

Генетика, 2021, T. 57, № 5, стр. 508-515
Роль генов фосфоламбана (PLN), триадина (TRDN) и джунктина (ASPH) в развитии сократительной дисфункции миокарда
Э. Ф. Муслимова 1, *, Т. Ю. Реброва 1, Д. С. Кондратьева 1, С. А. Афанасьев 1
1 Научно-исследовательский институт кардиологии, Томский национальный
исследовательский медицинский центр Российской академии наук
634012 Томск, Россия
* E-mail: muslimovef@yandex.ru
Поступила в редакцию 09.06.2020
После доработки 30.07.2020
Принята к публикации 25.08.2020
Аннотация
Обзор охватывает ряд исследований, посвященных оценке роли генов и белков фосфоламбана (PLN), триадина (TRDN), джунктина (ASPH) в развитии сократительной дисфункции миокарда. Представлены результаты, отражающие влияние изменений в структуре и экспрессии указанных генов на функционирование Ca2+-транспортирующей системы саркоплазматического ретикулума кардиомиоцитов.
Развитие хронической сердечной недостаточности (ХСН) у лиц с заболеваниями сердечно-сосудистой системы является серьезным фактором, ограничивающим качество жизни в трудоспособном возрасте и препятствующим активному долголетию. Признается, что существующих знаний недостаточно для понимания патогенеза сократительной дисфункции кардиомиоцитов и для разработки новых способов профилактики и лечения ХСН [1, 2]. С этой позиции изучение молекулярных механизмов регуляции внутриклеточного гомеостаза ионов кальция Ca2+ может выявить новые мишени для направленного терапевтического воздействия [3].
Хорошо известно, что состоятельность обмена Ca2+ в саркоплазматическом ретикулуме (СР) во время цикла сокращения–расслабления играет ключевую роль в реализации сократительной функции кардиомиоцитов. Одними из наиболее важных и изученных Ca2+-транспортирующих белков СР являются Ca2+-АТФаза SERCA2a, осуществляющая перенос ионов кальция из миоплазмы через мембрану СР, кальсеквестрин CASQ2, связывающий большую часть Ca2+, поступающего в СР из миоплазмы во время диастолы, и рианодиновые рецепторы RyR2, освобождающие Ca2+ из СР во время систолы [4, 5]. Отмечено, что нарушение экспрессии и активности данных белков имеет серьезные последствия для нормальной работы сердца, провоцирует развитие сердечной недостаточности и нарушений ритма сердца [6–8].
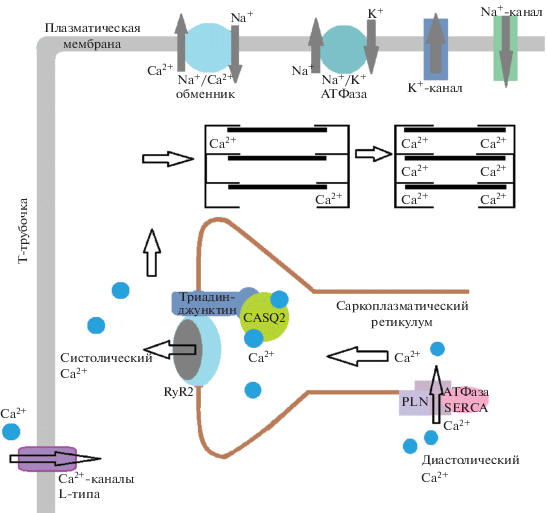
Однако нормальное функционирование Ca2+-транспортирующей системы СР зависит и от других белков, которые регулируют работу SERCA2a, CASQ2, RyR2 (рис. 1). К таким белкам, в первую очередь, относятся фосфоламбан, триадин и джунктин. Фосфоламбан – основной регулятор активности SERCA2a, а триадин и джунктин обеспечивают взаимодействие между RyR2 и CASQ2 [9, 10]. Следовательно, нарушения в функционировании и содержании фосфоламбана, триадина и джунктина также могут играть значимую роль в формировании ХСН. Кроме того, изменение любого компонента Ca2+-транспортирующей системы в кардиомиоцитах может привести к нарушению процесса возбудимости, электрической дисфункции миокарда и аритмогенезу [11]. Одними из основных факторов, определяющих представленность в клетке тех или иных белков и влияющих на их функциональную состоятельность, являются уровень экспрессии соответствующих генов, а также их полиморфные варианты.
Рис. 1.
