

Генетика, 2022, T. 58, № 10, стр. 1164-1176
Клинико-генетические особенности врожденного синдрома удлиненного интервала QT
А. Е. Постригань 1, *, Н. П. Бабушкина 1, Л. И. Свинцова 2, И. В. Плотникова 2, Н. А. Скрябин 1
1 Научно-исследовательский институт медицинской генетики, Томский национальный исследовательский медицинский центр Российской академии наук
634050 Томск, Россия
2 Научно-исследовательский институт кардиологии, Томский национальный исследовательский медицинский центр Российской академии наук
634012 Томск, Россия
* E-mail: postrigan.anna@medgenetics.ru
Поступила в редакцию 04.04.2022
После доработки 30.05.2022
Принята к публикации 31.05.2022
- EDN: ECKAQA
- DOI: 10.31857/S001667582210006X
Аннотация
Синдром удлиненного интервала QT представляет собой редкое заболевание, обусловленное нарушениями работы ионных каналов сердца, основными проявлениями которого являются удлинение интервала QT на ЭКГ и желудочковые аритмии, которые могут стать причиной внезапной сердечной смерти в молодом возрасте. Диагностика и лечение данного синдрома представляют собой актуальную проблему, так как заболевание гетерогенно и для назначения эффективной терапии необходимо проведение молекулярно-генетической диагностики для точного определения типа этой патологии. В данном обзоре рассмотрены клинико-генетические особенности различных типов врожденного синдрома удлиненного интервала QT.
Согласно данным Всемирной Организации Здравоохранения, сердечно-сосудистые заболевания (ССЗ) остаются главной причиной смертности, несмотря на постоянное совершенствование методов их диагностики и лечения. В Российской Федерации смертность от ССЗ представляет собой острую проблему, поскольку является одной из наиболее высоких в мире и составляет 614 на 100 000 жителей в год [1]. Синдром удлиненного интервала QT (СУИQT) представляет собой наследственное заболевание, характеризующееся удлинением интервала QT на ЭКГ, приступами потери сознания на фоне возникновения полиморфной желудочковой тахикардии torsadesdepointes, которая может перейти в фибрилляцию желудочков и стать причиной внезапной сердечной смерти (ВСС) [1, 2].
СУИQT может быть как врожденным, так и приобретенным. Причинами возникновения приобретенного СУИQT могут являться нарушения электролитного баланса (такие как гипокалиемия, гипокальциемия, гипомагниемия) и употребление ряда лекарственных препаратов (антипсихотики, антидепрессанты, антиаритмики, антибиотики, диуретики, противогрибковые, противоопухолевые и др.) [2].
Распространенность СУИQT в популяции составляет, по последним данным, около 1 : 2000–1 : : 3000 новорожденных, однако эти данные включают в себя только подтвержденные на ЭКГ случаи, фактическая частота гораздо выше. Основным диагностическим критерием СУИQT является удлинение корригированного интервала QT (QTc), который определяется по формуле Базетта для оценки величины интервала QT относительно частоты сердечных сокращений. Кроме того, для диагностики СУИQT требуется анализ записи 24-часового холтеровского мониторирования и тестов с физической нагрузкой, сбор клинического и семейного анамнеза (при этом особого внимания заслуживают случаи внезапных смертей близких родственников в молодом возрасте), выявление патогенных генетических вариантов у пациента и членов его семьи с помощью молекулярно-генетического тестирования [3]. Трудности диагностики синдрома обусловлены неспецифической клинической картиной, высокой частотой скрытой формы, при которой невозможно диагностировать синдром при помощи ЭКГ, сложностью и высокой стоимостью молекулярно-генетического тестирования.
У большинства пациентов первое аритмическое событие возникает в течение первых 20 лет жизни, однако возраст манифестации может варьировать в зависимости от типа синдрома [4]. Особую группу пациентов с СУИQT представляют собой дети до года: почти 10% младенцев, внезапно умирающих в первый год жизни, являются носителями генетических вариантов, вызывающих СУИQT [5]. Без генетического тестирования смерть младенца, внезапно умершего в первые месяцы жизни, как правило, трактуется как случай синдрома внезапной детской смерти, соответственно распространенность СУИQT может быть несколько занижена и в этой группе. Поэтому необходимо широко внедрять скрининг ЭКГ в первый месяц жизни для выявления младенцев с СУИQT и выделения их в группу риска по ВСС [5].
МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКАЯ ХАРАКТЕРИСТИКА ВРОЖДЕННЫХ ФОРМ СУИQT
В настоящее время идентифицировано 16 различных типов врожденного синдрома удлиненного интервала QT. В основе патогенеза всех типов СУИQT лежат нарушения работы ионных каналов, в результате которых происходят патологические изменения скорости ионных токов, также изменяется продолжительность потенциала действия (табл. 1).
Таблица 1.
Формы синдрома удлиненного интервала QT
| Тип СУИQT | Хромосомный локус | Ген | Изменение ионного тока |
|---|---|---|---|
| СУИQT1 | 11p15.5 | KCNQ1 | Снижение калиевого тока |
| СУИQT2 | 7p35-36 | KCNH2 | Снижение калиевого тока |
| СУИQT3 | 3p21-24 | SCN5A | Усиление натриевого тока |
| СУИQT4 | 4p25-27 | ANK2 | Снижение кальциевого, натриевого и калиевого токов |
| СУИQT5 | 21q22.1-22.2 | KCNE1 | Снижение калиевого тока |
| СУИQT6 | 21q22.1-22.2 | KCNE2 | Снижение калиевого тока |
| СУИQT7 | 17q23.1-q24.2 | KCNJ2 | Снижение калиевого тока |
| СУИQT8 | 12q13.3 | CACNA1C | Усиление кальциевого тока |
| СУИQT9 | 3p25.3 | CAV3 | Усиление натриевого тока |
| СУИQT10 | 11q23.3 | SCN4B | Усиление натриевого тока |
| СУИQT11 | 7q21-q22 | AKAP9 | Снижение калиевого тока |
| СУИQT12 | 20q11.21 | SNTA1 | Усиление натриевого тока |
| СУИQT13 | 11q24.3 | KCNJ5 | Снижение калиевого тока |
| СУИQT14 | 14q32.11 | CALM1 | Снижение кальциевого тока |
| СУИQT15 | 2p21 | CALM2 | Снижение кальциевого тока |
| СУИQT16 | 19q13.32 | CALM3 | Снижение кальциевого тока |
Известно, что клетки сердца обладают способностью к генерации и распространению потенциала действия (ПД). ПД формируется благодаря движению ионов через клеточную мембрану, в результате которого клетка переходит из состояния покоя в активированное состояние (фаза деполяризации) и обратно к мембранному потенциалу покоя (фаза реполяризации). Все эти процессы тесно опосредуются множеством ионных каналов и регуляторных белков: формирование фаз является следствием синергической активации и инактивации нескольких потенциал-зависимых ионных каналов, обеспечивающих избирательное проникновение ионов (натриевых, калиевых и кальциевых) [6]. Малейшее изменение генерации, распространения импульсов или продолжительности ПД является основой нарушений сердечного ритма. Интервал QT отражает продолжительность общей электрической активности желудочков, включая деполяризацию и реполяризацию, а его удлинение – замедленную и асинхронную реполяризацию миокарда желудочков [1].
