

Генетика, 2021, T. 57, № 7, стр. 832-841
Спектр и новые патогенные варианты в гене RS1 у российских больных Х-сцепленным ретиношизисом
А. А. Степанова 1, *, Е. А. Иванова 1, В. В. Кадышев 1, А. В. Поляков 1
1 Медико-генетический научный центр им. академика Н.П. Бочкова
115478 Москва, Россия
* E-mail: cany@yandex.ru
Поступила в редакцию 26.08.2020
После доработки 13.10.2020
Принята к публикации 14.10.2020
Аннотация
Х-сцепленный ювенильный ретиношизис – наследственное заболевание глаз, относящееся к группе ретинальных дистрофий. В данной работе представлены результаты поиска патогенных вариантов в гене RS1 у 48 неродственных мужчин с диагнозом “Х-сцепленный ювенильный ретиношизис”. Молекулярно-генетический диагноз установлен у 77% пробандов. Выявлено 27 различных патогенных вариантов, в том числе восемь новых, ранее неописанных. Спектр мутаций в гене RS1 у российских больных характеризуется высоким разнообразием и отсутствием мажорных мутаций.
Х-сцепленный ювенильный ретиношизис (XLRS; MIM #312700) – наиболее частая причина ювенильной макулярной и ретинальной дегенерации у пациентов мужского пола. Дебют заболевания приходится на первую–вторую декаду жизни и характеризуется развитием прогрессирующих спицеподобных кистозных полостей в центральной зоне сетчатки и характерными изменениями на электроретинограмме (ЭРГ) (снижением b-волны). Фовеальный шизис (расщепление сетчатки), выглядящий как складки, исходящие из фовеа в виде колеса, является характерным признаком XLRS и наблюдается у 98–100% больных. Однако со временем этот признак может стать менее отчетливым [1, 2]. Периферический ретиношизис наблюдается примерно у 50% больных с умеренным сужением границ полей зрения [3].
Первый симптом заболевания – снижение остроты зрения, не поддающееся коррекции. Прогрессирование патологического процесса возможно вплоть до 40–45 лет [2]. На состояние зрительной функции у больных также оказывают влияние возникающие осложнения, такие как витреоретинальные кровоизлияния, неоваскулярная глаукома, хориоидальный склероз и, в редких случаях, атрофия хориоретинального слоя сетчатки, приводящая к слепоте.
Помимо офтальмоскопических изменений глазных доньев могут присутствовать косоглазие, нистагм, осевая гиперметропия, нарушение цветового зрения (красно-зеленая дисхроматопсия) и фовеальная эктопия [4].
Клинический диагноз в некоторых случаях нелегко предположить из-за широкого спектра фенотипов, которые включают дегенеративные изменения центральной сетчатки и вторичные осложнения. Даже среди членов одной семьи степень тяжести заболевания может варьировать от легкой до тяжелой формы болезни [5, 6].
Женщины, являющиеся гетерозиготными носительницами мутаций в гене RS1, обычно не имеют никаких клинических признаков болезни [7]. Однако в публикации M. Saldana [8] описана девочка с клинической картиной XLRS и вариантом c.305G>A (p.Arg102Gln) в гетерозиготном состоянии, других мутаций в гене RS1 выявлено не было, исследование неравномерной иннактивации Х-хромосомы провести не удалось.
Частота распространения ретиношизиса составляет от 1 : 5000 до 1 : 25 000 мужчин во всем мире [9].
Причина XLRS – мутации в гене RS1 [10]. Ген RS1 (MIM *300839) был идентифицирован в 1997 г., содержит шесть экзонов и кодирует специфичный для сетчатки белок ретиношизин, состоящий из 224 аминокислотных остатков, с молекулярной массой 24 кДа. Структура зрелого ретиношизина состоит из N-концевой лидерной последовательности (аминокислотные остатки 1–23), Rs1-домена (аминокислотные остатки 24–63), высококонсервативного дискоидинового домена (аминокислотные остатки 64–219) и С-терминального участка (аминокислотные остатки 220–224) (рис. 1). Лидерная последовательность расщепляется в просвете эндоплазматического ретикулума сигнальной пептидазой, затем обработанный белок собирается в дисульфид-связанный гомооктамерный комплекс для секреции из клеток. Октамер ретиношизина представлен в виде зубчатой кольцевой структуры, в которой домены Rs1 обращены внутрь, тогда как домены дискоидина и особенно их выпуклые шипы обращены наружу. На поверхности клетки два октамера ретиношизина собираются, образуя гексадекамер [11] (рис. 2).

Рис. 2.
Трехмерная структура ретиношизина [11]. а – мономер ретиношизина; б – двойная октамерная структура ретиношизина; в – два октамера ретиношизина образуют гексадекамер. Домены Rs1 показаны в виде гребня (синяя стрелка).