Схема Ca2+-транспортирующей системы саркоплазматического ретикулума кардиомиоцитов с обозначением ключевых белков: Ca2+-АТФазы SERCA, фосфоламбана (PLN), рианодиновых рецепторов RyR, кальсеквестрина (CASQ2), триадина (triadin), джунктинa (junctin) (в соответствии с [4, 7, 9]).
В данном обзоре мы постарались представить результаты различных исследований, посвященных полиморфным вариантам и особенностям экспрессии генов фосфоламбана, триадина и джунктина при сердечно-сосудистой патологии. Также в обзоре отражены данные работ по изучению влияния изменений в уровне экспрессии генов триадина, джунктина и фосфоламбана на работу Ca2+-транспортирующей системы саркоплазматического ретикулума кардиомиоцитов.
ФОСФОЛАМБАН
Фосфоламбан регулирует активность SERCA2a в кардиомиоцитах. Показано, что дефосфорилированная форма фосфоламбана, существующая в основном в виде мономера, связывает и ингибирует активность SERCA2a [12]. В этом состоянии у SERCA2а снижается сродство к ионам Ca2+. В результате нарушается удаление свободных ионов Ca2+ из цитозоля и становится невозможным полноценное расслабление, а затем и сокращение кардиомиоцитов и миокарда в целом [10, 13]. Активность белка критически регулируется фосфорилированием цАМФ-зависимой протеинкиназой и Ca2+-CaM-зависимой протеинкиназой в ответ на бета-адренергическую стимуляцию.
У человека белок фосфоламбан кодируется геном PLN (MIM 172405, ENSG00000198523), локализованным на хромосоме 6 (6q22.31). Структура гена включает два экзона и охватывает 13.42 тпн [14, 15]. Сравнительные исследования показали, что ген PLN человека структурно близок аналогичному гену кролика, крысы и мыши [16]. Однако значимость фосфоламбана в регулировании сократительной функции сердца имеет некоторые видовые различия. Так, мыши с дефицитом фосфоламбана за счет полного удаления кодирующей области гена фенотипически выглядели нормально, а их пульс и кровяное давление значимо не отличались от животных дикого типа. Более того, в условиях изолированной перфузии сердце мыши с дефицитом фосфоламбана характеризовалось даже повышенными значениями сократительных показателей [17]. Однако в отличие от мышей для человека необходимо поддержание нормальной экспрессии гена PLN. Нарушение работы гена может приводить к летальной сердечной недостаточности [18].
Наглядным примером незаменимой роли фосфоламбана для функционирования Ca2+-транспортирующей системы миокарда человека можно считать мутацию T116G в гене PLN, при которой отмечено снижение его экспрессии и отсутствие детектируемого белка в сердечной мышце. Следовательно, при такой мутации не было регулирующего воздействия фосфоламбана на SERCA2a. И если у гетерозиготных носителей наблюдалась гипертрофия без снижения сократительной способности, то у гомозиготных носителей уже в возрасте 16 и 27 лет развились дилатационная кардиомиопатия (ДКМП) и сердечная недостаточность, требующая трансплантации сердца [18]. Кроме того, показано, что пациенты с тяжелой ХСН (III–IV функциональный класс) и фракцией выброса левого желудочка (ЛЖ) менее 35% характеризовались сниженной экспрессией гена PLN в ткани ЛЖ по сравнению с более легкими больными с нормальной функцией ЛЖ. При этом экспрессия гена, кодирующего SERCA2a, также была снижена, а экспрессия гена мозгового натрийуретического пептида BNP, маркера сердечной недостаточности, повышена [19].
Согласно базе данных OMIM (MIM 172405), для клинической практики большое значение имеют изменения в структуре гена PLN, так как они могут приводить к наследственным формам кардиомиопатии. Для таких пациентов характерны увеличенные размеры камеры и нарушения сократительной функции сердца уже в 20–30 лет. Ярким примером является упомянутая ранее замена T116G, при которой происходят изменение стоп-кодона и снижение экспрессии гена PLN [18]. Ещe один пример – миссенс-мутация, обусловливающая замену аргинина на цистеин (R9C) в цитозольном домене фосфоламбана, которая приводила к аутосомно-доминантной ДКМП с сердечной недостаточностью. Средний возраст на момент смерти у пациентов составил 25–30 лет. Показано, что мутантный белок не ингибирует непосредственно SERCA2a [20]. Мутация R9C приводит к стабилизации пентамерной формы белка вследствие образования дисульфидного мостика между цитоплазматическими доменами отдельных субъединиц, следовательно нарушается диссоциация пентамера на мономеры [21].