Мутации в генах ионных каналов кардиомиоцитов вызывают либо их функциональные изменения, либо полное прекращение работы. Для разных типов СУИQT характерны различия в тяжести течения и клинических проявлений, в зависимости от того, какой именно канал повреждается. Установлено, что риск сердечных событий значительно выше у пациентов с СУИQT 1-го и 2-го типов, чем среди лиц с СУИQT типа 3 [7]. Однако несмотря на меньшую встречаемость аритмических событий в группе пациентов с СУИQT 3-го типа, у них выявлена большая летальность. Так, в исследовании W. Zareba с соавт. было показано, что смерть во время синкопе, сопровождающегося желудочковой аритмией, была зарегистрирована у 20% у пациентов с СУИQT 3 и только в 4% случаев – у пациентов с СУИQT 1-го и 2-го типов [7].
Большая часть форм СУИQT вызывается патогенными или вероятно патогенными нуклеотидными заменами в генах, кодирующих различные субъединицы потенциал-зависимых калиевых, кальциевых, натриевых каналов, а также в генах белков-модификаторов работы этих каналов.
ФОРМЫ СУИQT, ОБУСЛОВЛЕННЫЕ НАРУШЕНИЕМ РАБОТЫ КАЛИЕВЫХ КАНАЛОВ
Большая часть типов СУИQT вызывается снижением калиевого тока (табл. 1), причем в основном – мутациями в генах потенциал-зависимых калиевых каналов (KCNQ1, KCNH2, KCNE1, KCNE2, KCNJ2, KCNJ5).
СУИQT1 является наиболее частой формой – до 30–35% от всех случаев врожденного синдрома удлиненного QT наследуется по аутосомно-доминантному типу. Эта форма обусловлена патогенными/вероятно патогенными вариантами в гене, кодирующем калиевый потенциал-зависимый канал подсемейства Q (KCNQ1) [3]. Ген KCNQ1 локализован в коротком плече 11-й хромосомы, содержит 19 экзонов и охватывает более 400 тыс. пар нуклеотидов (тпн) [8]. Белок KCNQ1 является порообразующей субъединицей канала IKs (рис. 1) и осуществляет регуляцию продолжительности потенциала действия миокарда [9]. Внешний калиевый ток, генерируемый комплексом белков KCNQ1/KCNE1, является одним из токов реполяризации, способствующих прекращению ПД [9]. В настоящее время описано более 600 вариантов нуклеотидной последовательности в гене KCNQ1, вызывающих СУИQT1, от локализации конкретной замены в структуре ионного канала зависит вероятность сердечных приступов [3]. Так, локализация в структуре С-петли представляет наивысший риск остановки сердца или ВСС [3]. Один из наиболее частых вариантов – однонуклеотидная замена A341V. Это миссенс-мутация, при которой происходит замена аланина на валин в положении 341 в трансмембранном домене S6, связанная с тяжелым течением СУИQT1 [10]. У носителей данного варианта описано более раннее проявление симптомов, большая продолжительность интервала QTc и высокая вероятность ВСС при бессимптомной форме. P. Brink с соавт. связывали вышеописанные проявления со снижением у носителей величины реполяризующего тока IKs примерно на 50%, также было установлено, что большинство эпизодов аритмии с летальным исходом у пациентов с СУИQT1 связано с физическими нагрузками и эмоциональным стрессом [11].
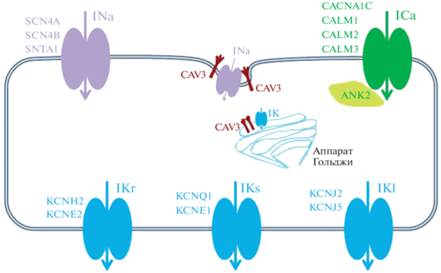
Рис. 1.
Схематичное расположение ионных каналов в кардиомицете. INa – входящий натриевый ток, осуществляется через потенциал-зависимые натриевые каналы, локализованные в цитоплазматической мембране кардиомиоцита; SCN4A, SCN4B, SNTA1 – белковые субъединицы натриевых каналов; CAV3 – белок кавеолин, локализован в кавеолах в цитоплазматической мембране и аппарате Гольджи; ICa – входящий кальциевый ток, осуществляется через потенциал-зависимые кальциевые каналы, локализованные в цитоплазматической мембране кардиомиоцита; CACNA1C – белковая субъединица кальциевого канала; CALM1, CALM2, CALM3 – белки-регуляторы работы кальциевых каналов; ANK2 – белок, стабилизирующий работу всех типов ионных каналов; IKr – быстрый калиевый ток задержанного выпрямления; KCNH2, KCNE2 – белковые субъединицы калиевого канала; IKs – медленный калиевый ток задержанного выпрямления; KCNQ1, KCNE1 – белковые субъединицы калиевого канала; IK1 – калиевый ток аномального выпрямления; KCNJ2, KCNJ5 – белковые субъединицы калиевого канала [75 ] .
СУИQT2 – второй по распространенности подтип синдрома, наследуется по аутосомно-доминатному типу и встречается у 25–30% пациентов [3]. Причиной заболевания являются патогенные и вероятно патогенные варианты нуклеотидной последовательности в гене KCNH2 (HERG). Ген локализован в длинном плече седьмой хромосомы, содержит 16 экзонов длиной от 100 до 553 пар нуклеотидов (пн) [12]. Ген кодирует порообразующую субъединицу калиевого канала задержанного выпрямления (hERG), который играет существенную роль в окончательной реполяризации потенциала действия желудочков сердца (рис. 1). Калиевый канал hERG состоит из четырех идентичных альфа-субъединиц, образующих поры в цитоплазматической мембране, каждая субъединица включает шесть трансмембранных альфа-спиралей. Спираль S4 содержит положительно заряженный аргинин или лизин и действует как чувствительный к напряжению датчик, который позволяет каналу изменять свою конформацию между проводящим и непроводящим состояниями (так называемое “стробирование”) [13]. Стробирование представляет собой процесс перехода ионного канала между открытым и закрытым состояниями. Когда ионные каналы находятся в закрытом состоянии, они непроницаемы для ионов и не проводят электрический ток. В открытом состоянии каналы, напротив, проводят электрический ток и позволяют определенным ионам (в зависимости от типа канала) проходить через них и таким образом попадать через плазматическую мембрану в клетки. В норме за счет механизма стробирования осуществляется естественное подавление аритмий. Нарушение этого процесса объясняет увеличение частоты ВСС у пациентов c СУИQT2 [14]. M. Sanguinetti с соавт. изучили биофизические свойства калиевого канала IKr и его чувствительность к различным фармакологическим агентам, а также показали связь белка KCNH2 с возникновением лекарственно индуцированных аритмий. Лекарственная блокировка KCNH2 и калиевого канала IKr в сочетании с гипокалиемией провоцируют желудочковую аритмию пируэтного типа – такую же, как при врожденной форме СУИQT2 [15]. Формирование дефектного белка при врожденном СУИQT2 приводит к нарушению функции канала: в зависимости от типа замены калиевый ток может как уменьшаться и вызывать удлинение интервала QT, так и увеличиваться, что приводит к укорочению интервала QT и другому наследственному заболеванию – синдрому короткого интервала QT [16]. СУИQT2 проявляется на поверхностной электрокардиограмме в виде раздвоенного асимметричного низкоамплитудного зубца T [3]. Показано, что СУИQT2 представляет наибольшую опасность для женщин в первые девять месяцев после родов, это связано с действием нескольких триггеров. Провоцирующими факторами возникновения жизнеугрожающих аритмий являются резкие громкие звуки, испуг, стресс и физическая нагрузка. Приступы аритмии могут возникать как во время нагрузки, так и в покое [3].