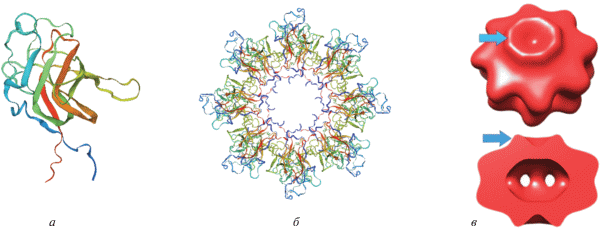
Дискоидиновые домены, впервые идентифицированные в белке дискоидина I Dictyostelium discoidium [12], в настоящее время обнаружены во многих секретируемых и трансмембранных белках, включая факторы свертывания крови, рецепторах тирозинкиназы и белках, участвующих в развитии нервной системы [13]. В некоторых белках дискоидиновый домен вовлечен в клеточную адгезию и передачу сигналов клетками через белок-белковые, белково-углеводные или белково-липидные взаимодействия [14]. Функционируя как белок клеточной адгезии, ретиношизин играет решающую роль в поддержании синаптической структуры сетчатки. Во взрослой сетчатке ретиношизин в основном экспрессируется в фоторецепторах палочек и колбочек, при секреции локализуется на поверхности фоторецепторов наружной сетчатки и биполярных клеток внутренней сетчатки [15].
На сегодняшний день в базе данных HGMD®Professional версия 2020.1 описано более 303 различных патогенных вариантов в гене RS1, вызывающих заболевание Х-сцепленный ретиношизис. Из них около 40% – мутации, приводящие к прекращению синтеза белка, LOF (loss of function), включая мутации сайта сплайсинга, нонсенс-мутации, мутации, приводящие к сдвигу рамки считывания. Оставшиеся 60% представляют собой миссенс-мутации, которые в основном влияют на секрецию ретиношизина и приводят к внутриклеточной деградации белка [16]. Мутации располагаются в гене неравномерно. Наибольшее число мутаций находится в экзонах 4, 5 и 6, кодирующих дискоидиновый домен. В большинстве случаев патогенез заболевания объясняется отсутствием зрелого белка ретиношизина на мембранах сетчатки. Однако при некоторых патогенных вариантах ретиношизин секретируется на поверхность клетки, тогда патогенез объясняется: 1) отсутствием октамеризации и неспецифическим связыванием с мембраной (замены p.Phe108Cys и p.Arg182Cys), 2) двойной октамеризацией без связывания с мембраной (p.Arg141His), 3) двойной октамеризацией и неспецифическим связыванием с мембраной (p. Arg141Gly, p.His207Gln и p.Arg209His) [17].
МАТЕРИАЛЫ И МЕТОДЫ
Для анализа были получены образцы крови от 48 неродственных больных мужского пола из различных регионов России. Больные были обследованы врачами-клиницистами и направлены в лабораторию ДНК-диагностики ФГБНУ “МГНЦ” с 2015 по 2020 г. для подтверждения диагноза (предварительный диагноз – ретиношизис).
Выделение геномной ДНК из лейкоцитов периферической крови выполняли с помощью набора реактивов Wizard® Genomic DNA Purification Kit (Promega, США) по протоколу производителя.
У всех больных проводился поиск патогенных вариантов методом прямого автоматического секвенирования по Сенгеру всей кодирующей последовательности и экзон-интронных соединений гена RS1 (NM_000330.3). Праймеры были выбраны из фланкирующих экзоны гена нуклеотидных последовательностей (табл. 1).
Таблица 1.
Последовательность праймеров и условия амплификации
| Экзон | Последовательность праймеров | Длина фрагмента, пн | Условия амплификации | ||
|---|---|---|---|---|---|
| T отжига, °C | MgCl2, мм | число циклов | |||
| 1 | RS1 1F GCCAGGGCTCAACTTAATCC | 218 | 60 | 1 | 30 |
| RS1 1R CAGGCTATATTCCTATTTATCAAC | |||||
| 2 | RS1 2F CCAGAAGGGGTGTTGGTAGC | 194 | |||
| RS1 2R CAAAAGTACTATGCATGTACATTACAGCC | |||||
| 3 | RS1 3F CACAGTTGCCTTTGACCGTGAC | 252 | |||
| RS1 3R GCGTTCAGGGGGTTAATTCAGC | |||||
| 4 | RS1 4F GTTTTGTTATTTCAGTCACCTGGTGC | 286 | |||
| RS1 4R GCCACGCTGGTAGAGAGGC | |||||
| 5 | RS1 5F GGGCTTTTTGCAGACATGCAC | 303 | |||
| RS1 5R GTCCCAGAGGGTGCGAGC | |||||
| 6 | RS1 6F CTCCCCGATGTGATGGTGAC | 244 | |||
| RS1 6R CCCGCTCTGTGCCAGTCAC | |||||
Для секвенирования по Сенгеру использовали фрагменты ДНК, полученные в ходе ПЦР, с применением набора реактивов ABI Dye Terminator, version 1 (Applied Biosystems, США) с последующим анализом на приборе 3130 ABI genetic analyzer (Applied Biosystems). Полученные хроматограммы анализировали с помощью программы Chromas version 2 (Technelysium). Название обнаруженным изменениям в гене RS1 присваивалось в соответствии с международной номенклатурой HGVS (http://www.hgvs.org/mutnomen/), использовалась референсная последовательность кДНК, представленная на портале NCBI (http:// www.ncbi.nlm.nih.gov/nuccore): NM_000330.3.