Мутация R9C является примером нарушения структуры гена PLN, приводящего к патологии сердца за счет отсутствия ингибирующего влияния белка на SERCA2a. Другой вариант – делеция по аргинину R14Del в кодирующей области гена PLN – приводил к кардиомиопатии за счет необратимого хронического ингибирования SERCA2a измененным белком. В семье, в которой была обнаружена мутация, к среднему возрасту у гетерозиготных лиц (гомозигот по патологичному аллелю не выявлено) развились дилатация ЛЖ, сократительная дисфункция и эпизодические желудочковые аритмии. В эксперименте на трансгенных мышах был показан сходный эффект избыточной экспрессии мутантного белка, приводящей к преждевременной смерти [22].
Ещe один вариант – AF177763.1: g.203A>C в области промотора PLN – приводил к увеличению экспрессии гена. У гетерозиготных носителей этого варианта описана сердечная недостаточность в возрасте 18–44 лет с низкой фракцией выброса 22 ± 9% [23]. Известны и другие варианты гена PLN (табл. 1), связанные с развитием сердечной недостаточности и кардиомиопатий [24–26].
Таблица 1.
Варианты гена фосфоламбана, связанные с развитием кардиомиопатии и сердечной недостаточности
| Вариант | Эффект | Патология | Ссылки |
|---|---|---|---|
| rs111033560, T116G, L39X | Снижение экспрессии гена, белок не регулирует работу SERCA2a | Дилатационная кардиомиопатия | [18] |
| RCV000022714, -42C-G | Снижение экспрессии гена | Гипертрофическая кардиомиопатия | [25] |
| p.Glu2Ter, c.4G>T | Снижение экспрессии белка у гетерозигот, отсутствие у гомозиготы | Дилатационная кардиомиопатия | [26] |
| RCV000022715, -36A-C | Увеличение экспрессии гена | Сердечная недостаточность в молодом возрасте | [23] |
| RCV000022713, -77A-G | Увеличение экспрессии гена | Гипертрофическая кардиомиопатия | [24] |
| rs111033559, C25T, R9C | Белок не ингибирует SERCA2a | Дилатационная кардиомиопатия | [20, 21] |
| rs397516784, c.40_42del, R14Del | Белок необратимо ингибирует работу SERCA2a | Дилатационная кардиомиопатия | [22, 27] |
Проводились исследования, направленные на выявление связи вариаций числа копий ДНК (copy number variation, CNV) в регионе, затрагивающем ген PLN, с патологией сердца [26, 28]. Так, проводился анализ CNVs у детей с врожденными пороками сердца (ВПС) и была выявлена дупликация 6q22.31 (chr6: 118 693 553–119 050 523, 360 тпн). Однако вариант не был унаследован от родителей, и его клиническая значимость остается неизвестной [28]. В исследовании I. Mademont-Soler et al. [29] при скрининге CNVs выявили у двух пациентов с гипертрофической кардиомиопатией делеции всей кодирующей области гена PLN. Первый пациент имел делецию c.1-7587_159 + + 190del7936, включающую часть интрона и второго экзона гена. Вторая делеция PLN не была дополнительно охарактеризована. Эти варианты были классифицированы как патогенные [29].
Процесс поиска и более подробного изучения вариантов гена PLN не прекращается. Развитие технологий генной терапии позволяет надеяться на появление в будущем эффективных подходов к профилактике развития ХСН и лечению пациентов с нарушениями в структуре столь значимого для функционирования Ca2+-транспортирующей системы гена.
ТРИАДИН И ДЖУНКТИН
Белки триадин и джунктин обеспечивают взаимодействие между RyR2 и CASQ2 в рамках единой Ca2+-транспортирующей системы СР кардиомиоцитов [9, 30]. При этом установлено, что это взаимодействие является чувствительным к концентрации ионов Ca2+ в СР. В условиях низкой концентрации ионов кальция триадин и джунктин формируют прочный комплекс с белками CASQ2 и RyR2. В этом состоянии выброс Ca2+ из СР через рианодиновые рецепторы невозможен. Если же Ca2+ в СР слишком много, то этот комплекс менее прочен, благодаря чему RyR2 могут открыться и происходит выброс ионов в миоплазму.