СУИQT5 обусловлен патогенными и вероятно патогенными заменами в гене KCNE1. Тип наследования – аутосомно-доминантный. Ген кодирует регуляторную субъединицу калиевых потенциал-зависимых каналов подсемейства Е, содержит три экзона (рис. 1) [12]. Белок KCNE1 состоит из 129 аминокислотных остатков, трансмембранного домена и фланкирующей С-концевой последовательности. Бета-субъединицы белка KCNE1 связываются с альфа-субъединицами KCNQ1 и белком KCNH. Образующиеся в результате объединения калиевые каналы задержанного выпрямления индуцируют селективную проницаемость для ионов калия за счет деполяризации мембраны, таким образом белок KCNE1 является одним из центральных регуляторов частоты сердечных сокращений и сердечного ритма [17]. Впервые связь синдрома удлиненного интервала QT с KCNE1 продемонстрировали L. Bianchi с соавт. в 1995 г., этими же авторами СУИQT5 был выделен как отдельная форма синдрома. Было показано, что патогенные замены в гене KCNE1 приводят к дефекту белка, который, в свою очередь, продуцирует калиевые токи с измененным стробированием и уменьшенными амплитудами, а также подавляет токи KCNQ1 и KCNH2, что в итоге может приводить к более тяжелому клиническому фенотипу [18]. В дальнейших исследованиях у части пациентов с лекарственно индуцированным СУИQT были обнаружены миссенс-мутации в гене KCNE1 в гетерозиготной форме, отсутствующие у здоровых людей [19].
СУИQT6 встречается достаточно редко (в 1–2% случаев) [12, 20]. СУИQT 6-го типа обусловлен патогенными/вероятно патогенными вариантами нуклеотидной последовательности в гене KCNE2, наследуется по аутосомно-доминантному типу. Ген кодирует белок KCNE2 (регуляторная субъединица 2 подсемейства E калиевых потенциал-зависимых каналов), содержит два экзона. KCNE2 представляет собой интегральную мембранную субъединицу, которая, как и KCNE1, объединяется с порообразующим белком KCNH2 и оказывает влияние на его функции (рис. 1). Ген KCNE2 расположен в длинном плече 21-й хромосомы, в том же регионе, что и ген KCNE1. Гены KCNE2 и KCNE1 сходны между собой – их открытые рамки считывания идентичны на 34%, что указывает на их общее происхождение [21]. Каналы, образованные мутантными субъединицами KCNE2 и KCNH2, показали более медленную активацию, более быструю инактивацию и повышенную чувствительность к лекарствам [21].
СУИQT7 обусловлен патогенными вариантами в гене KCNJ2 и наследуется по аутосомно-доминантному типу. Ген KCNJ2 кодирует интегральный мембранный белок KCNJ2 (калиевый канал внутреннего выпрямления подсемейства J) (рис. 1). KCNJ2 формирует входящий калиевый ток, участвует в генерации ПД нейронов и кардиомиоцитов. Активность и функционирование каналов критически зависят от взаимодействия с фосфатидилинозитол-4,5-бисфосфатом [22]. Мутации KCNJ2 приводят к нарушению этого взаимодействия, в результате чего калиевый ток снижается. Снижение калиевого тока вызывает удлинение терминальной фазы ПД миокарда, формируются спонтанные аритмии [23]. Ген KCNJ2 картирован в длинном плече хромосомы 17, содержит два экзона [24]. Патогенные замены в этом гене приводят к синдрому Андерсена–Тавила, который характеризуется наличием у пациента триады симптомов: эпизоды периодического паралича, признаки дисморфогенеза, желудочковые нарушения ритма сердца, а также удлинение интервала QT [25].
СУИQT13 обусловлен патогенными вариантами в гене KCNJ5. Ген локализован в длинном плече 11-й хромосомы, состоит из 500 тпн, содержит два экзона [26]. Наиболее широко экспрессируется в миокарде и скелетных мышцах, кодирует белок калиевого канала внутреннего выпрямления подсемейства J (рис. 1) [27, 28]. Канал чувствителен к G-белкам, активируется путем их взаимодействия с цитоплазматическими N и C концами. Белки KCNJ высокоселективны к ионам калия, могут образовывать высокоактивные гетеромультимеры путем спаривания друг с другом [29]. Y. Yang с соавт. была также установлена связь замены G387R с синдромом удлиненного интервала QT. Данный вариант нуклеотидной последовательности локализован в высококонсервативном остатке, ингибирует работу калиевых каналов внутреннего выпрямления путем задержки реполяризации желудочков, что приводит к значительному снижению проводимости и удлинению интервала QT. ЭКГ при СУИQT13 отличается от прочих типов синдрома наличием выступающего зубца U. Вариант G387R был идентифицирован в китайской семье с СУИQT, причем как у индивидов с клиническими проявлениями, так и у бессимптомных носителей, что указывает на наличие неполной пенетрантности [27].
Синдром Джервела и Ланге-Нильсена (JLNS) является относительно редкой формой синдрома удлиненного интервала QT, представляет собой аутосомно-рецессивное заболевание, клинически проявляющееся глубокой врожденной нейросенсорной тугоухостью и выраженным удлинением интервала QTc [3]. JLNS возникает в результате гомозиготных или компаунд-гетерозиготных мутаций в генах KCNQ1 или KCNE1. JLNS представляет собой наиболее тяжелую и прогностически неблагоприятную форму СУИQT: около 90% пациентов имеют выраженную симптоматику, ВСС наступает более чем в 25% случаев, несмотря на терапию бета-адреноблокаторами. Кроме того, JLNS манифестирует в достаточно раннем возрасте, у 15% пациентов приступы случаются уже в первый год жизни, у 50% – в течение первых трех лет, к 18 годам симптомы присутствуют у 90% [30].
СУИQT11 также характеризуется изменением калиевого тока, обусловленным патогенными заменами в гене AKAP9. Данный тип синдрома наследуется по аутосомно-доминантному типу. Ген AKAP9 локализован в длинном плече седьмой хромосомы, содержит 51 экзон и включает более 170 тпн [31]. Ген кодирует якорный белок киназы А, основная функция которого состоит в удержании холоферментов в определенных клеточных компартментах путем связывания с протеинкиназой А. В результате замен в AKAP9 (преимущественно миссенс-мутаций) происходит изменение процессов фосфорилирования калиевого потенциал-зависимого канала и нарушение его работы [32]. Впервые связь гена AKAP9 с СУИQT11 была показана L. Chen с соавт. – исследователи идентифицировали миссенс-мутацию на C-конце KCNQ1-связывающего домена в гене AKAP9 [33]. AKAP9, кроме того, может выступать в роли модификатора клинического течения СУИQT 1-го типа [34]. Для пациентов с сочетанным наличием мутаций в генах KCNQ1 и AKAP9 характерны более выраженные клинические проявления в виде более значимого удлинения интервала QT и увеличения частоты возникновения аритмических событий. Интересно, что в данном случае мутации в гене AKAP9 были локализованы в интронах [34].
ФОРМЫ СУИQT, ОБУСЛОВЛЕННЫЕ НАРУШЕНИЕМ РАБОТЫ НАТРИЕВЫХ КАНАЛОВ
Изменение работы натриевых каналов приводит к развитию СУИQT типов 3, 9, 10 и 12 (табл. 1), причем типы 3 и 10 вызываются патогенными или вероятно патогенными вариантами в генах, кодирующих субъединицы натриевого потенциал-зависимого канала.