Популяционная частота выявленных неописанных ранее вариантов была оценена с использованием выборки проектов “1000 геномов”, ESP6500 и Genome Aggregation Database (gnomAD) (v2.1.1).
Клиническая значимость (патогенности) выявленных неописанных ранее вариантов нуклеотидной последовательности была оценена на основании российских рекомендаций для интерпретации данных, полученных методами массового параллельного секвенирования (MPS).
Патогенность трех новых миссенс-мутаций была предсказана in silico четырьмя программами.
РЕЗУЛЬТАТЫ
В ходе исследования из 48 неродственных больных ретиношизисом были выявлены патогенные и вероятно патогенные варианты у 37 пробандов, у 11 пробандов генетическая причина заболевания не установлена.
Всего было выявлено 27 различных мутаций, включая 16 миссенс, три нонсенс, три мутации сайта сплайсинга, одну делецию со сдвигом рамки считывания, три делеции целого экзона и одну мутацию в инициирующем кодоне.
Наибольшее количество мутаций встретилось в экзоне 6 (20), включая две делеции всего экзона, далее по убыванию в экзоне 5 (7), экзоне 3 и его акцепторном сайте сплайсинга (4), в экзоне 4 (3), в экзоне 1 (1), делеция экзона 2 (1) и мутация в донорном сайте сплайсинга экзона 2 (1) (табл. 2, рис. 3).
Таблица 2.
Выявленные варианты в гене RS1
| Вариант | Эффект | Экзон/ интрон | Число поврежденных хромосом | Доля, % | Источник |
|---|---|---|---|---|---|
| c.3G>A | p.M1? | 1 | 1 | 2.7 | [18] |
| c.(87+1_88-1)_(113+1_114-1)del | Делеция экзона 2 | 2 | 1 | 2.7 | ? |
| c.141_142insAT | p.Ala48Metfs*79 | 3 | 1 | 2.7 | Не описан |
| с.120C>A | р.Cys40* | 3 | 1 | 2.7 | [10] |
| с.184+2T>A | Splicing | in3 | 1 | 2.7 | Не описан |
| c.185-1G>C | Splicing | in3 | 1 | 2.7 | [19] |
| с.185-1G>T | Splicing | in3 | 1 | 2.7 | Не описан |
| c.274T>G | p.Trp92Gly | 4 | 1 | 2.7 | [20] |
| c.304C>T | p.Arg102Trp | 4 | 1 | 2.7 | [10] |
| c.305G>A | p.Arg102Gln | 4 | 1 | 2.7 | [19] |
| c.337C>T | p.Leu113Phe | 5 | 1 | 2.7 | [19] |
| с.365G>A | p.Trp122* | 5 | 1 | 2.7 | Не описан |
| c.422G>A | p.Arg141His | 5 | 1 | 2.7 | [19] |
| c.421C>T | p.Arg141Cys | 5 | 2 | 5.4 | [19] |
| c.425G>A | p.Cys142Tyr | 5 | 1 | 2.7 | Не описан |
| c.511G>C | p.Gly171Arg | 5 | 1 | 2.7 | » |
| c.554C>A | p.Thr185Lys | 6 | 1 | 2.7 | [21] |
| с.577C>T | p.Pro193Ser | 6 | 2 | 5.4 | [22] |
| c.587C>T | p.Ser196Phe | 6 | 1 | 2.7 | Не описан |
| c.589C>T | p.Arg197Cys | 6 | 1 | 2.7 | [19] |
| c.598C>T | p.Arg200Cys | 6 | 1 | 2.7 | [19] |
| c.608C>T | p.Pro203Leu | 6 | 4 | 10.8 | [19] |
| c.637C>T | p.Arg213Trp | 6 | 4 | 10.8 | [19] |
| c.638G>A | р.Arg213Gln | 6 | 3 | 8.1 | [23] |
| c.657C>A | p.Cys219* | 6 | 1 | 2.7 | Не описан |
| NG_008659.2:g.(?_34948)_(37416_?)del | Делеция экзона 6 | 6 | 1 | 2.7 | » |
| NG_008659.2:g.(?_34948)_(37416_?)del | Делеция экзона 6 | 6 | 1 | 2.7 | » |
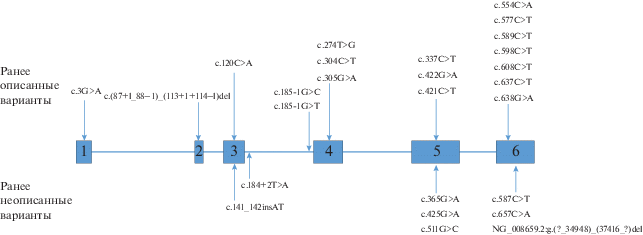
Пять из ранее описанных вариантов встретились более чем один раз: c.608C>T (p.Pro203Leu), c.637C>T (p.Arg213Trp), c.638G>A (р.Arg213Gln), с.577C>T (p.Pro193Ser), c.421C>T (p.Arg141Cys).