Установлено, что у человека белок триадин кодируется геном TRDN (MIM 603283, ENSG00000186439), локализованным в 6-й (6q22.31) хромосоме [31]. Экзон 1 содержит инициирующий кодон, а экзон 41 содержит стоп-кодон и 1.3 тпн нетранслируемой последовательности. Для гена TRDN (420.612 тпн) характерен альтернативный сплайсинг, что обеспечивает синтез нескольких изоформ белка, по-разному представленных в скелетной и сердечной мышцах [32]. Для сердца специфична изоформа триадина CT1 (Cardiac Triadin), она же Trisk 32 (TRIadin Skeletal). Транскрипт (ENST00000546248.5) длиной в 1.179 пн содержит восемь экзонов [33].
Белок джунктин кодируется геном ASPH (MIM 600582, ENSG00000198363), который локализован в 8-й (8q12.3) хромосоме. Особенностью этого гена (214.037 тпн, 25 экзонов) является то, что кроме джунктина он определяет синтез белков аспартат бета-гидроксилазы и junctate. Для синтеза ASPH задействованы экзоны 1, 3, 5 и 8–16, тогда как для джунктина используются экзоны 2, 3, 5 и 6, а для junctate – экзоны 2–5 и 8–16. Белки имеют значительные различия в своих C-концевых доменах, что определяет их особенности в функциональных свойствах. При этом джунктин экспрессируется только в сердечной и скелетных мышцах и является частью Ca2+-транспортирующей системы СР, будучи связанным с кальсеквестрином [34, 35].
Триадин и джунктин обеспечивают взаимодействие между RyR2 и CASQ2, контролируя таким образом процесс выброса Ca2+ из СР в миоплазму при возбуждении кардиомиоцитов [9]. Следовательно есть основания считать, что уровень экспрессии как триадина, так и джунктина – один из критических факторов, влияющих на эффективность СР как компартмента, обеспечивающего внутриклеточные осцилляции ионов Ca2+ в ходе цикла сокращение–расслабление [36]. Сравнительное исследование трех трансгенных линий мышей показало, что сверхэкспрессия либо джунктина, либо триадина вызывает снижение экспрессии комплементарного белка и RyR2. В работе [37] были использованы линия мышей с повышенной экспрессией белка триадина, линия с повышенной экспрессией джунктина и линия с повышенной экспрессией обоих белков. Повышенная экспрессия триадина сопровождалась снижением содержания джунктина, и наоборот, однако добиться сочетанного выраженного увеличения содержания белков триадина и джунктина не удалось даже у мышей с увеличенной экспрессией обоих генов. У таких животных содержание белка триадина было в 2.9 раза выше, чем у мышей дикого типа, в то время как содержание белка джунктина оставалось на уровне дикого типа. Во всех трех линиях трансгенных мышей оказалась снижена экспрессия рианодиновых рецепторов, а вот экспрессия фосфоламбана и Serca2a не изменялась. Частота сердечных сокращений также была ниже во всех трех линиях мышей во время физической нагрузки и во время восстановления после тренировки по сравнению с мышами дикого типа. Но у мышей с повышенной экспрессией обоих белков стресс или физическая нагрузка были способны провоцировать желудочковую тахикардию при нормальных размерах ЛЖ и адекватной систолической функции сердца [37]. Повышенная в 5 раз экспрессия гена Trdn1, сопровождавшаяся увеличенным содержанием белка триадина, оказалась сопряжена со сниженными более чем наполовину уровнями экспрессии белков рианодиновых рецепторов и джунктина без изменения экспрессии кальсеквестрина, фосфоламбана и Serca2a у трансгенных мышей. У таких животных описаны гипертрофия сердца, нарушение расслабления и ослабление сократимости при увеличении нагрузки давлением. При этом максимальное укорочение кардиомиоцитов со сверхэкспрессией Trdn1 было снижено, к тому же трансгенным миоцитам потребовалось больше времени для расслабления, чем контрольным миоцитам [38]. При исследовании биологического материала человека также было обнаружено, что пациенты с ишемической и дилатационной кардиомиопатией отличались значительно повышенной экспрессией гена TRDN по сравнению с результатами, полученными при исследовании здорового миокарда лиц, погибших в результате травмы головы или субарахноидального кровоизлияния [39].