СУИQT3 обусловлен патогенными вариантами в гене SCN5A и наследуется по аутосомно-доминантному типу. Ген локализован в коротком плече хромосомы 3 и состоит из 28 экзонов, охватывающих примерно 80 тпн, экспрессируется преимущественно в кардиомиоцитах [35]. SCN5A кодирует альфа-субъединицу натриевого потенциал-зависимого канала (рис. 1). В миокарде этот белок отвечает за начальный подъем потенциала действия на электрокардиограмме. P. Bennett с соавт. описали молекулярный механизм СУИQT3: в результате дефекта каналов происходит их колебание между функциональным и неактивным режимами стробирования, в результате чего во время деполяризации мембран формируется устойчивый входящий натриевый ток, вызывающий удлинение фазы ПД кардиомиоцитов (более 50 мс) и удлинение интервала QT [36]. N. Makita с соавт. предположили, что клинические проявления СУИQT3, обусловленные дефектом SCN5A, зависят от типа наследования и воздействия модификаторов. Согласно наблюдениям авторов, одного мутантного аллеля SCN5A может быть недостаточно для возникновения тяжелого клинического фенотипа. Установлено, что в результате совместного наследования варианта D1275N в гене SCN5A и замены в промоторе белка Сх40 (коннексин 40), мутации в котором приводят к удлинению интервала PQ, наблюдаются более выраженные клинические проявления [37]. Кроме того, были описаны варианты нуклеотидной последовательности в гене SCN5A с противоположным эффектом – миссенс-мутация W1421X связана с аутосомно-доминантным наследованием аритмий и ВСС, в то же время вариант R1193Q вызывал усиление функции натриевых каналов и компенсировал негативные эффекты замены W1421X [38]. На ЭКГ СУИQT3 может проявляться в виде удлиненного изоэлектрического интервала, предшествующего относительно нормальной морфологии зубца. Приступы аритмии у пациентов с СУИQT 3-го типа происходят без явных триггеров, на фоне брадикардии, как в покое, так и во сне.
К развитию СУИQT10 приводят патогенные варианты в гене SCN4B. SCN4B кодирует бета-субъединицу натриевого потенциал-зависимого канала, представляющую собой трансмембранный белок, образующий дисульфидные связи с SCN2A и участвующий таким образом в изменении кинетики натриевых каналов (рис. 1) [39]. Ген SCN4B локализован в длинном плече 11-й хромосомы и содержит пять экзонов протяженностью в 19.5 тпн [39]. В гене описаны патогенные миссенс-мутации, вызывающие 8-кратное увеличение тока натрия. В результате происходят удлинение интервала QT и фибрилляция предсердий [40]. Тип наследования – аутосомно-доминантный.
СУИQT12 обусловлен патогенными заменами в гене SNTA1, наследуется по аутосомно-доминантному типу. Ген состоит из 24 тпн, содержит восемь экзонов и кодирует периферический мембранный белок синтрофин альфа 1 – наиболее распространенную изоформу синтрофина, локализующуюся в тканях сердца и скелетных мышцах, в меньшей степени в других тканях (рис. 1) [41, 42]. N-концевой домен этого белка взаимодействует с C-концом порообразующей альфа-субъединицы натриевого канала SCN5A. K. Ueda с соавт. идентифицирована миссенс-мутация в гене SNTA1 (A390V – замена аланина на валин в высококонсервативном домене белка, приводящая к усилению позднего натриевого тока). Вариант A390V вызывает нарушение взаимодействия белка SNTA1 с белками SCN5A, PMCA4b и эндотелиальной синтазой оксида азота, в результате чего происходит усиление позднего натриевого тока, что является характерной для СУИQT биофизической дисфункцией. У описанного авторами пациента корригированный интервал QT составлял 529 мс, наблюдались обмороки [43]. G. Wu с соавт. у трех неродственных пациентов с диагнозом СУИQT описана замена A257G в гене SNTA1, в результате которой натриевые каналы также демонстрировали патологическое усиление функции [44]. Интересно, что данная замена в сочетании с вариантом P74L была зарегистрирована у клинически здоровых индивидов; функциональные исследования показали, что P74L нивелирует патологические эффекты A257G [45].
СУИQT 9 связывают с патогенными вариантами в гене CAV3, наследование происходит по аутосомно-доминантному типу. Ген CAV3 локализован в коротком плече 3-й хромосомы, содержит два экзона. CAV3 кодирует белок кавеолин 3, который представляет собой каркасный белок и функционирует как компонент плазматических мембран кавеол – субкомпартментов плазматических мембран, представляющих собой инвагинации диаметром от 50 до 100 нм, находящиеся в большинстве типов клеток (рис. 1) [46]. Основная функция кавеол – эндоцитоз, который запускается при утрате клеткой контактов с соседними клетками или межклеточным матриксом и ингибировании серин/треониновых фосфатаз [47, 48]. Патогенные варианты, идентифицированные в CAV3, вызывают нарушение олигомеризации кавеолина-3 и образования кавеол на плазматической мембране мышечных клеток, что в большинстве случаев приводит к различным мышечным патологиям – миодистрофиям и миопатиям [49]. M. Vatta с соавт. в гене CAV3 были описаны четыре гетерозиготные миссенс-мутации, приводящие к образованию дефектного кавеолина-3 и увеличению натриевого тока в 2–3 раза [50]. Кроме того, L. Cronk с соавт. описали связь мутаций кавеолина-3 с синдромом внезапной детской смерти, у носителей патогенных генетических вариантов было выявлено повышение интенсивности натриевых токов в 5 раз по сравнению с контрольной группой [51].
ФОРМЫ СУИQT, ОБУСЛОВЛЕННЫЕ НАРУШЕНИЕМ РАБОТЫ КАЛЬЦИЕВЫХ КАНАЛОВ
Изменение работы кальциевых каналов приводит к патологическим фенотипам СУИQT8, 14, 15, 16 (табл. 1), вызываемых мутациями в генах кальмодулинов и субъединицах потенциал-зависимого кальциевого канала.
СУИQT8 вызывается патогенными вариантами в гене CACNA1C. Тип наследования данного типа синдрома – аутосомно-доминантный. Ген кодирует белок CACNA1C – альфа-1 субъединицу потенциал-зависимого кальциевого канала (рис. 1). Деполяризация мембраны запускает процесс активации чувствительных к напряжению кальциевых каналов и каскад ключевых клеточных реакций, таких как сокращение, секреция, возбуждение и передача электрических импульсов [52]. Токи L-типа, создаваемые потенциал-зависимыми кальциевыми каналами, блокируются производными 1,4-дигидропиридина (DHP), таким образом каналы, ответственные за эти токи, называются DHP-чувствительными. Эти каналы представляют собой комплекс из пяти субъединиц: альфа-1, альфа-2, бета, гамма и дельта, каждая из которых содержит, по крайней мере, шесть трансмембранных доменов (S1–S6), которые соединены линкерами переменной длины [53]. Белок CACNA1C в миокарде состоит из 2180 аминокислот и имеет расчетную молекулярную массу 243.6 кДа. [54]. Ген CACNA1C расположен в коротком плече 12-й хромосомы, его длина примерно 150 тпн и содержит 44 инвариантных и шесть альтернативных экзонов. Существует предположение, что кальциевые каналы человека L-типа регулируются посредством генерации множества вариантов сплайсинга мРНК (некоторые из них тканеспецифичны), а также посредством экспрессии различных изоформ генов [55]. Патогенные варианты в гене CACNA1C, помимо СУИQT, приводят к синдрому Тимоти и синдрому Бругада [12, 56]. СУИQT8 вызываются миссенс-мутациями в гетерозиготной форме, которые приводят к увеличению экспрессии рецепторов кальциевых каналов L-типа на поверхности клетки и усилению входящего кальциевого тока, что в итоге может увеличивать продолжительность ПД миокарда и способствовать возникновению ранней деполяризации. В результате формируется фенотип СУИQT со склонностью к потенциально летальным аритмиям [57].