Выявлено восемь ранее неописанных однонуклеотидных замен: c.141_142insAT (p.Ala48Metfs*79), с.185-1G>T, с.184+2T>A, с.365G>A (p.Trp122*), c.425G>A (p.Cys142Tyr), c.511G>C(p.Gly171Arg), c.587C>T (p.Ser196Phe), c.657C>A (p.Cys219*). А также нами были выявлены крупные делеции у троих пациентов: у двоих делеция экзона 6 (NG_008659.2:g.(?_34948)_(37416_?) del) и у одного больного делеция экзона 2 (c.(87+1_88-1)_ (113+1_114-1)del). В базе данных HGMD®Professional описаны делеции, затрагивающие экзон 2, однако не описано ни одной крупной делеции, включающей весь экзон 6. В настоящем исследовании не были определены точки разрыва, и неизвестно была ли наблюдаемая нами делеция экзона 2 идентична известным делециям экзона 2 на уровне ДНК и одинаковые ли делеции экзона 6.
ОБСУЖДЕНИЕ
Нами была установлена генетическая причина заболевания у 77% больных с направляющим (предварительным) диагнозом “Х-сцепленный ретиношизис”. У 11 больных не была выявлена генетическая причина заболевания, что может свидетельствовать как о вариантах, расположенных в интронных и регуляторных областях гена, так и о том, что заболевание не связано с геном RS1 и ретиношизис у этих больных является симптомом другого заболевания. Более высокая выявляемость мутаций в других популяциях (табл. 3) также подтверждает версию о более строгих клинических критериях отбора пациентов для постановки диагноза “X-сцепленный ретиношизис”. Несмотря на вышеизложенное, информативность настоящего исследования достаточно высокая, что свидетельствует о характерной клинической картине у междисциплинарно обследованных пациентов и о том, что большая часть патогенных вариантов в гене RS1 хорошо выявляется методом секвенирования по Сенгеру.
В исследуемой группе пациентов были идентифицированы практически все типы мутаций гена RS1. Большинство мутаций были миссенс (59%, 16/27), что согласуется с предыдущими публикациями [15, 16]. Значительная доля вариантов (30%, 8/27), не считая крупных делеций, ранее не была описана.
Варианты, выявленные в исследовании, не были распределены случайным образом по гену. Большинство из обнаруженных нами вариантов (18/27) были расположены в экзонах 4–6, кодирующих высококонсервативный дискоидиновый домен белка RS1, который считается критическим для функционирования ретиношизина (табл. 4). В целом полученные результаты согласуются с ранее проведенными исследованиями (HGMD), небольшие различия по экзонам можно объяснить величиной исследуемой выборки.
Таблица 4.
Распределение патогенных вариантов по гену RS1
| Патогенные варианты в гене RS1 | Настоящее исследование | База данных HGMD | |||
|---|---|---|---|---|---|
| число вариантов | доля, % | число вариантов | доля, % | ||
| Точковые замены и маленькие делеции/инсерции | Экзон 1 | 1 | 3.7 | 14 | 4.6 |
| Экзон 2 | 0 | 0.0 | 6 | 2.0 | |
| Экзон 3 | 2 | 7.4 | 14 | 4.6 | |
| Экзон 4 | 3 | 11.1 | 65 | 21.5 | |
| Экзон 5 | 6 | 22.2 | 77 | 25.4 | |
| Экзон 6 | 9 | 33.3 | 68 | 22.4 | |
| Мутации сайта сплайсинга | 3 | 11.1 | 25 | 8.3 | |
| Крупные делеции/инсерции/перестройки | 3 | 11.1 | 34 | 11.2 | |
Наиболее частыми мутациями в изученной выборке больных стали миссенс-замены экзона 6 – c.608C>T (p.Pro203Leu) и c.637C>T (p.Arg213Trp). Каждая из этих замен встретилась у четырех больных. Доля составила 10.8%. Данные замены не являются частыми в других популяциях.
Вариант c.608C>T (p.Pro203Leu) встретился 2 раза у японских [29] и испанских [30] больных, по одному разу у индийских [31], финских [21] и канадских [32] больных. У испанских больных с помощью анализа гаплотипов было доказано независимое происхождение мутаций [30].
Миссенс-замена p.Arg213Trp вызвана транзицией С на Т в 637-м положении гена RS1, приводящей к аминокислотной замене аргинина на триптофан в 213-м положении белка. Эта миссенс-замена меняет положительно заряженный аргинин на неполярный триптофан, тем самым изменяя заряд белка. Предполагается, что вариант c.637C>T может мешать сворачиванию белка, приводя к аномальной конформации и задержке мутантного белка во внутриклеточном пространстве, что приводит к отсутствию ретиношизина в сетчатке у пораженных мужчин и к фенотипу ретиношизиса [33]. Вариант c.637C>T (p.Arg213Trp) также встречается у больных из различных популяций [6, 31, 34], но ни в одной из популяций не является мажорным.
