

Генетика, 2021, T. 57, № 9, стр. 1073-1081
Связь метилирования ДНК в гене YJEFN3 с когнитивным дефицитом при расстройствах шизофренического спектра
М. В. Алфимова 1, *, Н. В. Кондратьев 1, А. К. Голов 1, В. Г. Каледа 1, Л. И. Абрамова 1, В. Е. Голимбет 1
1 Научный центр психического здоровья
115522 Москва, Россия
* E-mail: m.alfimova@gmail.com
Поступила в редакцию 14.10.2020
После доработки 13.11.2020
Принята к публикации 01.12.2020
Аннотация
В данной работе проанализировано метилирование каждого из цитозинов на фрагменте гена YJEFN3, расположенного в одном из локусов риска шизофрении. Цель исследования состояла в том, чтобы определить, связано ли метилирование данного фрагмента с уровнем когнитивного дефицита больных и в какой степени оно зависит от гаплотипа риска. Образцы периферической крови 70 больных c расстройствами шизофренического спектра и 72 здоровых, прошедших когнитивное тестирование, исследовали с помощью метода бисульфитного секвенирования единичных молекул ДНК в реальном времени. Обнаружена отрицательная корреляция между метилированием CpG-сайта с координатой chr19:19532203 (Illumina ID: cg08623644) и индексом когнитивного функционирования пациентов. Метилирование cg08623644 было ассоциировано с гаплотипом риска в объединенной выборке и у здоровых. У больных наблюдалось ослабление этой ассоциации. Полученные результаты позволяют предположить, что метилирование cg08623644 может быть периферическим маркером молекулярных процессов, связанных с тяжестью течения заболевания, что требует дальнейшего изучения.
Консорциум по психиатрической геномике в полногеномных мегаисследованиях (PGC2 GWAS) выявил более 100 локусов, значимо ассоциированных с шизофренией [1, 2]. Большинство из них ранее не рассматривались в качестве функциональных кандидатов для шизофрении и не изучались в связи с ее проявлениями. Для раскрытия патогенеза заболевания необходимо понимание роли в нем этих участков ДНК как на уровне последствий для работы генома, так и на уровне симптомов и синдромов.
Предполагается, что в каждом локусе риска может действовать целый ряд генетических вариантов, которые влияют на заболевание в разной степени и разными способами, в том числе регулируя экспрессию генов посредством метилирования ДНК [3]. Анализ локусов количественных признаков (QTL) указывает на значительное обогащение наборов QTL для метилирования (mQTL) и экспрессии (eQTL) в мозге индексными полиморфизмами PGC2 GWAS шизофрении [4]. Учитывая это, оценка метилирования ДНК (меДНК) представляется перспективным направлением исследования молекулярных механизмов, ведущих от индексных локусов GWAS шизофрении к проявлениям заболевания. В частности, такой подход может оказаться продуктивным для раскрытия биологических основ когнитивного дефицита больных [5], одного из ключевых синдромов шизофрении, определяющего тяжесть ее течения и уровень адаптации пациента [6].
В данной работе, в рамках проекта исследования связи меДНК и когнитивных нарушений при шизофрении, мы проанализировали фрагмент одного из индексных локусов PGC2 GWAS [1], совпадающих с локусами дифференциального метилирования, найденными в полнометиломных исследованиях периферической крови [7] и постмортальных образцов мозга больных [8], а именно локуса 47 (chr19: 19263213–19547213, GRCh38/hg38). Для изучения был выбран фрагмент ДНК (chr19: 19532136–19533050) внутри гена YJEFN3 (YjeF N-terminal domain-containing protein 3, также известный как AIBP2). Критерии выбора фрагментов подробно описаны ранее [9]. В частности учитывали наличие однонуклеотидных полиморфизмов (SNP), расположенных внутри сайтов связывания транскрипционных факторов или внутри сайтов, образующих CpG (CpG-SNP), поскольку они могут оказывать влияние на экспрессию генов и метилирование соседних цитозинов.
Исследованный фрагмент (рис. 1) содержит участки генома, метилирование которых по-разному ассоциировано с экспрессией и транскрипцией генов, а именно: части интронов, CG-островок, экзон, границы экзон-интрон (UCSC genomic browser [10]). В нем присутствуют кластер гиперчувствительности к ДНКазе I и чувствительные к метилированию ДНК сайты связывания транскрипционных факторов, включая многофункциональный транскрипционный фактор CTCF. Локализованные в пределах фрагмента четыре общих SNP являются eQTL в разных тканях, в том числе в мозге и крови, и локусами, влияющими на альтернативный сплайсинг (sQTL), как самого гена YJEFN3, так и ряда близлежащих генов (табл. 1) [11].