СУИQT типов 14, 15, 16 связаны с патогенными вариантами в генах кальмодулинов – CALM1, CALM2 и CALM3. Ген CALM1 картирован в длинном плече 14-й хромосомы, CALM2 локализован в коротком плече хромосомы 2, CALM3 – в длинном плече хромосомы 19 [58]. Все три варианта синдрома, ассоциированные с генами кальмодулинов, наследуются по аутосомно-доминантному типу. Все три гена имеют уникальные нуклеотидные последовательности, но кодируют идентичные 149-аминокислотные белки кальмодулина с четырьмя петлями связывания кальция [59]. Кальмодулин осуществляет регуляцию множества кальций-зависимых процессов в организме, в частности работу ионных каналов CaV1.2 (ген CACNA1C) и NaV1.5 (SCN5A), локализованных в сердце (рис. 1) [59]. Регуляция осуществляется посредством взаимодействия с кальмодулин-зависимой протеинкиназой I (CAMK1), степень активности которой напрямую зависит от строения консервативных доменов кальмодулина (замены в N-концевых участках могут снижать активность CAMK1 на 80%) [60]. Также идентифицирован консервативный сайт связывания кальмодулина, играющий ключевую роль в регуляции синаптической активности кальциевых каналов, в зависимости от остаточной концентрации ионов кальция [61]. L. Crotti с соавт. было описано две миссенс-мутации в гене CALM1 – D130G и F142L, вызывающие синдром удлиненного интервала QT. Оба варианта возникли de novo, клинически проявлялись удлинением интервала QT и желудочковыми аритмиями [62]. Позднее авторы проанализировали анамнестические данные 74 носителей патогенных и вероятно патогенных вариантов в генах CALM1, CALM2 или CALM3. У 64 (86.5%) пациентов присутствовали патологические симптомы, а 10-летняя кумулятивная смертность составила 27%. Наиболее частыми диагнозами были синдром удлиненного интервала QT (49%) и катехоламинергическая полиморфная желудочковая тахикардия (28%); другие диагнозы включали идиопатическую фибрилляцию желудочков (10%), внезапную необъяснимую смерть (5%) и перекрывающийся фенотип (4%). Большинство патогенных вариантов (80%) затрагивали аминокислотные остатки в Ca-связывающих петлях. Авторы отметили, что варианты кальмодулина, ведущие к симптоматике СУИQT, демонстрируют значительное снижение активности кальций-зависимого канала L-типа, что приводит к задержке реполяризации [62]. Также была обнаружена миссенс-мутация (F90L) в CALM1, приводящая к удлинению интервала QT после физических нагрузок и фибрилляции желудочков [63]. Патогенные варианты в гене CALM2 были описаны N. Makita с соавт.: у пяти неродственных пациентов с синдромом удлиненного интервала QT идентифицировали пять миссенс-мутаций, возникших de novo. Функциональный анализ обнаруженных вариантов продемонстрировал значительное снижение аффинности связывания кальция с кальмодулином [37].
СУИQT4 характеризуется нарушением фукционирования трех типов каналов – кальциевых, натриевых и калиевых (табл. 1); вызывается патогенными/вероятно патогенными вариантами нуклеотидной последовательности в гене ANK2. СУИQT4 наследуется по аутосомно-доминантному типу. Ген локализован в длинном плече 4-й хромосомы, содержит 46 экзонов [64]. ANK2 кодирует белок анкирин-B, функция которого заключается в связывании интегральных мембранных белков со спектрин-актиновым цитоскелетом (рис. 1). Семейство анкиринов играет важную роль в процессах клеточной пролиферации, подвижности и формировании специализированных мембранных доменов. Большинство анкиринов состоит из трех структурных доменов: амино-концевого, содержащего несколько анкириновых повторов; центральной области с высококонсервативным доменом связывания спектрина и регуляторного домена на карбокси-конце, который наименее консервативен и подвержен изменениям. В кардиомиоцитах анкирин-B играет роль стабилизатора натриевых каналов. P. Mohler с соавт. впервые описали связь гена ANK2 с СУИQT 4-го типа и молекулярный механизм формирования патологии: мутации анкирина-B приводят к нарушению клеточной организации натриевого насоса, натрий/кальциевого ионообменного канала и инозитол-1,4,5-трифосфатных рецепторов. Это снижает общий уровень белка, а также приводит к изменению передачи ионов кальция в кардиомиоцитах, что вызывает экстрасистолию и аритмию. Помимо молекулярного механизма была описана клиническая картина семьи, в которой СУИQT4, обусловленный носительством патогенного варианта E1425G в гене ANK2, был диагностирован у 25 человек (11 мужчин и 14 женщин). У пациентов отмечалась синусовая брадикардия, узловой выскальзывающий ритм, эпизоды фибрилляции предсердий, причем нарушения ритма у четырех индивидов были диагностированы уже во внутриутробном периоде [65]. Позднее этим же коллективом авторов были описаны патогенные генетические варианты ANK2 у неродственных пациентов. Было установлено, что для пациентов с мутациями анкирина-В характерны различные виды нарушений ритма сердца, включая брадикардию, синусовую аритмию, идиопатическую фибрилляцию желудочков, катехоламинергическую полиморфную желудочковую тахикардию и соответственно риск внезапной смерти. Однако удлиненный интервал QT не был постоянным признаком, указывая на то, что дисфункция анкирина-B отличается от классических синдромов удлиненного QT. Также исследователи предположили, что различные степени потери функции анкирина-B являются модуляторами возникновения жизнеугрожающих аритмических событий [66].
Помимо рассмотренных выше 16 типов СУИQT существуют сочетанные формы. Интересное исследование было проведено P. Westenskow с соавт. [67]. Они проанализировали гены KCNQ1, KCNH2, SCN5A, KCNE1 и KCNE2 у 252 пробандов с СУИQT и идентифицировали патогенные либо вероятно патогенные варианты у 20 человек. При этом гомозиготное носительство было зарегистрировано у одного человека, 18 же индивидов являлись либо носителями двух патогенных вариантов в одном гене (моногенные составные гетерозиготы), либо компаунд-гетерозиготами [67]. Было установлено, что у пациентов с двумя патогенными вариантами продолжительность интервала QTc была выше и в 3.5 раза повышалась вероятность остановки сердца (у всех 20 пробандов в группе с двумя генетическими вариантами были зарегистрированы аритмические события). Авторы пришли к выводу, что двуаллельные моно- или дигенные мутации вызывают тяжелый фенотип и нередко встречаются при синдроме удлиненного интервала QT. Подобные исследования демонстрируют важность проведения молекулярной диагностики всех генов, мутации в которых приводят к СУИQT, даже после выявления у пациента одного патогенного или вероятно патогенного варианта нуклеотидной последовательности.