Однако у мышей с нокаутированным геном Trdn1, наряду с отсутствием белка триадина, снижается и содержание белков Casq2 и джунктина. При этом даже небольшое снижение содержания кальсеквестрина увеличивает утечку диастолического Ca2+, что приводит к аритмиям [40]. Показано, что скорость высвобождения Ca2+ из СР была значительно снижена, а диастолический Ca2+ и содержание Ca2+ в СР были увеличены в миоцитах Trdn–/– по сравнению с Trdn+/+ миоцитами. Стоит отметить, что у мышей Trdn–/– структурный анализ указывает на гораздо более слабую ассоциацию Casq2 с комплексом RyR2. Это может способствовать снижению скорости диффузии Ca2+ в просвете СР и высвобождения Ca2+, наблюдаемого в миоцитах [41]. В миокарде мышей, дефицитных по гену джунктина, не выявлено каких-либо значительных изменений уровней RyR, триадина или кальсеквестрина, а также в активности транспорта Ca2+ по сравнению с диким типом. Но в миоцитах Asph–/– амплитуда индуцированного кофеином высвобождения Са2+ была увеличена, что указывает на более высокое содержание Ca2+ СР, а время высвобождения Ca2+ было сокращено. В то же время у трансгенных мышей со сверхэкспрессией джунктина выявлены противоположные эффекты, т.е. сниженная сократительная функция. Важно отметить, что животные Asph–/– быстро погибали из-за аритмий, вызванных спонтанным высвобождением ионов Ca2+ из СР [42].
Для гена RYR2 и гена CASQ2 описаны варианты, сопряженные с нарушениями ритма сердца [3]. В связи с этим вполне ожидаемо, что и для гена TRDN были обнаружены варианты, носительство которых оказалось ассоциировано с риском развития аритмий и внезапной сердечной смерти [36]. Так, выявлены варианты гена TRDN (табл. 2), являющиеся основой для развития катехоламинергической полиморфной желудочковой тахикардии (КПЖТ) [43, 44]. Так, один из этих вариантов – миссенс-мутация T59R (c.176C<G в экзоне 2) – влияет на стабильность белка, а другой – нонсенс-мутацию Q205X (c.613C>T в экзоне 8) – приводит к преждевременной терминации синтеза белка. Также описана делеция (c.53_56delACAG) в экзоне 2 гена TRDN, вызвавшая сдвиг рамки считывания у двухлетнего пациента, умершего от остановки сердца из-за КПЖТ. Авторы, описавшие варианты гена TRDN, у этих же пациентов предприняли анализ структуры гена ASPH, кодирующего джунктин, но каких-либо вариантов, связанных с тахикардией, им обнаружить не удалось [43]. Обнаружена ассоциация варианта rs361508 в 3'-нетранслируемой области гена TRDN с риском внезапной сердечной смерти у пациентов с ХСН. Предположительно данный вариант может приводить к снижению экспрессии гена или к изменению связи с другими белками-партнерами. В рамках этой же работы проводился анализ вариантов rs4507756, rs7003147, rs6759 гена ASPH, но для них не выявлено связи с внезапной сердечной смертью [45].
Таблица 2.
Варианты гена TRDN, связанные с развитием сердечной недостаточности и аритмий
| Вариант | Эффект | Патология | Ссылки |
|---|---|---|---|
| rs397515459, c.176C>G, T59R | Нарушение стабильности, быстрая деградация белка | КПЖТ | [43] |
| rs397515458, c.613C>T, Q205X | Преждевременный стоп-кодон, короткий транскрипт | КПЖТ | [43] |
| rs768049331, c.53_56del |
Сдвиг рамки считывания, преждевременный стоп-кодон, потеря основной части белка | КПЖТ, синдром удлиненного интервала Q-T | [43, 46] |
| c.del 572_576 | Преждевременный стоп-кодон | Синдром удлиненного интервала Q-T | [46] |
| c.613C>T, p.Q205* | Преждевременный стоп-кодон | КПЖТ | [44] |
| c.22 + 29A>G | Нарушение сплайсинга, более длинный транскрипт, преждевременный стоп-кодон | КПЖТ | [44] |
| rs361508, c.*62G>A | Предположительно изменение экспрессии гена или связи с другими белками | Риск внезапной сердечной смерти у пациентов с ХСН | [45] |
Таким образом, исследование генов Ca2+-транспортирующей системы СР кардиомиоцитов может способствовать выявлению лиц с повышенным риском сердечной недостаточности и внезапной сердечной смерти. Однако особенности в экспрессии генов TRDN и ASPH и соответствующих белков у людей, в том числе в условиях патологии, по-прежнему остаются малоизученными. Исследования на биоптатах сердца человека могли бы прояснить, какие изменения в уровне экспрессии наиболее значимо влияют на функцию Ca2+-транспортирующей системы СР и риск неблагоприятного течения ХСН.