У троих больных встретился вариант c.638G>A, приводящий к миссенс-замене аргинина на глутамин также в 213-м положении белка. Аллельная частота выявленного варианта составила 6.25%. В настоящем исследовании c.638G>A встретился у больных из различных этнических групп, что свидетельствует в пользу того, что повторяющиеся случаи встречаемости варианта объясняются “горячими” точками гена, а не эффектом основателя. Ранее вариант c.638G>A 1–2 раза встретился в испанской [30], китайской [35], японской [29] популяциях.
Миссенс-замена p.Pro193Ser возникает вследствие транзиции С на T в 577-м положении гена RS1. В публикации Y.V. Sergeev с соавт. [33] показано, что замена пролина на серин в 193-м положении белка влияет на стойкость конформации белка, а не на изменение вторичной структуры, и, как следствие, ожидается, что белок будет секретироваться из клетки. Данный вариант описан у китайского больного с “мягким” течением болезни [22].
На двух хромосомах нами был выявлен вариант c.421C>T. Транзиция С на Т в 421-м положении гена приводит к аминокислотной замене аргинина на цистеин в 141-м положении белка – p.Arg141Cys. Между цистеинами в белке создаются дисульфидные мостики; появляясь в 141-м положении новый цистеин является конкурентом для Cys142 и может образововать дисульфидную связь с Cys110, влияя на стабильность конформации [33]. В материалах консорциума ретиношизиса 1998 г. наличие данного варианта было показано у немецкого больного, двух датских, двух французских больных [19], а также он встретился у пяти больных из Великобритании [36] и из США [37].
Остальные ранее описанные патогенные варианты в настоящем исследовании встретились по одному разу: c.3G>A (p.M1?), с.120C>A (р.Cys40*), c.274T>G (p.Trp92Gly), c.304C>T (p.Arg102Trp), c.305G>A (p.Arg102Gln), c.337C>T (p.Leu113Phe), c.422G>A (p.Arg141His), c.554C>A (p.Thr185Lys), c.589C>T (p.Arg197Cys), c.598C>T (p.Arg200Cys), c.657C>A (p.Cys219*), c.185-1G>C, делеция экзона 2 – c.(87+1_88-1)_(113+1_114-1)del).
Все эти варианты ранее встречались в различных популяциях мира с различной частотой. Однако ни один из этих вариантов не являлся мажорным в какой-либо выборке больных с превалирующей частотой. Только вариант c.304C>T (p.Arg102Trp) встретился с достаточно высокой частотой у больных из Великобритании на шести хромосомах (17%) [36] и у китайских больных на трех хромосомах (10%) [35].
Все ранее неописанные как патогенные варианты встретились у больных по одному разу (табл. 5). Для делеций экзона 6 (NG_008659.2: g.(?_34948)_ (37416_?)del) не были определены точки разрыва, поэтому нельзя сказать повторяющийся ли этот вариант или нет. Пять из восьми точковых вариантов были выявлены в “горячих” экзонах гена – в экзонах 5 и 6, крупные делеции также затрагивали экзон 6. Один вариант встретился в экзоне 3, один – в донороном сайте сплайсинга экзона 3 (с.184+2T>A) и один – в акцепторном сайте сплайсинга экзона 4 (с.185-1G>T).
Таблица 5.
Ранее неописанные варианты в RS1 и их патогенность
| Вариант | Эффект | Экзон/ интрон | Число хромосом | Критерии патогенности | Патогенность |
|---|---|---|---|---|---|
| c.141_142insAT | p.Ala48Metfs*79 | 3 | 1 | PVS1, PM2 | Вероятно патогенный |
| с.185-1G>T | Splicing | in3 | 1 | PVS1, PM2 | » |
| с.184+2T>A | Splicing | in3 | 1 | PVS1, PM2 | » |
| с.365G>A | p.Trp122* | 5 | 1 | PVS1, PM1, PM2 | Патогенный |
| c.425G>A | p.Cys142Tyr | 5 | 1 | PM1, PM2, PM5, PP2, PP3 | Вероятно патогенный |
| c.511G>C | p.Gly171Arg | 5 | 1 | PM1, PM2, PP2, PP3 | » |
| c.587C>T | p.Ser196Phe | 6 | 1 | PM1, PM2, PP2, PP3 | » |
| c.657C>A | p.Cys219* | 6 | 1 | PVS1, PM1, PM2 | Патогенный |
| NG_008659.2:g.(?_34948)_ (37416_?)del | Делеция экзона 6 | 6 | 1 | PVS1, PM1, PM2 | » |
| NG_008659.2:g.(?_34948)_ (37416_?)del | Делеция экзона 6 | 6 | 1 | PVS1, PM1, PM2 | » |
Инсерция двух нуклеотидов c.141_142insAT в экзоне 3 приводит к сдвигу рамки считывания и образованию преждевременного стоп-кодона в 79-м положении белка. Выявленный вариант нуклеотидной последовательности не зарегистрирован в контрольных выборках “1000 геномов”, ESP6500 и gnomAD (The Genome Aggregation Database). Согласно рекомендациям по интерпретации данных [38] выявленный вариант нуклеотидной последовательности следует расценивать как вероятно патогенный (PVS1, PM2).