Рис. 1.
Схематическое изображение изученного фрагмента ДНК в геномном браузере UCSC [по 10]. Вверху – локус 47 PGC2 GWAS шизофрении с указанием положения изученного фрагмента в гене YJEFN3. Внизу – изученный фрагмент YJEFN3 с указанием расположения общих SNP, cg08623644, CG-островка (CpG: 24) и сайта связывания CTCF. На верхнем рисунке экзоны в генах обозначены вертикальными линиями, на нижнем – прямоугольником.
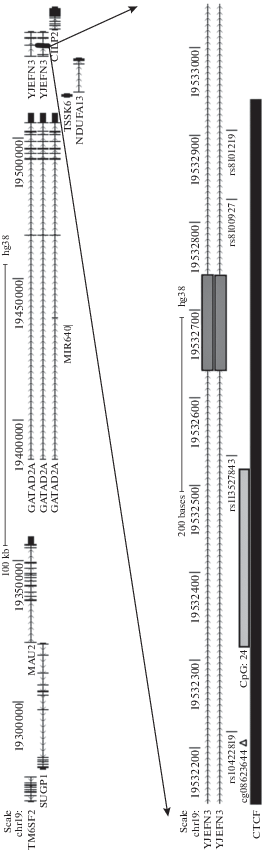
Таблица 1.
Характеристика полиморфных сайтов (SNP), образующих гаплотип
| SNP | Координата на хромосоме 19 | Описание | MAF | LD с rs2905426 | eQTL | |
|---|---|---|---|---|---|---|
| кровь | мозг | |||||
| rs10422819 (CpG-SNP) | 19532219 | Интрон, C>A | A: 32% | D': 1 R2: 0.95 | GATAD2A, LPAR2, MAU2, NDUFA13, TSSK6 |
CILP2, YJEFN3 |
| rs113527843 | 19532534 | Интрон, A>T | T: 17% | D': 1 R2: 0.41 |
GATAD2A, GMIP, LPAR2, NDUFA13, TSSK6, YJEFN3 | CILP2 |
| rs8100927 | 19532827 | Интрон, C>G | G: 17% | D': 1 R2: 0.41 |
GMIP, LPAR2, NDUFA13, TSSK6, YJEFN3 | CILP2 |
Примечание. Координаты даны согласно GRCh38/hg38. MAF – частота минорного аллеля в европейских популяциях по данным проекта 1000 геномов [18]. SNP rs2905426 – теговый SNP локуса 47 PGC2 GWAS [1]. LD – неравновесие по сцеплению в европейских популяциях [18]. eQTL – локусы количественных признаков для экспрессии перечисленных генов по данным GTEx v8 [11], для гена CILP2 – в базальных ганглиях, для YJEFN3 – в коре мозга.
Цель исследования состояла в том, чтобы определить: 1) связано ли меДНК данного фрагмента в периферической крови с уровнем когнитивного дефицита больных шизофренией и 2) в какой степени уровень метилирования в данном регионе зависит от локальной структуры ДНК, а именно от наличия гаплотипа риска заболевания. Учитывая, что среднее метилирование промотора/тела гена реже коррелирует с экспрессией гена, чем метилирование единичных CpG [12], основной анализ проводили на уровне индивидуальных вариативно метилированных цитозинов. Применяли метод бисульфитного секвенирования единичных молекул ДНК в реальном времени (single-molecule real-time bisulfite sequencing, SMRT-BS [13]), позволяющий изучать метилирование длинных фрагментов ДНК с разрешением до индивидуальных цитозинов и определять гаплотипы непосредственно из данных секвенирования.
МАТЕРИАЛЫ И МЕТОДЫ
Выборку формировали с использованием базы данных лаборатории клинической генетики (НЦПЗ, Москва) на основе критериев, более подробно описанных ранее [14]. Согласно самоотчету, испытуемые имели полное среднее или высшее образование, были русскими, не младше 18 и не старше 45 лет и не страдали заболеваниями, связанными с нарушением интеллектуальных процессов.
Выборку составили 70 психотических больных, находившихся в момент исследования на стационарном лечении в стабильном состоянии, позволявшем провести когнитивное тестирование (средний возраст 27.41 ± 6.71 лет, 49% женщин), и 72 здоровых испытуемых без наследственной отягощенности психозами (средний возраст 26.63 ± ± 6.72 лет, 53% женщин). Большинство пациентов (n = 63) страдали шизофренией (F20 согласно Международной классификации болезней 10-го пересмотра). Выборка также включала лиц с острым психотическим (F23, n = 3) и шизоаффективным расстройством (F25, n = 4). Все испытуемые дали письменное информированное согласие на участие в исследовании. Проведение исследования одобрено Этическим комитетом НЦПЗ.
