) в виде хитиновых шипов, окружающих ротовое отверстие мшанок.
В целом ДНК-метабаркодинг по короткому фрагменту СОI позволяет оценить локальное разнообразие фауны Лиственничного залива, отмечены представители 42 родов, 31 семейства, 9 типов. Разнообразие донного населения в 2019 г. сопоставимо с таковым в прошлом столетии [4]. Результаты проведенных исследований указывают на необходимость создания базы данных референсных последовательностей СОI байкальских гидробионтов. Полученные последовательности могут быть использованы для совершенствования системы мониторинга оз. Байкал, для исследования связей таксономического состава и обилия гидробионтов с трендами природно-климатических факторов.
Работа выполнена по проекту Российского фонда фундаментальных исследований № 19-05-00398_а; частично сбор материала проведен в рамках проекта № 0345–2019–0004 (АААА-А16-116122110060-9) по теме: “Комплексное исследование эволюционных процессов у бентосных организмов в условиях резких изменений экосистемы Байкала”; секвенирование образцов проведено в ЦКП “Геномика” СО РАН (ИХБФМ СО РАН).
Все применимые международные, национальные и/или институциональные принципы ухода и использования животных были соблюдены.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Hampton S.E., Mc Gowan S., Ozersky T. et al. Recent ecological change in ancient lakes // Limnology and Oceanography. 2018. V. 63. № 5. P. 2277–2304.
Hsieh C.H., Sakai Y., Ban S. et al. Eutrophication and warming effects on long-term variation of zooplankton in Lake Biwa // Biogeosciences. 2011. V. 8. № 5. P. 1383–1399.
Cohen A.S., Gergurich E.L., Kraemer B.M. et al. Climate warming reduces fish production and benthic habitat in Lake Tanganyika, one of the most biodiverse freshwater ecosystems // Proc. Natl Acad. Sci. USA. 2016. V. 113. № 34. P. 9563–9568.
Аннотированный список фауны озера Байкал и его водосборного бассейна: В 2 томах. Т. 1. Озеро Байкал. Новосибирск: Наука, 2004. 1679 с.
Шимараев М.Н., Куимова Л.Н., Синюкович В.Н., Цехановский В.В. О проявлении на Байкале глобальных изменений климата в ХХ столетии // ДАН. 2002. № 3. С. 397–400.
Hajibabaei M., Shokralla S., Zhou X. et al. Environmental barcoding: a next-generation sequencing approach for biomonitoring applications using river benthos // PLoS One. 2011. V. 6. P. e17497.
Janzen D.H., Hajibabaei M., Burns J.M. et al. Wedding biodiversity inventory of a large and complex Lepidoptera fauna with DNA barcoding // Philosoph. Transactions Royal Soc. London. Series B. Biol. Sci. 2005. V. 360. P. 1835–1845.
Porazinska D.L., Giblin-Davis R.M., Faller L. et al. Evaluating high-throughput sequencing as a method for metagenomic analysis of nematode diversity // Mol. Ecol. Resources. 2009. V. 9. P. 1439–1450.
Aylagas E., Borja Á., Rodríguez-Ezpeleta N. Environmental status assessment using DNA metabarcoding: Towards a genetics based marine biotic index (gAMBI) // PLoS One. 2014. V. 9. P. e90529.
Elbrecht V., Vamos E.E., Meissner K. et al. Assessing trengths and weaknesses of DNA metabarcoding-based macroinvertebrate identification for routine stream monitoring // Methods Ecol. Evol. 2017. № 8. P. 1265–1275.
Kuntke F., de Jonge N., Hesselsøe M., Nielsen J.L. Stream water quality assessment by metabarcoding of invertebrates // Ecol. Indicators. 2020. V. 111. e 105982.
Hajibabaei M., Singer G.A., Clare E.L., Hebert P.D.N. Design and applicability of DNA arrays and DNA barcodes in biodiversity monitoring // BMC Biology. 2007. V. 5. P. 24.
Meusnier I., Singer G.A., Landry J.F. et al. A universal DNA mini-barcode for biodiversity analysis // BMC Genomics. 2008. V. 9. P. 214.
Leray M., Yang J.Y., Meyer C.P. et al. A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: application for characterizing coral reef fish gut contents // Frontiers in Zoology. 2013. V. 10. P. 34.
Doyle J.J., Dickson E. Preservation of plant samples for DNA restriction endonuclease analysis // Taxon. 1987. V. 36. P. 715–722.
Geller J.B., Meyer C.P., Parker M. et al. Redesign of PCR primers for mitochondrial cytochrome c oxidase subunit I for marine invertebrates and application in all-taxa biotic surveys // Mol. Ecol. Resources. 2013. V. 13. № 5. P. 851–861.
Edgar R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads // Nature Methods. 2013. V. 10. P. 996–998.
Edgar R.C. Search and clustering orders of magnitude faster than BLAST // Bioinformatics. 2010. V. 26. № 19. P. 2460–2461.
Edgar R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon reads // bioRxiv. 2016. https://doi.org/10.1101/081257
Edgar R.C. SINTAX, a Simple Non-Bayesian Taxonomy Classifier for 16S and ITS Sequences // bioRxiv. 2016. https://doi.org/10.1101/074161
Machida R.J., Leray M., Ho S.-L., Knowlton N. Metazoan mitochondrial gene sequence reference datasets for taxonomic assignment of environmental samples // Scientific Data. 2017. V. 4. P. 170027.
Hsieh T.C., Ma K.H., Chao A. iNEXT: an R package for rarefaction and extrapolation of species diversity (Hill numbers) // Methods Ecol. Evol. 2016. V. 7. P. 1451–1456.
Hebert P.D.N., Cywinska A., Ball S.L., De Waard J.R. Biological identifications through DNA barcodes // Proc. Royal Soc. London. Series B. 2003. V. 270. P. 313–321.
Folmer O., Black M.B., Hoeh W.R. et al. DNA primer for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates // Mol. Marine Biol. Biotechnol. 1994. V. 3. P. 294–299.
Ekrem T., Stur E., Hebert P.D.N. Females do count: Documenting Chironomidae (Diptera) species diversity using DNA barcoding // Organisms Diversity and Evolution. 2010. № 10. P. 397–408.
Meyer C.P. Molecular systematics of cowries (Gastro-poda: Cypraeidae) and diversification patterns in the tropics // Biol. J. Linnean Society. 2003. V. 79. P. 401–459.
Derycke S., Vanaverbeke J., Rigaux A. et al. Exploring the use of cytochrome oxidase c subunit 1 (COI) for DNA barcoding of free living marine nematodes // PLoS One. 2010. V. 5. P. e13716.
Кравцова Л.С., Потемкина Т.Г., Механикова И.В. и др. Пространственное распределение бентосных сообществ беспозвоночных животных в южной котловине озера Байкал // Зоол. беспозвоночных. 2006. Т. 3. Вып. 1. С. 65–76.
Kravtsova L.S., Kamaltynov R.M., Karabanov E.B. et al. Macrozoobenthic communities of underwater landscapes in the shallow-water zone of southern Lake Baikal // Hydrobiologia. 2004. V. 522. P. 193–205.
Haenel Q., Holovachov O., Jondelius U. et al. NGS-based biodiversity and community structure analysis of meiofaunal eukaryotes in shell sand from Hållö Island, Smögen, and soft mud from Gullmarn Fjord, Sweden // Biodiversity Data J. 2017. V. 5. P. e12731.
Дополнительные материалы отсутствуют.