Варианты с.185-1G>T и с.184+2T>A находятся в канонических сайтах сплайсинга. Данные варианты нуклеотидной последовательности не зарегистрированы в контрольных выборках “1000 геномов”, ESP6500 и gnomAD (The Genome Aggregation Database). Согласно рекомендациям по интерпретации данных [38] выявленные варианты нуклеотидной последовательности следует расценивать как вероятно патогенные (PVS1, PM2).
Замены с.365G>A и c.657C>A приводят к образованию преждевременного стоп-кодона – p.Trp122* и p.Cys219* соответственно. Замены расположены в “горячих” экзонах гена RS1, не зарегистрированы в контрольных выборках “1000 геномов”, ESP6500 и gnomAD (The Genome Aggregation Database). Согласно рекомендациям по интерпретации данных [38] выявленные варианты нуклеотидной последовательности следует расценивать как патогенные (PVS1, PM1, PM2).
К миссенс-заменам p.Cys142Tyr, p.Gly171Arg, p.Ser196Phe приводят варианты нуклеотидной последовательности c.425G>A, c.511G>C, c.587C>T соответственно. Варианты расположены в “горячих” экзонах гена RS1. Выявленные варианты не зарегистрированы в контрольных выборках “1000 геномов”, ESP6500 и gnomAD (The Genome Aggregation Database). В гене RS1 миссенс-варианты обычно вызывают заболевание, а доброкачественные изменения встречааются редко [36]. Алгоритмы предсказания патогенности MutationTaster [39], FATHMM [40], LRT [41], DEOGEN2 [42] оценивают данные варианты как патогенные. Миссенс-вариант c.425G>A (p.Cys142Tyr) приводит к изменению аминокислоты в том же положении, в котором ранее были описаны варианты c.424T>C (p. Cys142Arg) [33] и c.426T>G (Cys142Trp) как патогенные [26]. Таким образом, согласно рекомендациям по интерпретации данных [38] выявленные варианты нуклеотидной последовательности следует расценивать как вероятно патогенные (PM1, PM2, PP2, PP3, для c.425G>A еще PM5).
Делеции NG_008659.2:g.(?_34948)_(37416_?)del “горячего” экзона 6 приводят к отсутствию фрагмента высококонсервативного дискоидинового домена. Выявленные варианты не зарегистрированы в контрольной выборке gnomAD (The Genome Aggregation Database). Согласно рекомендациям по интерпретации данных [38] выявленные варианты нуклеотидной последовательности следует расценивать как патогенные (PVS1, PM1, PM2).
Таким образом, все выявленные нами варианты нуклеотидной последовательности гена RS1, ранее неописанные как патогенные, мы классифицировали как патогенные и вероятно патогенные.
Полученные данные показывают, что для российских больных XLRS не характерно наличие “мажорных” мутаций в гене RS1, что свойственно практически всем популяциям. Только у финских больных патогенный вариант c.214G>A (p.Glu72Lys) встречается у 70% больных [21], что объясняется изолированностью популяции.
В настоящее время разработка генной терапии для лечения XLRS достигла стадии клинических испытаний [43] и знание точного генетического дефекта у конкретного пациента позволит применить эффективное патогенетическое лечение. Выявление мутаций, вызывающих болезнь у пациентов с XLRS, также необходимо для медико-генетического консультирования (прогноза заболевания, прогноза потомства, возможности предимлантационной диагностики) и разработки корректного лечебно-профилактического алгоритма. Поиск патогенных вариантов методом прямого автоматического секвенирования по Сенгеру является необходимым и достаточным инструментом для установления генетической причины заболевания XLRS.
Работа выполнена в рамках Государственного задания Минобрнауки России.
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики.
От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
George N.D., Yates J.R., Moore A.T. Clinical features in affected males with X-linked retinoschisis // Arch. Ophthalmol. 1996. V. 114. P. 274–280.
Kellner U., Brummer S., Foerster M.H., Wessing A. X‑linked congenital retinoschisis // Graefe’s Arch. Ophthalmol. 1990. V. 228(5). P. 432–437.
Tantri A., Vrabec T.R., Cu-Unjieng A. et al. X-linked retinoschisis: A clinical and molecular genetic review // Surv. Ophthalmol. 2004. V. 49(2). P. 214–230. https://doi.org/10.1016/j.survophthal.2003.12.007
Mooy C.M., Van Den Born L.I., Baarsma S. et al. Hereditary X-linked juvenile retinoschisis: a review of the role of Müller cells // Arch. Ophthalmol. 2002. V. 120(7). P. 979–984.