Представленная выше клинико-генетическая характеристика структуры синдрома удлиненного интервала QT является классической. Мутации, которые были определены как “причинные” для развития патологии, в большинстве своем были открыты в 1990–2000-е гг. [19–21]. Вместе с тем пересмотр клинической значимости описанных ранее нуклеотидных замен привел к несколько неожиданным результатам: в результате работы международного многоцентрового исследования LQTS ClinGenWorking более чем для половины генов СУИQT доказательства их “причинности” были признаны спорными или недостаточными [68]. Подтверждена клиническая значимость генов KCNQ1, KCNH2, SCN5A (приводящих к развитию СУИQT 1-го, 2-го и 3-го типов соответственно; типичный СУИQT (табл. 1)), CALM1, CALM2, CALM3 (СУИQT типов 14, 15 и 16; СУИQT с атипичными признаками). Для гена CACNA1C (СУИQT8) доказательства сочтены умеренными [68]. К генам с ограниченными доказательствами их этиологической роли были отнесены KCNE1, KCNJ2 (5-й и 7-й типы; доказаны четкие причинно-следственные связи с патологией) и CAV3 (9-й тип).
Этиологическая роль остальных генов была признана спорной [68]. Парадоксально, но недостаточными были признаны именно генетические доказательства. Так, гены AKAP9, KCNE2, SCN4B, SNTA1 были идентифицированы как “причинные” для СУИQT при реализации кандидатного подхода (не на полногеномном анализе, который стал применяться значительно позже), поэтому не подтверждены достаточными статистическими данными. Заключение об этиологической роли генов ANK2 и KCNJ5 было сделано на основании анализа сцепления в больших семьях, без учета популяционной распространенности выявленных нуклеотидных замен и возможной роли расположенных вблизи генов [68]. Неизвестно, удастся ли получить дополнительные доказательства этиологической роли данных генов в будущем, однако обращает на себя внимание следующее: СУИQT в целом является редким заболеванием, причем на долю первых трех форм приходится до 80% всех случаев. Именно для генов, являющихся “причинными” для первых трех форм, в исследовании LQTS ClinGenWorking доказательства их этиологической значимости признаны наиболее убедительными. Не стоит исключать вероятность того, что именно ввиду малочисленности семей с патогенными вариантами в генах со спорной клинической значимостью для них не было получено достаточно убедительных доказательств.
ТАКТИКА ЛЕЧЕНИЯ
Установлено, что эффективность лечения больных зависит от конкретного молекулярно-генетического варианта синдрома. Основные методы лечения СУИQT – терапия бета-адреноблокаторами, левосторонняя симпатэктомия, имплантация кардиовертера-дефибриллятора, геноспецифическая терапия. Основная цель терапии – предотвращение рецидивов тахикардии torsade de pointes и профилактика ВСС. Первой линией терапии СУИQT является терапия бета-адреноблокаторами. Носителям генетической мутации без клинических проявлений и с нормальным интервалом QT также рекомендован прием бета-адреноблокаторов в профилактических целях. Это подчеркивает необходимость молекулярно-генетической диагностики СУИQT у родственников пациентов с целью обнаружения бессимптомного носительства и предотвращения ВСС [69]. Эффективность бета-адреноблокаторов установлена при различных молекулярно-генетических вариантах и предотвращает развитие жизнеугрожающей аритмии у 81% пациентов с СУИQT 1-го типа, у 59% – с СУИQT 2-го типа и 50% – с СУИQT 3-го типа [2]. В течение многих лет существовало ошибочное представление о том, что блокаторы β-адренорецепторов бесполезны и потенциально опасны для пациентов с СУИQT 3-го типа. В настоящее время эффективность препаратов доказана и для этой категории пациентов [4]. Понимание механизма действия мутаций в гене SCN5A привело к предложению использовать блокатор натриевых каналов мексилетин для укорочения интервала QT у пациентов с СУИQT3. Этот первый пример генно-специфической терапии оказался успешным и был подтвержден недавними исследованиями. Если при фармакологической пробе с мексилетином QTc укорачивается более чем на 40 мс у пациентов с исходным QTc > 500 мс, этот препарат следует добавлять к терапии СУИQT 3-го типа [70]. Кроме того, мексилетин может быть эффективным и у части пациентов с СУИQT 2-го типа [71]. В дополнение к этим препаратам возможно назначение пациентам с СУИQT 2-го типа калий-сберегающих диуретиков с целью повышения содержания калия в сыворотке крови и коррекции продолжительности интервала QT [72]. В лечении СУИQT 3-го типа успешно применяется флекаинид. У пациентов с мутацией D1790G в гене SCN5A на фоне терапии отмечалось уменьшение продолжительности интервала QT и подавление альтернации зубца Т [72].
Признанными немедикаментозными методами лечения СУИQT являются симпатическая денервация левых отделов сердца и имплантация кардиовертера-дефибриллятора. Левосторонняя симпактэктомия (ЛСЭ) увеличивает порог желудочковой фибрилляции и продолжительность рефрактерного периода миокарда желудочков, уменьшает продолжительность интервала QT и снижает количество синкопальных эпизодов. Наиболее эффективна ЛСЭ при СУИQT 1-го типа. ЛСЭ рекомендуется пациентам, у которых рецидивы желудочковой тахикардии сохраняются, несмотря на прием максимальных доз бета-блокаторов или в связи с их плохой переносимостью [69].
ИКД-терапия в сочетании с бета-блокаторами показана больным, пережившим остановку сердца (класс I), а также тем, у кого сохраняются синкопальные состояния, связанные с желудочковой тахикардией на фоне приема бета-блокаторов (класс IIа). Имплантация кардиовертера-дефибриллятора в сочетании с приемом бета-адреноблокаторов рекомендована также пациентам из группы высокого риска, в которую входят пациенты с QTc > 500 мс (класс IIb), и носителям двух и более мутаций (в том числе пациентам с синдромом Джервелла–Ланге-Нильсена и Тимоти) [73].
Все больные с диагнозом СУИQT должны находиться под наблюдением кардиолога с оценкой динамики риска ВСС не реже, чем один раз в год.
Важной составляющей терапии является профилактика приступов – исключение триггеров и препаратов, удлиняющих интервал QT. Так, пациенты с типом 1 СУИQT подвергаются повышенному риску возникновения аритмического события всякий раз, когда увеличивается симпатическая активность, например во время эмоционального или физического стресса, особенно плавания. У пациентов со 2-ым типом жизнеугрожающая аритмия может возникнуть при воздействии внезапных шумов, особенно если они находятся в состоянии покоя, или при внезапном пробуждении [74]. Пациенты с СУИQT 3-го типа в первую очередь подвержены риску в состоянии покоя или во сне [73].
В заключение хотелось бы отметить, что максимальная эффективность терапии может быть достигнута только при наличии полной диагностической картины – результатов ЭКГ, данных молекулярно-генетического тестирования, анамнеза (включая семейный). Молекулярно-генетическая диагностика особенно важна как в подборе оптимальной терапии, так и с точки зрения профилактики, учитывая тот факт, что триггеры приступов желудочковой тахикардии различаются в зависимости от типа синдрома. Очевидно также, что необходимо расширение диагностических возможностей и более доскональное изучение менее распространенных типов СУИQT для целенаправленного подбора терапии и выявления специфических триггеров.
Работа выполнена при поддержке Министерства науки и высшего образования Российской Федерации (Федеральная научно-техническая программа развития генетических технологий на 2019–2027 гг., соглашение № 075-15-2021-1061, РФ 193021X0029).
Настоящая статья не содержит каких-либо исследований с использованием в качестве объекта животных.
Настоящая статья не содержит каких-либо исследований с участием в качестве объекта людей.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Шляхто Е.В., Арутюнов Г.П., Беленков Ю.Н. и др. Национальные рекомендации по определению риска и профилактике внезапной сердечной смерти (2-е изд.). М., 2018. 247 p.