Каждый участник выполнил шесть когнитивных тестов для оценки эпизодической памяти, регуляторно-исполнительных функций и социальных когниций. Как подробно описано ранее [14], индивидуальный индекс когнитивного функционирования вычисляли путем усреднения выраженных в Т-баллах показателей этих тестов.
Геномная ДНК была выделена из 1 мл периферической крови с помощью стандартных процедур. Статус метилирования определяли методом SMRT-BS на основе бисульфитной конверсии и высокопроизводительного секвенирования третьего поколения PacBio: PacBio RSII, P6/C4 chemistry, CCS reads. Метод был ранее модифицирован и включал подавление ПЦР на коротких матрицах с помощью идентичных адаптеров на концах праймеров (“panhandle” SMRT-BS [15]). Метод и его модификация, а также процедуры подготовки и контроля качества данных секвенирования и определения гаплотипов подробно описаны ранее [15, 16]. Для создания библиотеки использовали праймеры на обратную цепь ДНК: прямой –
GCAGTCGAACATGTAGCTGACTCAGGTCACGGTGTTTAAGATTTAGGAGGTGGTAG
и обратный –
GCAGTCGAACATGTAGCTGACTCAGGTCACTCAAACACTACAACCTTAAAAACCCA.
Всего было получено 4005 прочтений. В дальнейший анализ включали образцы с количеством прочтений не менее пяти. Для каждого испытуемого вычисляли долю метилирования индивидуальных CpG/CpH как отношение прочтений с неконвертированным цитозином к общему количеству прочтений для данного CpG/CpH.
Статистический анализ проводили с помощью свободно распространяемой программы JASP 0.12.2 [17]. В дополнение к анализу цитозинов, показавших вариативное метилирование – от 0.2 до 0.8, оценили средние значения метилирования СpG для участков ДНК с различным геномным значением. Для транскрипта 1 гена YJEFN3 (NCBI Reference Sequence: NM_198537.4; chr19: 19528911–19537581) это были часть интрона 2, расположенная вокруг CpG-SNP перед CG-островком, сам островок (chr19: 19532316–19532518), экзон 3 и прилегающая к экзону часть интрона 3. Эти же участки представляли собой части интронов 1 и 2 и экзон 2 транскрипта 2 (NM_001190328.2).
Связь метилирования с когнитивным индексом оценивали методом пошаговой линейной регрессии, отдельно в группах больных и здоровых. Для этого у гетерозигот уровень метилирования усредняли по двум аллелям. Использовали иерархический подход: полную модель – с показателями метилирования всех вариативно метилированных CpG – сравнивали с нулевой моделью, включающей в качестве предикторов только пол и возраст. Аналогичный анализ проводили для участков ДНК. Изменение доли объясняемой дисперсии когнитивного индекса (ΔR2) при введении в модель метилирования служило показателем размера эффекта метилирования на когнитивный дефицит. Анализ с помощью программы G*Power 3.1.9.7 показал, что при мощности 80% и α = 0.05 выборка в 70 больных достаточна для выявления средних эффектов метилирования индивидуальных CpG на количественный признак(Cohen’s f2 ≥ 0.12, или ΔR2 ≥ 0.10) с контролем пола и возраста.
Учитывая особенности распределения показателей меДНК, аллель-специфичное метилирование (ASM) оценивали с помощью непараметрического теста Краскела–Уоллиса. Сравнивали три распространенных в европейских популяциях гаплотипа, образованных полиморфными сайтами rs10422819, rs113527843 и rs8100927 [18]. Для учета множественности сравнений значимыми считали различия, прошедшие порог FDR < 0.05. Post-hoc анализ проводили c помощью теста Данна.
РЕЗУЛЬТАТЫ
Пациенты и контроль не различались по возрасту, полу и среднему количеству прочтений. Среднее количество прочтений для больных и здоровых было равно 27.96 ± 23.44 и 28.44 ± 26.14 соответственно. Группы оказались идентичны по доле курящих (34%), потенциальному конфаундеру в исследованиях меДНК. У больных был достоверно снижен когнитивный показатель: 38.47 ± 6.76 против 50.25 ± 5.17 Т-баллов у контроля, t = 11.69, d.f. = 140, p < 0.001, Cohen’s d = 1.96.