Eksandh L.C., Ponjavic V., Ayyagari R. et al. Phenotypic expression of juvenile X-linked retinoschisis in Swedish families with different mutations in the XLRS1 gene // Arch. Ophthalmol. 2000. V. 118(8). P. 1098–1104. https://doi.org/10.1001/archopht.118.8.1098
Xu J., Gu H., Ma K. et al. R213W mutation in the retinoschisis 1 gene causes X-linked juvenile retinoschisis in a large Chinese family // Mol. Vis. 2010. V. 12. P. 1593–1600.
Deutman A.F. The Hereditary Dystrophies of the Posterior Pole of the Eye. Amsterdam: Von Gorcum and Company, 1971. 484 p.
Saldana M., Thompson J., Monk E. et al. X-linked retinoschisis in a female with a heterozygous RS1 missense mutation // Am. J. Med. Genet. 2007. V. 143A. P. 608–609. https://doi.org/10.1002/ajmg.a.31568
Sikkink S.K., Biswas S., Parry N.R. et al. X-linked retinoschisis: an update // J. Med. Genet. 2007. V. 44. P. 225–232.
Sauer C.G., Gehrig A., Warneke-Wittstock R. et al. Positional cloning of the gene associated with X-linked juvenile retinoschisis // Nat. Genet. 1997. V. 17. P. 164–170.
Tolun G., Vijayasarathy C., Huang R. et al. Paired octamer rings of retinoschisin suggest a junctional model for cell-cell adhesion in the retina // PNAS. 2016. V. 113(19). P. 5287–5292. https://doi.org/10.1073/pnas.1519048113
Poole S., Firtel R.A., Lamar E., Rowekamp W. Sequence and expression of the discoidin I gene family in Dictyostelium discoideum // J. Mol. Biol. 1981. V. 153(2). P. 273–289. https://doi.org/10.1016/0022-2836(81)90278-3
Baumgartner S., Hofmann K., Chiquet-Ehrismann R., Bucher P. The discoidin domain family revisited: New members from prokaryotes and a homology-based fold prediction// Protein Sci. 1998. V. 7(7). P. 1626–1631. https://doi.org/10.1002/pro.5560070717
Pratt K., Shen B., Takeshima K. et al. Structure of the C2 domain of human factor VIII at 1.5 Å resolution // Nature. 1999. V. 402. P. 439–442. https://doi.org/10.1038/46601
Wu W.W., Molday R.S. Defective discoidin domain structure, subunit assembly, and endoplasmic reticulum processing of retinoschisin are primary mechanisms responsible for X-linked retinoschisis // J. Biol. Chem. 2003. V. 278. P. 28139–28146. https://doi.org/10.1074/jbc.M302464200
Molday R.S., Kellner U., Weber B.H. X-linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms // Prog. Retin. Eye Res. 2012. V. 31(3). P. 195–212. https://doi.org/10.1016/j.preteyeres.2011.12.002
Plössl K., Schmid V., Straub K. et al. Pathomechanism of mutated and secreted retinoschisin in X-linked juvenile retinoschisis // Exp. Eye Res. 2018. V. 177. P. 23–34. https://doi.org/10.1016/j.exer.2018.07.021
Xiao Y., Liu X., Tang L. et al. X-linked retinoschisis: Phenotypic variability in a Chinese family // Sci. Rep. 2016. V. 29(6). P. 20118. https://doi.org/10.1038/srep20118
Functional implications of the spectrum of mutations found in 234 cases with X-linked juvenile retinoschisis (XLRS) // Hum. Mol. Genet. 1998. V. 7(7). P. 1185–1192. https://doi.org/10.1093/hmg/7.7.1185
Lamey T., Laurin S., Chelva E., De Roach J. Genotypic analysis of X-linked retinoschisis in Western Australia // Retinal Degenerative Diseases. Advances in Experimental Medicine and Biology / Eds Anderson R., Hollyfield J., LaVail M. N.Y.: Springer, 2010. V. 664. P. 283–291.
Huopaniemi L., Rantala A., Forsius H. et al. Three widespread founder mutations contribute to high incidence of X-linked juvenile retinoschisis in Finland // Eur. J. Hum. Genet. 1999. V. 7(3). P. 368–376. https://doi.org/10.1038/sj.ejhg.5200300
Chan W.M., Choy K.W., Wang J. et al. Two cases of X‑linked juvenile retinoschisis with different optical coherence tomography findings and RS1 gene mutations // Clin. & Exp. Ophthalmol. 2004. V. 32. P. 429–432. https://doi.org/10.1111/j.1442-9071.2004.00820.x
Hotta Y., Fujiki K., Hayakawa M. et al. Japanese juvenile retinoschisis is caused by mutations of the XLRS1 gene // Hum. Genet. 1998. V. 103. P. 142–144. https://doi.org/10.1007/PL00008705
Hewitt A.W., FitzGerald L.M., Scotter L.W. et al. Genotypic and phenotypic spectrum of X-linked retinoschisis in Australia // Clin. Exp. Ophthalmol. 2005. V. 33(3). P. 233–239. https://doi.org/10.1111/j.1442-9071.2005.01018.x
Skorczyk A., Krawczyński M.R. Four novel RS1 gene mutations in Polish patients with X-linked juvenile retinoschisis // Mol. Vis. 2012. V. 18. P. 3004–3012.