Ильдарова Р.А., Школьникова М.А., Ильдарова Р.А. и др. Современная тактика ведения пациентов молодого возраста с синдромом удлиненного интервала QT: от ранней диагностики к имплантации кардиовертера дефибриллятора и мониторингу маркеров риска внезапной смерти // Сиб. журн. клинич. и эксперим. медицины. 2015. P. 28–35.
Wallace E., Howard L., Liu M. et al. Long QT syndrome: Genetics and future perspective // Pediatr. Cardiol. 2019. V. 40. № 7. P. 1419–1430. https://doi.org/10.1007/S00246-019-02151-X
Schwartz P.J., Ackerman M.J., Antzelevitch C. et al. Inherited cardiac arrhythmias // Nat. Rev. Dis. Prim. NIH Public Access. 2020. V. 6. № 1. P. 58. https://doi.org/10.1038/S41572-020-0188-7
Tfelt-Hansen J., Winkel B.G., Grunnet M. et al. Cardiac channelopathies and sudden infant death syndrome // Cardiology. 2011. V. 119. № 1. P. 21–33. https://doi.org/10.1159/000329047
Garcia-Elias A., Benito B. Ion channel disorders and sudden cardiac death // Int. J. Mol. Sci. 2018. V. 19. № 3. https://doi.org/10.3390/IJMS19030692
Zareba W., Moss A.J., Schwartz P.J. et al. Influence of the genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group // N. Engl. J. Med. 1998. V. 339. № 14. P. 960–965. https://doi.org/10.1056/NEJM199810013391404
Neyroud N., Richard P., Vignier N. et al. Genomic organization of the KCNQ1 K+ channel gene and identification of C-terminal mutations in the long-QT syndrome // Circ. Res. 1999. V. 84. № 3. P. 290–297. https://doi.org/10.1161/01.RES.84.3.290
Dixit G., Dabney-Smith C., Lorigan G.A. et al. The membrane protein KCNQ1 potassium ion channel: Functional diversity and current structural insights HHS public access author manuscript // Biochim. Biophys. Acta Biomembr. 2020. V. 1862. № 5. P. 183148. https://doi.org/10.1016/j.bbamem.2019.183148
Mikuni I., Torres C.G., Bienengraeber M.W. et al. Partial restoration of the long QT syndrome associated KCNQ1 A341V mutant by the KCNE1 β-subunit // Biochim. Biophys. Acta. NIH Public Access. 2011. V. 1810. № 12. P. 1285. https://doi.org/10.1016/J.BBAGEN.2011.07.018
Brink P.A., Crotti L., Corfield V. et al. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population // Circulation. 2005. V. 112. № 17. P. 2602–2610. https://doi.org/10.1161/CIRCULATIONAHA.105.572453
Splawski I., Shen J., Timothy K.W. et al. Genomic structure of three long QT syndrome genes: KVLQT1, HERG, and KCNE1 // Genomics. 1998. V. 51. № 1. P. 86–97. https://doi.org/10.1006/GENO.1998.5361
Trudeau M.C., Warmke J.W., Ganetzky B. et al. HERG, a human inward rectifier in the voltage-gated potassium channel family // Science. 1995. V. 269. № 5220. P. 92–95. https://doi.org/10.1126/SCIENCE.7604285
Smith P.L., Baukrowitz T., Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel // Nature. 1996. V. 379. № 6568. P. 833–836. https://doi.org/10.1038/379833A0
Sanguinetti M.C., Jiang C., Curran M.E. et al. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel // Cell. 1995. V. 81. № 2. P. 299–307. https://doi.org/10.1016/0092-8674(95)90340-2
Bjerregaard P. Diagnosis and management of short QT syndrome // Hear. Rhythm. Elsevier. 2018. V. 15. № 8. P. 1261–1267. https://doi.org/10.1016/J.HRTHM.2018.02.034
McDonald T.V., Yu Z., Ming Z. et al. A minK-HERG complex regulates the cardiac potassium current I(Kr) // Nature. 997. V. 388. № 6639. P. 289–292. https://doi.org/10.1038/40882
Bianchi L., Shen Z., Dennis A.T. et al. Cellular dysfunction of LQT5-minK mutants: Abnormalities of IKs, IKr and trafficking in long QT syndrome // Hum. Mol. Genet. 1999. V. 8. № 8. P. 1499–1507. https://doi.org/10.1093/HMG/8.8.1499
Paulussen A.D.C., Gilissen R.A.H.J., Armstrong M. et al. Genetic variations of KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 in drug-induced long QT syndrome patients // J. Mol. Med. (Berl.). 2004. V. 82. № 3. P. 182–188. https://doi.org/10.1007/S00109-003-0522-Z
Millat G., Chevalier B., Restier-Miron L. et al. Spectrum of pathogenic mutations and associated polymorphisms in a cohort of 44 unrelated patients with long QT syndrome // Clin. Genet. 2006. V. 70. № 3. P. 214–227. https://doi.org/10.1111/J.1399-0004.2006.00671.X
Abbott G.W., Sesti F., Splawski I. et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia // Cell. 1999. V. 97. № 2. P. 175–187. https://doi.org/10.1016/S0092-8674(00)80728-X
Lopes C.M.B., Zhang H., Rohacs T. et al. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies // Neuron. 2002. V. 34. № 6. P. 933–944. https://doi.org/10.1016/S0896-6273(02)00725-0
Tristani-Firouzi M., Jensen J.L., Donaldson M.R. et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome) // J. Clin. Invest. 2002. V. 110. № 3. P. 381–388. https://doi.org/10.1172/JCI15183
Derst C., Karschin C., Wischmeyer E. et al. Genetic and functional linkage of Kir5.1 and Kir2.1 channel subunits // FEBS Lett. 2001. V. 491. № 3. P. 305–311. https://doi.org/10.1016/S0014-5793(01)02202-5
Pérez-Riera A.R., Barbosa-Barros R., Samesina N. et al. Andersen–Tawil syndrome: A comprehensive review // Cardiol. Rev. 2021. V. 29. № 4. P. 165–177. https://doi.org/10.1097/CRD.0000000000000326
Wickman K., Seldin M.F., Gendler S.J. et al. Partial structure, chromosome localization, and expression of the mouse Girk4 gene // Genomics. 1997. V. 40. № 3. P. 395–401. https://doi.org/10.1006/GENO.1997.4599
Yang Y., Yang Y., Liang B. et al. Identification of a Kir3.4 mutation in congenital long QT syndrome // Am. J. Hum. Genet. 2010. V. 86. № 6. P. 872–880. https://doi.org/10.1016/J.AJHG.2010.04.017
Kokunai Y., Nakata T., Furuta M. et al. A Kir3.4 mutation causes Andersen–Tawil syndrome by an inhibitory effect on Kir2.1 // Neurology. 2014. V. 82. № 12. P. 1058–1064. https://doi.org/10.1212/WNL.0000000000000239
He C., Zhang H., Mirshahi T. et al. Identification of a potassium channel site that interacts with G protein betagamma subunits to mediate agonist-induced signaling // J. Biol. Chem. 1999. V. 274. № 18. P. 12517–12524. https://doi.org/10.1074/JBC.274.18.12517
Matsuda S., Ohnuki Y., Okami M. et al. Jervell and Lange-Nielsen syndrome with novel KCNQ1 and additional gene mutations // Hum. Genome Var. Nature Publ. Group. 2020. V. 7. № 1. https://doi.org/10.1038/S41439-020-00121-X