Частота гаплотипов rs10422819–rs113527843–rs8100927 в группе больных составила: AAC – 0.23, ATG – 0.14, CAC – 0.63; у здоровых: AAC – 0.215, ATG – 0.215, CAC – 0.57, при частотах в европейской популяции [18]: AAC – 0.15, ATG – 0.17, CAC – 0.68. Различия между больными и здоровыми по распределению гаплотипов не были значимы. Среди больных было 24 (0.34) гетерозиготы, среди здоровых – 26 (0.36). Гаплотип ATG был определен как гаплотип риска шизофрении на основании данных проекта 1000 геномов [18] о сцеплении в европейских популяциях образующих гаплотип полиморфных сайтов с теговым полиморфизмом rs2905426 индексного локуса 47 PGC2 GWAS шизофрении (табл. 1).
Всего на изученном фрагменте ДНК находится 263 цитозина, из которых 43 образуют CpG-сайты. СpH-сайты были в основном неметилированы. Средний уровень метилирования геномных участков соответствовал их функциям (табл. 2). Обнаружено девять CpG с вариативным уровнем метилирования, четыре из которых находились вокруг CpG-SNP rs10422819, один располагался на границе интрона 2 и экзона 3, остальные – компактно в самом экзоне (координаты сайтов даны в табл. 2). Согласно тесту Манна–Уитни курение не влияло на метилирование в этих сайтах и участках.
Таблица 2.
Средние значения (стандартные отклонения) уровней метилирования CpG-сайтов и участков в объединенной выборке в зависимости от гаплотипа rs10422819–rs113527843–rs8100927
| Сайт (участок) YJEFN3 | Среднее (SD) | KW, p | Тест Данна, p | ||||
|---|---|---|---|---|---|---|---|
| AAC N = 43 |
ATG N = 34 |
CAC N = 115 |
AAC/ATG | AAC/CAC | ATG/CAC | ||
| 19532203 cg08623644 |
0.75 (0.13) | 0.63 (0.20) | 0.72 (0.15) | 0.011* | 0.002 | 0.087 | 0.012 |
| 19532220 | <0.01 | <0.01 | 0.63 (0.14) | <0.001* | ns | <0.001 | <0.001 |
| 19532250 | 0.21 (0.16) | 0.28 (0.15) | 0.28 (0.18) | 0.021 | 0.012 | 0.004 | ns |
| 19532259 | 0.15 (0.13) | 0.20 (0.14) | 0.22 (0.15) | 0.028 | ns | 0.004 | ns |
| 19532560 | 0.27 (0.19) | 0.28 (0.17) | 0.28 (0.16) | ns | |||
| 19532688 | 0.72 (0.16) | 0.71 (0.16) | 0.69 (0.16) | ns | |||
| 19532699 | 0.23 (0.13) | 0.24 (0.16) | 0.22 (0.15) | ns | |||
| 19532711 | 0.56 (0.20) | 0.54 (0.24) | 0.48 (0.19) | ns | ns | 0.006 | 0.032 |
| 19532726 | 0.23 (0.15) | 0.24 (0.19) | 0.21 (0.13) | ns | |||
| Инт2-CGI | 0.25 (0.06) | 0.25 (0.08) | 0.40 (0.08) | <0.001* | ns | <0.001 | <0.001 |
| CGI | 0.01 (0.01) | 0.02 (0.01) | 0.01 (0.01) | ns | |||
| Экзон 3 | 0.71 (0.07) | 0.71 (0.07) | 0.69 (0.06) | ns | |||
| Интрон 3 | 0.90 (0.07) | 0.91 (0.07) | 0.89 (0.06) | ns | |||
Примечание. SD – стандартное отклонение, ns – различия не значимы, p > 0.05. * – различия, значимые при FDR < 0.05. В колонке “Сайт” для отдельных вариативно метилированных CpG даны координаты на хромосоме 19 (hg38). Int2-CGI – фрагмент интрона 2 транскрипта 1 YJEFN3, расположенный перед CG-островком – вокруг CpG-SNP; CGI – островок в интроне 2. KW – критерий Краскела–Уоллиса.
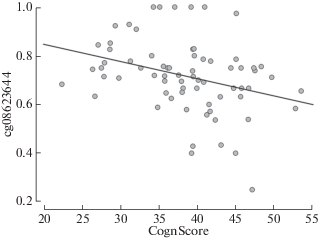
Единственным значимым предиктором когнитивного дефицита больных (p = 0.010, ΔR2 = 0.093) оказался уровень метилирования в СpG-сайте с координатой chr19: 19532203, который обозначен как проба cg08623644 на микрочипах Illumina Infinium Human-Methylation450, использовавшихся в большинстве полнометиломных исследований шизофрении (см. обзор [19]). Корреляция между метилированием и когнитивным индексом была негативной: значение коэффициента регрессии = = –14.09, 95% bca CI = –22.37...–3.53 (95%-ные доверительные интервалы [CI] определяли с помощью бутстреп-метода с использованием 5000 бутстреп-выборок, применяли байесовскую коррекцию). Таким образом, более низкому уровню метилирования соответствовали более высокие значения когнитивного индекса (рис. 2). Последующий анализ показал, что вклад метилирования cg08623644 в дисперсию когнитивного индекса оставался значимым (p = 0.021; ΔR2 = 0.085) при контроле генотипа и курения. Для данного анализа генотип кодировали как 0, 1 или 2 в зависимости от количества аллелей с гаплотипом риска. Он не влиял на когнитивный индекс. У здоровых не выявлено связей метилирования с уровнем когнитивного функционирования.