Hiriyanna Kelaginamane T., Bingham E.L., Yashar Beverly M. et al. Novel mutations in XLRS1 causing retinoschisis, including first evidence of putative leader sequence change // Hum. Mut. 1999. P. 14(5). V. 423–427.
Mashima Y., Shinoda K., Ishida S. et al. Identification of four novel mutations of the XLRS1 gene in Japanese patients with X-linked juvenile retinoschisis // Hum. Mutat. 1999. V. 13(4). P. 338. https://doi.org/10.1002/(SICI)1098-1004(1999)13:4<338:: AID-HUMU16>3.0.CO;2-0
Hu Q.R., Huang L.Z., Chen X.L. et al. Genetic analysis and clinical features of X-linked retinoschisis in Chinese patients // Sci. Rep. 2017. V. 7. P. 44060. https://doi.org/10.1038/srep44060
Kondo H., Oku K., Katagiri S. et al. Novel mutations in the RS1 gene in Japanese patients with X-linked congenital retinoschisis // Hum. Genome Var. 2019. V. 8(6). P. 3. https://doi.org/10.1038/s41439-018-0034-6
Riveiro-Alvarez R., Trujillo-Tiebas M.J., Gimenez-Pardo A. et al. Correlation of genetic and clinical findings in Spanish patients with X-linked juvenile retinoschisis // Invest. Ophthalmol. Vis. Sci. 2009. V. 50(9). P. 4342–4350. https://doi.org/10.1167/iovs.09-3418
Suganthalakshmi B., Shukla D., Rajendran A. et al. Genetic variations in the hotspot region of RS1 gene in Indian patients with juvenile X-linked retinoschisis // Mol. Vis. 2007. V. 19(13). P. 611–617.
Gerth C., Zawadzki R.J., Werner J.S., Héon E. Retinal morphological changes of patients with X-linked retinoschisis evaluated by Fourier-domain optical coherence tomography // Arch. Ophthalmol. 2008. V. 126(6). P. 807–811. https://doi.org/10.1001/archopht.126.6.807
Sergeev Y.V., Caruso R.C., Meltzer M.R. et al. Molecular modeling of retinoschisin with functional analysis of pathogenic mutations from human X-linked retinoschisis // Hum. Mol. Genet. 2010. V. 19(7). P. 1302–1313. Epub 2010 Jan 8https://doi.org/10.1093/hmg/ddq006
Sieving P.A., Bingham E.L., Kemp J. et al. Juvenile X‑linked retinoschisis from xlrs1 arg213trp mutation with preservation of the electroretinogram scotopic b-wave // Am. J. Ophthalmol. 1999. V. 128. P. 179–184.
Chen J., Xu K., Zhang X. et al. Novel mutations of the RS1 gene in a cohort of Chinese families with X-linked retinoschisis // Mol. Vis. 2014. V. 31(20). P. 132–139.
Pimenides D., George N.D., Yates J.R. et al. X-linked retinoschisis: Clinical phenotype and RS1 genotype in 86 UK patients // J. Med. Genet. 2005. V. 42(6). P. e35. https://doi.org/10.1136/jmg.2004.029769
Stone E.M., Andorf J.L., Whitmore S.S. et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease // Ophthalmology. 2017. V. 124(9). P. 1314–1331. https://doi.org/10.1016/j.ophtha.2017.04.008
Рыжкова О.П., Кардымон О.Л., Прохорчук Е.Б. и др. Руководство по интерпретации данных последовательности ДНК человека, полученных методами массового параллельного секвенирования (MPS) // Мед. генетика. 2019. Т. 18(2). С. 3–23. https://doi.org/10.25557/2073-7998.2019.02.3-23
Schwarz J.M., Rodelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations // Nat. Methods. 2010. V. 7. P. 575–576.
Shihab H.A., Gough J., Cooper D.N. et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models // Hum. Mut. 2013. V. 34. P. 57–65.
Chun S., Fay J.C. Identification of deleterious mutations within three human genomes // Genome Res. 2009. V. 19. P. 1553–1561.
Raimondi D., Tanyalcin I., Ferté J. et al. DEOGEN2: prediction and interactive visualization of single amino acid variant deleteriousness in human proteins // Nucl. Acids Res. 2017. V. 45(W1). P. W201–W206.
Hafler BP. Clinical progress in inherited retinal degenerations: Gene therapy clinical trials and advances in genetic sequencing // Retina. 2017. V. 37(3). P. 417–423. https://doi.org/10.1097/IAE.0000000000001341
Дополнительные материалы отсутствуют.