ЗАКЛЮЧЕНИЕ
В обзоре представлены данные исследований, посвященных изучению генов, кодирующих фосфоламбан, триадин и джунктин, в контексте сократительной функции миокарда и ее нарушений. В частности, показаны результаты, указывающие на значимую роль изменений структуры и экспрессии генов PLN, TRDN и ASPH в обеспечении нормальной работы Ca2+-транспортирующей системы СР и развитии сердечной недостаточности и аритмий.
Обзор подготовлен в рамках НИР НИИ кардиологии АААА-А15-115123110026-3-0.
Настоящая статья не содержит каких-либо исследований с использованием в качестве объекта животных.
Настоящая статья не содержит каких-либо исследований с участием в качестве объекта людей.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Тепляков А.Т., Березикова Е.Н., Шилов С.Н. и др. Оценка роли полиморфизма гена матриксной металлопротеиназы-3 в развитии хронической сердечной недостаточности // Терапевтический архив. 2015. Т. 87. № 4. С. 8–12. https://doi.org/10.17116/terarkh20158748-12
Свеклина Т.C., Шустов С.Б., Козлов В.А. и др. Протеомный анализ в диагностике хронической сердечной недостаточности с нормальной фракцией выброса // Вестник Северо-Западного гос. мед. ун‑та им. И.И. Мечникова. 2019. Т. 11. № 2. С. 79–90. https://doi.org/10.17816/mechnikov201911279-90
Реброва Т.Ю., Муслимова Э.Ф., Кондратьева Д.С. и др. Роль полиморфных вариантов генов Cа2+-АТФазы 2A (ATP2A2), рианодиновых рецепторов (RYR2) и кальсеквестрина (CASQ2) в развитии сердечной недостаточности // Генетика. 2018. Т. 54. № 6. С. 597–602. https://doi.org/10.7868/S0016675818060024
Faggioni M., Knollmann B.C. Calsequestrin 2 and arrhythmias // Am. J. Physiol.-Heart and Circulatory Physiol. 2012. V. 302. № 6. P. H1250–H1260. https://doi.org/10.1152/ajpheart.00779.2011
Dulhunty A.F., Beard N.A., Hanna A.D. Regulation and dysregulation of cardiac ryanodine receptor (RyR2) open probability during diastole in health and disease // J. Gen. Physiol. 2012. V. 140. № 2. P. 87–92. https://doi.org/10.1085/jgp.201210862
Salazar-Cantú A., Pérez-Treviño P., Montalvo-Parra D. et al. Role of SERCA and the sarcoplasmic reticulum calcium content on calcium waves propagation in rat ventricular myocytes // Archiv. Biochem. Biophys. 2016. V. 604. P. 11–19. https://doi.org/10.1016/j.abb.2016.05.018
Zarain-Herzberg A., García-Rivas G., Estrada-Avilés R. Regulation of SERCA pumps expression in diabetes // Cell Calcium. 2014. V. 56. № 5. P. 302–310. https://doi.org/10.1016/j.ceca.2014.09.005
Муслимова Э.Ф., Реброва Т.Ю., Кондратьева Д.С. и др. Экспрессия гена Са2+-АТФазы SERCA2a (ATP2A2) и гена рианодиновых рецепторов (RYR2) у больных хронической сердечной недостаточностью // Генетика. 2020. Т. 56. № 7. С. 819–825. https://doi.org/10.31857/S0016675820070103
Bers D.M. Macromolecular complexes regulating cardiac ryanodine receptor function // J. Mol. Cell. Cardiol. 2004. V. 37. № 2. P. 417–429. https://doi.org/10.1016/j.yjmcc.2004.05.026
Asahi M., Otsu K., Nakayama H. et al. Cardiac-specific overexpression of sarcolipin inhibits sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA2a) activity and impairs cardiac function in mice // PNAS. 2004. V. 101. № 25. P. 9199–9204. https://doi.org/10.1073/pnas.0402596101
Shemarova I.V., Nesterov V.P. Evolution of Ca2+-signaling mechanisms: the role of Ca2+ in regulation of specialized cardiomyocyte functions in chronic heart diseases // J. Evol. Biochem. and Physiol. 2014. V. 50. № 6. P. 420–427. https://doi.org/10.1134/S0022093014060027
Park W.J., Oh J.G. SERCA2a: a prime target for modulation of cardiac contractility during heart failure // BMB Reports. 2013. V. 46. № 5. P. 237–243. https://doi.org/10.5483/BMBRep.2013.46.5.077
Yokoe S., Asahi M. Phospholamban is downregulated by pVHL-mediated degradation through oxidative stress in failing heart // Int. J. Mol. Sci. 2017. V. 18. № 11. P. 2232. https://doi.org/10.3390/ijms18112232
Fujii J., Zarain-Herzberg A., Willard H.F. et al. Structure of the rabbit phospholamban gene, cloning of the human cDNA, and assignment of the gene to human chromosome 6 // The J. Biol. Chem. 1991. V. 266. P. 11669–11675.