Рис. 2.
Связь между когнитивным индексом и уровнем метилирования cg08623644 (CpG_19532203) в группе больных шизофренией. На оси х расположены значения когнитивного индекса, выраженные в Т‑баллах, на оси y – доля метилирования CpG.
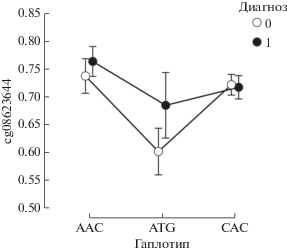
Результаты оценки ASM в объединенной выборке представлены в табл. 2. Как и ожидалось, имела место отчетливая зависимость меДНК от гаплотипа непосредственно в CpG-SNP: в случае аллеля А CpG сайт отсутствовал. Соответственно, при гаплотипах AAC и ATG, в отличие от гаплотипа CAC, метилирования практически не наблюдалось. Аналогичные различия между гаплотипами получены для среднего уровня метилирования всей части интрона 2, расположенной вокруг CpG-SNP, перед CG-островком. Помимо этого, значимое ASM отмечено для cg08623644. В этом сайте метилирование было снижено при гаплотипе риска ATG относительно двух других гаплотипов – AAC и CAC. Раздельный анализ групп показал, что у больных ASM в cg08623644 в отличие от здоровых не выражено: в группе больных p = = 0.296, в группе здоровых p = 0.028 (рис. 3). При этом логистическая регрессия (с контролем пола и возраста) не выявила значимой связи уровня метилирования с диагнозом в этом или каком-либо другом вариативно метилированном CpG. Группы также не различались по средним уровням метилирования участков. Для того, чтобы проверить, не является ли это следствием неодинакового покрытия для разных испытуемых, дополнительно провели попарные сравнения метилирования у больных и здоровых с помощью теста Вилкоксона для связанных выборок. Пары аллелей “пациент-контроль” (n = 76) подобрали по количеству прочтений и гаплотипу. Как и логистическая регрессия, тест Вилкоксона не выявил значимых различий по метилированию между больными и здоровыми.
ОБСУЖДЕНИЕ
В настоящей работе мы оценили уровень метилирования индивидуальных цитозинов и участков с различными геномными функциями на фрагменте ДНК длиной около 1 kb, расположенном внутри индексного локуса 47 PGC2 GWAS шизофрении и представляющего собой часть гена YJEFN3, а также ассоциацию меДНК на данном участке с когнитивным дефицитом больных. Полученные данные свидетельствуют о наличии ассоциации между метилированием CpG-сайта с координатой chr19: 19532203 (Illumina ID: cg08623644) и индексом когнитивного функционирования пациентов. Более низкие, т.е. более близкие к нормативным, значения метилирования в этом сайте соответствовали большей сохранности когнитивных функций. В группе здоровых подобная корреляция отсутствовала, что указывает на ее специфичность для шизофрении. Этот же сайт показал аллель-специфичное метилирование, связанное с гаплотипом риска шизофрении: метилирование было снижено при гаплотипе риска относительно других гаплотипов в объединенной выборке, а также в группе здоровых. У больных выявлено ослабление связи между метилированием cg08623644 и гаплотипом, хотя значимых отличий по метилированию от здоровых не наблюдалось.
Общая гипотеза, лежащая в основе поиска ассоциации меДНК в регионах GWAS шизофрении с симптомами болезни, заключается в том, что меДНК может зависеть от генотипа и опосредовать причинно-следственную связь между структурой ДНК и проявлениями заболевания. Предполагается, что это имеет место, если метилирование находится под влиянием генотипа, т.е. mQTL; в других случаях выявляемое при различных заболеваниях дифференциальное метилирование отражает не путь от структуры ДНК к заболеванию, а нисходящие влияния болезненного процесса или предшествующих ему средовых факторов риска на метилирование [20]. Ранее [7], в отсутствие дифференциального метилирования cg08623644 при шизофрении, байесовский анализ ко-локализации подтвердил вероятность существования общего каузального фактора – полиморфизма – для связи локуса 47 с шизофренией и для аллель-специфичного метилирования cg08623644. В последующих исследованиях было показано, что на метилирование cg08623644 влияет более 200 mQTL, включая расположенный на исследованном фрагменте rs113527843, но не ближайший CpG-SNP rs10422819 ([20]; https://ftp.ncbi.nlm.nih.gov/eqtl/ original_submissions/FHS_meQTLs/). Наши результаты относительно аллель-специфичного метилирования cg08623644 в общей выборке полностью согласуются с этими данными. Тем не менее, вопреки первоначальной гипотезе, полученные нами результаты в совокупности указывают на то, что метилирование в этом сайте при шизофрении, по-видимому, отражает средовые или стохастические факторы, запускающие или сопровождающие болезненный процесс, и не является “проводником” генетических влияний на фенотип.
Проведенное исследование не дает ответа на вопрос, маркером изменений работы какого именно гена или группы генов может быть повышение метилирования cg08623644. Регуляторный участок EH38E1945345, на границе которого находится cg08623644, определен в рамках ENCODE как дистальный энхансер (>2 kb от старта инициации транскрипции) [21], что позволяет рассматривать целый ряд генов в качестве мишеней. На основании проведенного нами анализа баз данных UCSC genomic browser [10], GTEx [11], Human Protein Atlas [22] и ORegAnno [23] и литературы о близлежащих генах (табл. 1), наиболее интересным кандидатом для дальнейших исследований представляется YJEFN3, поскольку он, единственный из этих генов, характеризуется выраженной экспрессией в мозге. Недавнее исследование [24] позволяет предположить, чтоYJEFN3 может быть вовлечен в патогенез шизофрении несколькими путями, в частности, как фактор ангиогенеза или как участник нарушенной при шизофрении регуляции NOTCH-сигнального пути [25].
Полученные результаты должны быть рассмотрены в свете имеющихся ограничений исследования. Во-первых, использованная выборка, возможно, не обладала достаточной мощностью для выявления восходящих от структуры ДНК – каузальных – эффектов метилирования на фенотип, поскольку предполагается, что их размер меньше, чем нисходящий эффект болезни на метилирование [20]. На основании имеющихся немногочисленных данных о связи метилирования с количественными поведенческими фенотипами (например, [26]) можно предположить, что эффекты (R2) составляют примерно 0.05 и, следовательно, требуют выборки размером более 150 человек для обеспечения 80% мощности. Второе ограничение связано с использованием периферической крови для поиска маркеров психических функций и их патологии, так как меДНК тканеспецифично. Для cg08623644 обнаружены значимые положительные корреляции между уровнями его метилирования в крови и ряде регионов мозга [27, 28] несмотря на то, что доля метилирования данного цитозина в крови (около 70%) значительно выше такового в мозге (около 30–40%). Эти данные получены для общей популяции и согласуются с представлениями о межтканевой конкордантности меДНК в таких CpG, где метилирование находится под существенным генетическим влиянием (например, [29]). В нашем исследовании у больных эффект гаплотипа на метилирование данного сайта в периферической крови был ослаблен, что затрудняет интерпретацию выявленных закономерностей применительно к работе мозга. Наконец, мы учитывали только ограниченный ряд негенетических факторов, связанных с заболеванием или образом жизни в целом, потенциально влияющих на метилирование. В частности, мы не могли включить в анализ эффекты антипсихотического лечения, поскольку все пациенты получали комплексную психофармакотерапию, различную на разных этапах заболевания. При этом следует отметить, что роль типичных нейролептиков, в частности галоперидола, в выявленной связи между метилированием индивидуального цитозина и тяжестью когнитивного дефицита представляется маловероятной, поскольку влияние галоперидола на метилом, по-видимому, носит генерализованный характер, способствуя гиперметилированию ДНК путем усиления экспрессии генов, кодирующих ДНК-метилтрасферазы и метилцитозин-связывающий доменный белок 2 [30]. В то же время атипичный антипсихотик клозапин вызывает разнонаправленные, в том числе нормализующие картину меДНК при шизофрении, изменения в метилировании нескольких десятков тысяч конкретных CpG [31, 32]. Назначение клозапина связано с резистентными, более тяжелыми формами шизофрении, и, возможно, улучшает когнитивное функционирование (см. обзор [33]). На этом основании можно было бы предположить роль клозапина в выявленной ассоциации уровня метилирования cg08623644 и когнитивного дефицита, например, как фактора, нормализующего оба признака. Однако среди генов, для которых к настоящему времени обнаружена выраженная связь лечения клозапином с метилированием, YJEFN3 отсутствует [31, 34].