Otsu K., Fujii J., Periasamy M. et al. Chromosome mapping of five human cardiac and skeletal muscle sarcoplasmic reticulum protein genes // Genomics. 1993. V. 17. № 2. P. 507–509. https://doi.org/10.1006/geno.1993.1357
McTiernan C.F., Frye C.S., Lemster B.H. et al. The human phospholamban gene: structure and expression // J. Mol. Cell. Cardiol. 1999. V. 31. № 3. P. 679–692. https://doi.org/10.1006/jmcc.1998.0904
Luo W., Grupp I.L., Harrer J. et al. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractilit and loss of beta-agonist stimulation // Circ. Res. 1994. V. 75. № 3. P. 401–409. https://doi.org/10.1161/01.RES.75.3.401
Haghighi K., Kolokathis F., Pater L. et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human // J. Clin. Invest. 2003. V. 111. № 6. P. 869–876. https://doi.org/10.1172/JCI17892
Vanderheyden M., Mullens W., Delrue L. et al. Myocardial gene expression in heart failure patients treated with cardiac resynchronization therapy responders versus nonresponders // J. Am. Coll. Cardiol. 2008. V. 51. № 2. P. 129–136. https://doi.org/10.1016/j.jacc.2007.07.087
Schmitt J.P., Kamisago M., Asahi M. et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban // Science. 2003. V. 299. № 5611. P. 1410–1413. https://doi.org/10.1126/science.1081578
Ha K.N., Masterson L.R., Hou Z. et al. Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase A // PNAS. 2011. V. 108. № 7. P. 2735–2740. https://doi.org/10.1073/pnas.1013987108
Haghighi K., Kolokathis F., Gramolini A.O. et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy // PNAS. 2006. V. 103. № 5. P. 1388–1393. https://doi.org/10.1073/pnas.0510519103
Haghighi K., Chen G., Sato Y. et al. A human phospholamban promoter polymorphism in dilated cardiomyopathy alters transcriptional regulation by glucocorticoids // Hum. Mutat. 2008. V. 29. № 5. P. 640–647. https://doi.org/10.1002/humu.20692
Minamisawa S., Sato Y., Tatsuguchi Y. et al. Mutation of the phospholamban promoter associated with hypertrophic cardiomyopathy // Biochem. Biophys. Res. Communicat. 2003. V. 304. № 1. P. 1–4. https://doi.org/10.1016/S0006-291X(03)00526-6
Medin M., Hermida-Prieto M., Monserrat L. et al. Mutational screening of phospholamban gene in hypertrophic and idiopathic dilated cardiomyopathy and functional study of the PLN –42C>G mutation // Eur. J. Heart Failure. 2007. V. 9. № 1. P. 37–43. https://doi.org/10.1016/j.ejheart.2006.04.007
Li Z., Chen P., Xu J. et al. A PLN nonsense variant causes severe dilated cardiomyopathy in a novel autosomal recessive inheritance mode // Intern. J. Cardiol. 2019. V. 279. P. 122–125. https://doi.org/10.1016/j.ijcard.2018.12.075
Karakikes I., Stillitano F., Nonnenmacher M. et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy // Nat. Commun. 2015. № 6. P. 6955. https://doi.org/10.1038/ncomms7955
Wu X., Li R., Fu F. et al. Chromosome microarray analysis in the investigation of children with congenital heart disease // BMC Pediatr. 2017. № 17. Article 117. https://doi.org/10.1186/s12887-017-0863-3
Mademont-Soler I., Mates J., Yotti R. et al. Additional value of screening for minor genes and copy number variants in hypertrophic cardiomyopathy // PLoS One. 2017. V. 12. № 8. e0181465. https://doi.org/10.1371/journal.pone.0181465
Chopra N., Knollmann B.C. Triadin regulates cardiac muscle couplon structure and microdomain Ca2+ signalling: a path towards ventricular arrhythmias // Cardiovascular Res. 2013. V. 98. № 2. P. 187–191. https://doi.org/10.1093/cvr/cvt023
Taske N.L., Eyre H.J., O’Brien R.O. et al. Molecular cloning of the cDNA encoding human skeletal muscle triadin and its localisation to chromosome 6q22–6q23 // FEBS J. 1995. V. 233. № 1. P. 258–265. https://doi.org/10.1111/j.1432-1033.1995.258_1.x