В целом исследование выявило сайт CpG_19532203 (cg08623644), метилирование которого у больных шизофренией коррелирует с уровнем когнитивного дефицита и в меньшей степени зависит от гаплотипа, чем у здоровых. Полученные результаты позволяют предположить, что метилирование cg08623644 может быть периферическим маркером молекулярных процессов, связанных с тяжестью течения заболевания, что требует дальнейшего изучения.
Исследование выполнено за счет гранта Российского научного фонда (проект № 16-15-00056).
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики.
От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci // Nature. 2014. V. 511. P. 421–427. https://doi.org/10.1038/nature13595
Pardiñas A.F., Holmans P., Pocklington A.J. et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection // Nat. Genet. 2018. V. 50. № 3. P. 381–389. https://doi.org/10.1038/s41588-018-0059-2
Cannon M.E., Mohlke K.L. Deciphering the emerging complexities of molecular mechanisms at GWAS loci // Am. J. Hum. Genet. 2018. V. 103. P. 637–653. https://doi.org/10.1016/j.ajhg.2018.10.001
Schulz H., Ruppert A., Herms S. et al. Genome-wide mapping of genetic determinants influencing DNA methylation and gene expression in human hippocampus // Nat. Commun. 2017. V. 8. Article 151. https://doi.org/10.1038/s41467-017-01818-4
Алфимова М.В., Кондратьев Н.В., Голимбет В.Е. Результаты и перспективы генетики когнитивных нарушений при шизофрении: эпигенетические подходы // Журн. неврологии и психиатрии им. С.С. Корсакова. 2017. Т. 117. № 2. С. 130–135. https://doi.org/10.17116/jnevro201711721130-135
Halverson T.F., Orleans-Pobee M., Merritt et al. Pathways to functional outcomes in schizophrenia spectrum disorders: Meta-analysis of social cognitive and neurocognitive predictors // Neurosci. Biobehav. Rev. 2019. V. 105. P. 212–219. https://doi.org/10.1016/j.neubiorev.2019.07.020
Hannon E., Dempster E., Viana J. et al. An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation // Genome Biol. 2016. V. 17. Article 176. https://doi.org/10.1186/s13059-016-1041-x
Viana J., Hannon E., Dempster E. et al. Schizophrenia-associated methylomic variation: molecular signatures of disease and polygenic risk burden across multiple brain regions // Hum. Mol. Genet. 2017. V. 26. P. 210–225. https://doi.org/10.1093/hmg/ddw373
Кондратьев Н.В., Алфимова М.В., Голимбет В.Е. Поиск мишеней для обнаружения связи между метилированием ДНК и нейрокогнитивным дефицитом при шизофрении // Журн. неврологии и психиатрии им. C.C. Корсакова. 2017. Т. 117. № 8. С. 72–75. https://doi.org/10.17116/jnevro20171178172-75
Haeussler M., Zweig A.S., Tyner C. et al. The UCSC Genome Browser database: 2019 update // Nucleic Acids Res. 2019. V. 47 (D1). P. D853–D858. https://doi.org/10.1093/nar/gky1095
GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (LDACC)–Analysis Working Group; Statistical Methods groups–Analysis Working Group et al. Genetic effects on gene expression across human tissues // Nature. 2017. V. 550. № 7675. P. 204–213. https://doi.org/10.1038/nature24277
Lioznova A.V., Khamis A.M., Artemov A.V. et al. CpG traffic lights are markers of regulatory regions in human genome // BMC Genomics. 2019. V. 20. Article 102. https://doi.org/10.1186/s12864-018-5387-1
Yang Y., Scott S.A. DNA methylation profiling using long-read single molecule real-time bisulfite sequencing (SMRT-BS) // Methods Mol. Biol. 2017. V. 1654. P. 125–134. https://doi.org/10.1007/978-1-4939-7231-9_8
Лежейко Т.В., Алфимова М.В. Эпигенетические исследования когнитивного дефицита при шизофрении: некоторые методологические проблемы // Журн. неврологии и психиатрии им. C.C. Корсакова. 2017. Т. 117. № 10. С. 76–80. https://doi.org/10.17116/jnevro201711710176-80
Kondratyev N., Golov A., Alfimova M. et al. Prediction of smoking by multiplex bisulfite PCR with long amplicons considering allele-specific effects on DNA methylation // Clin. Epigenetics. 2018. V. 10. Article 130. https://doi.org/10.1186/s13148-018-0565-1
Алфимова М.В., Кондратьев Н.В., Голов А.К., Голимбет В.Е. Метилирование промотора гена рилина в периферической крови и его связь с когнитивным функционированием больных шизофренией // Мол. биология. 2018. Т. 52. № 5. С. 782–792. https://doi.org/10.1134/S0026898418050026
JASP Team. JASP (Version 0.12.2) [Computer software]. 2020.https://jasp-stats.org/.
The 1000 Genomes Project Consortium. A global reference for human genetic variation // Nature. 2015. V. 526. P. 68–74. https://doi.org/10.1038/nature15393
Richetto J., Meyer U. Epigenetic modifications in schizophrenia and related disorders: Molecular scars of environmental exposures and source of phenotypic variability // Biol. Psychiatry. 2020. S0006-3223(20)31327-5. https://doi.org/10.1016/j.biopsych.2020.03.008
Huan T., Joehanes R., Song C. et al. Genome-wide identification of DNA methylation QTLs in whole blood highlights pathways for cardiovascular disease // Nat. Commun. 2019. V. 10. Article 4267. https://doi.org/10.1038/s41467-019-12228-z
ENCODE Project Consortium, Moore J.E., Purcaro M.J. et al. Expanded encyclopedias of DNA elements in the human and mouse genomes // Nature. 2020. V. 583. № 7818. P. 699–710. https://doi.org/10.1038/s41586-020-2493-4
Uhlén M., Fagerberg L., Hallström B.M. et al. Proteomics. Tissue-based map of the human proteome // Science. 2015. V. 347. № 6220. Article 1260419. https://doi.org/10.1126/science.1260419
Lesurf R., Cotto K.C., Wang G. et al. ORegAnno 3.0: A community-driven resource for curated regulatory annotation // Nucl. Ac. Res. 2016. V. 44 (D1). P. D126–D132. https://doi.org/10.1093/nar/gkv1203
Gu Q., Yang X., Lv J. et al. AIBP-mediated cholesterol efflux instructs hematopoietic stem and progenitor cell fate // Science. 2019. V. 363. P. 1085–1088. https://doi.org/10.1126/science.aav1749
Hoseth E.Z., Krull F., Dieset I. et al. Attenuated notch signaling in schizophrenia and bipolar disorder // Sci. Rep. 2018. V. 8. Article 5349. https://doi.org/10.1038/s41598-018-23703-w
Taylor R.M., Smith R., Collins C.E. et al. Global DNA methylation and cognitive and behavioral outcomes at 4 years of age: A cross-sectional study // Brain Behav. 2020. V. 10. e01579. https://doi.org/10.1002/brb3.1579
Hannon E., Lunnon K., Schalkwyk L., Mill J. Blood Brain DNA Methylation Comparison Tool. 2018. https://epigenetics.essex.ac.uk/bloodbrain/?probena-meg=cg08623644.
EdgarR., Jones M., Meaney M. et al. BECon: A tool for interpreting DNA methylation findings from blood in the context of brain // Transl. Psychiatry. 2017. V. 7. e1187. https://doi.org/10.1038/tp.2017.171
van den Oord E.J., Clark S.L., Xie L.Y. et al. A Whole methylome CpG-SNP association study of psychosis in blood and brain tissue // Schizophr. Bull. 2016. V. 42. P. 1018–1026. https://doi.org/10.1093/schbul/sbv182
Swathy B., Banerjee M. Haloperidol induces pharmacoepigenetic response by modulating miRNA expression, global DNA methylation and expression profiles of methylation maintenance genes and genes involved in neurotransmission in neuronal cells // PLoS One. 2017. V. 12. e0184209. https://doi.org/10.1371/journal.pone.0184209
Kinoshita M., Numata S., Tajima A. et al. Effect of Clozapine on DNA methylation in peripheral leukocytes from patients with treatment-resistant schizophrenia // Int. J. Mol. Sci. 2017. V. 18. Article 632. https://doi.org/10.3390/ijms18030632
Higgins-Chen A.T., Boks M.P., Vinkers C.H. et al. Schizophrenia and epigenetic aging biomarkers: Increased mortality, reduced cancer risk, and unique Clozapine Effects // Biol. Psychiatry. 2020. V. 88. P. 224–235. https://doi.org/10.1016/j.biopsych.2020.01.025
MacKenzie N.E., Kowalchuk C., Agarwal S.M. et al. Antipsychotics, metabolic adverse effects, and cognitive function in schizophrenia // Front. Psychiatry. 2018. V. 9. Article 622. https://doi.org/10.3389/fpsyt.2018.00622
Hannon E., Dempster E.L., Mansell G. et al. Large-scale analysis of DNA methylation identifies cellular alterations in blood from psychosis patients and molecular biomarkers of treatment-resistant schizophrenia // bioRxiv 2020.04.05.026211. https://doi.org/10.1101/2020.04.05.026211
Дополнительные материалы отсутствуют.