Thevenon D., Smida-Rezgui S., Chevessier F. et al. Human skeletal muscle triadin: gene organization and cloning of the major isoform, Trisk 51 // Biochem. Biophys. Res. Communicat. 2003. V. 303. № 2. P. 669–675. https://doi.org/10.1016/S0006-291X(03)00406-6
Marty I., Faur’e J., Fourest-Lieuvin A. et al. Triadin: what possible function 20 years later? // J. Physiol. 2009. V. 587. № 13. P. 3117–3121. https://doi.org/10.1113/jphysiol.2009.171892
Treves S., Feriotto G., Moccagatta L. et al. Molecular cloning, expression, functional characterization, chromosomal localization, and gene structure of junctate, a novel integral calcium binding protein of sarco(endo)plasmic reticulum membrane // The J. Biol. Chem. 2000. V. 275. P. 39555–39568. https://doi.org/10.1074/jbc.M005473200
Dinchuk J.E., Focht R.J., Kelley J.A. et al. Absence of post-translational aspartyl β-hydroxylation of epidermal growth factor domains in mice leads to developmental defects and an increased incidence of intestinal neoplasia // The J. Biol. Chem. 2001. V. 277. P. 12970–12977. https://doi.org/10.1074/jbc.M110389200
Wleklinski M.J., Kannankeril P.J., Knollmann B.C. Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia // The J. Physiology. 2020. V. 598. № 14. P. 2817–2834. Online Ver. https://doi.org/10.1113/JP276757
Kirchhof P., Klimas J., Fabritz L. et al. Stress and high heart rate provoke ventricular tachycardia in mice expressing triadin // J. Mol. Cell. Cardiol. 2007. V. 42. № 5. P. 962–971. https://doi.org/10.1016/j.yjmcc.2007.02.012
Kirchhefer U., Neumann J., Baba H.A. et al. Cardiac hypertrophy and impaired relaxation in transgenic mice overexpressing triadin 1 // The J. Biol. Chem. 2001. V. 276. P. 4142–4149. https://doi.org/10.1074/jbc.M006443200
Zhang L., Salgado-Somoza A., Vausort M. et al. A heart-enriched antisense long non-coding RNA regulates the balance between cardiac and skeletal muscle triadin // Biochim. Biophys. Acta (BBA) – Mol. Cell Res. 2018. V. 1865. № 2. P. 247–258. https://doi.org/10.1016/j.bbamcr.2017.11.002
Knollmann B.C. New roles of calsequestrin and triadin in cardiac muscle // The J. Physiology. 2009. V. 587. № 13. P. 3081–3087. https://doi.org/10.1113/jphysiol.2009.172098
Chopra N., Yang T., Asghari P. et al. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation–contraction coupling, and cardiac arrhythmias // PNAS. 2009. V. 106. № 18. P. 7636–7641. https://doi.org/10.1073/pnas.0902919106
Yuan Q., Fan G., Dong M. et al. Sarcoplasmic reticulum calcium overloading in junctin deficiency enhances cardiac contractility but increases ventricular automaticity // Circulation. 2007. V. 115. P. 300–309. https://doi.org/10.1161/CIRCULATIONAHA.106.654699
Roux-Buisson N., Cacheux M., Fourest-Lieuvin A. et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human // Hum. Mol. Genet. 2012. V. 21. № 12. P. 2759–2767. https://doi.org/10.1093/hmg/dds104
Rooryck C., Kyndt F., Bozon D. et al. New family with catecholaminergic polymorphic ventricular tachycardia linked to the triadin gene // J. Cardiovasc. Electrophysiol. 2015. V. 26. № 10. P. 1146–1150. https://doi.org/10.1111/jce.12763
Liu Z., Liu X., Yu H. et al. Common variants in TRDN and CALM1 are associated with risk of sudden cardiac death in chronic heart failure patients in chinese Han population // PLoS One. 2015. V. 10. № 7. P. e0132459. https://doi.org/10.1371/journal.pone.0132459
Altmann H.M., Tester D.J., Will M.L. et al. Homozygous/compound heterozygous triadin mutations associated with autosomal-recessive long-QT syndrome and pediatric sudden cardiac arrest. Elucidation of the triadin knockout syndrome // Circulation. 2015. V. 131. P. 2051–2060. https://doi.org/10.1161/CIRCULATIONAHA.115.015397
Дополнительные материалы отсутствуют.