Witczak O., Skålhegg B.S., Keryer G. et al. Cloning and characterization of a cDNA encoding an A-kinase anchoring protein located in the centrosome, AKAP450 // EMBO J. 1999. V. 18. № 7. P. 1858. https://doi.org/10.1093/EMBOJ/18.7.1858
Kurokawa J., Motoike H.K., Rao J. et al. Regulatory actions of the A-kinase anchoring protein Yotiao on a heart potassium channel downstream of PKA phosphorylation // Proc. Natl Acad. Sci. USA. 2004. V. 101. № 46. P. 16374. https://doi.org/10.1073/PNAS.0405583101
Chen L., Marquardt M.L., Tester D.J. et al. Mutation of an A-kinase-anchoring protein causes long-QT syndrome // Proc. Natl Acad. Sci. USA. 2007. V. 104. № 52. P. 20990–20995. https://doi.org/10.1073/PNAS.0710527105
De Villiers C.P., Van Der Merwe L., Crotti L. et al. AKAP9 is a genetic modifier of congenital long-QT syndrome type 1 // Circ. Cardiovasc. Genet. 2014. V. 7. № 5. P. 599–606. https://doi.org/10.1161/CIRCGENETICS.113.000580
Wang Q., Li Z., Shen J. et al. Genomic organization of the human SCN5A gene encoding the cardiac sodium channel // Genomics. 1996. V. 34. № 1. P. 9–16. https://doi.org/10.1006/GENO.1996.0236
Bennett P.B., Yazawa K., Makita N. et al. Molecular mechanism for an inherited cardiac arrhythmia // Nature. 1995. V. 376. № 6542. P. 683–685. https://doi.org/10.1038/376683A0
Makita N., Yagihara N., Crotti L. et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility // Circ. Cardiovasc. Genet. 2014. V. 7. № 4. P. 466–474. https://doi.org/10.1161/CIRCGENETICS.113.000459
Niu D.M., Hwang B., Hwang H.W. et al. A common SCN5A polymorphism attenuates a severe cardiac phenotype caused by a nonsense SCN5A mutation in a Chinese family with an inherited cardiac conduction defect // J. Med. Genet. 2006. V. 43. № 10. P. 817–821. https://doi.org/10.1136/JMG.2006.042192
Yu F.H., Westenbroek R.E., Silos-Santiago I. et al. Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2 // J. Neurosci. 2003. V. 23. № 20. P. 7577–7585. https://doi.org/10.1523/JNEUROSCI.23-20-07577.2003
Li R.G., Wang Q., Xu Y.J. et al. Mutations of the SCN4B-encoded sodium channel β4 subunit in familial atrial fibrillation // Int. J. Mol. Med. 2013. V. 32. № 1. P. 144–150. https://doi.org/10.3892/IJMM.2013.1355
Ahnt A.H., Freener C.A., Gussoni E. et al. The three human syntrophin genes are expressed in diverse tissues, have distinct chromosomal locations, and each bind to dystrophin and its relatives // J. Biol. Chem. 1996. V. 271. № 5. P. 2724–2730. https://doi.org/10.1074/JBC.271.5.2724
Adams M.E., Dwyer T.M., Dowler L.L. et al. Mouse alpha 1- and beta 2-syntrophin gene structure, chromosome localization, and homology with a discs large domain // J. Biol. Chem. 1995. V. 270. № 43. P. 25859–25865. https://doi.org/10.1074/JBC.270.43.25859
Ueda K., Valdivia C., Medeiros-Domingo A. et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex // Proc. Natl Acad. Sci. USA. 2008. V. 105. № 27. P. 9355–9360. https://doi.org/10.1073/PNAS.0801294105
Wu G., Ai T., Kim J.J. et al. Alpha-1-syntrophin mutation and the long-QT syndrome: A disease of sodium channel disruption // Circ. Arrhythm. Electrophysiol. 2008. V. 1. № 3. P. 193–201. https://doi.org/10.1161/CIRCEP.108.769224
Cheng J., Van Norstrand D.W., Medeiros-Domingo A. et al. LQTS-associated mutation A257G in α1-syntrophin interacts with the intragenic variant P74L to modify its biophysical phenotype // Cardiogenetics. 2011. V. 1. № 1. P. e13. https://doi.org/10.4081/CARDIOGENETICS.2011.E13
Minetti C., Sotgia F., Bruno C. et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy // Nat. Genet. 1998. V. 18. № 4. P. 365–368. https://doi.org/10.1038/NG0498-365
Parton R.G. Caveolae and caveolins // Curr. Opin. Cell Biol. 1996. V. 8. № 4. P. 542–548. https://doi.org/10.1016/S0955-0674(96)80033-0
Echarri A., Del Pozo M.A. Caveolae – mechanosensitive membrane invaginations linked to actin filaments // J. Cell Sci. 2015. V. 128. № 15. P. 2747–2758. https://doi.org/10.1242/JCS.153940
Gazzerro E., Sotgia F., Bruno C. et al. Caveolinopathies: From the biology of caveolin-3 to human diseases // Eur. J. Hum. Genet. 2010. V. 18. № 2. P. 137. https://doi.org/10.1038/EJHG.2009.103
Vatta M., Ackerman M.J., Ye B. et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome // Circulation. 2006. V. 114. № 20. P. 2104–2112. https://doi.org/10.1161/CIRCULATIONAHA.106.635268
Cronk L.B., Ye B., Kaku T. et al. Novel mechanism for sudden infant death syndrome: persistent late sodium current secondary to mutations in caveolin-3 // Hear. Rhythm. 2007. V. 4. № 2. P. 161–166. https://doi.org/10.1016/J.HRTHM.2006.11.030
Tsien R.W., Ellinor P.T., Horne W.A. Molecular diversity of voltage-dependent Ca2+ channels // Trends Pharmacol. Sci. 1991. V. 12. № 9. P. 349–354. https://doi.org/10.1016/0165-6147(91)90595-J
Perez-Reyes E., Wei X., Castellano A. et al. Molecular diversity of L-type calcium channels. Evidence for alternative splicing of the transcripts of three non-allelic genes // J. Biol. Chem. Elsevier. 1990. V. 265. № 33. P. 20430–20436. https://doi.org/10.1016/S0021-9258(17)30522-7
Schultz D., Mikala G., Yatani A. et al. Cloning, chromosomal localization, and functional expression of the alpha 1 subunit of the L-type voltage-dependent calcium channel from normal human heart // Proc. Natl Acad. Sci. USA. 1993. V. 90. № 13. P. 6228–6232. https://doi.org/10.1073/PNAS.90.13.6228
Soldatov N.M. Genomic structure of human L-type Ca2+ channel // Genomics. 1994. V. 22. № 1. P. 77–87. https://doi.org/10.1006/GENO.1994.1347
Antzelevitch C. Genetic basis of Brugada syndrome // Heart Rhythm. NIH Public Access. 2007. V. 4. № 6. P. 756. https://doi.org/10.1016/J.HRTHM.2007.03.015
Boczek N.J., Best J.M., Tester D.J. et al. Exome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome // Circ. Cardiovasc. Genet. 2013. V. 6. № 3. P. 279–289. https://doi.org/10.1161/CIRCGENETICS.113.000138
Rhyner J.A., Ottiger M., Wicki R. et al. Structure of the human CALM1 calmodulin gene and identification of two CALM1-related pseudogenes CALM1P1 and CALM1P2 // Eur. J. Biochem. 1994. V. 225. № 1. P. 71–82. https://doi.org/10.1111/J.1432-1033.1994.00071.X
Boczek N.J., Gomez-Hurtado N., Ye D. et al. Spectrum and prevalence of CALM1-, CALM2-, and CALM3-encoded calmodulin variants in long QT syndrome and functional characterization of a novel long QT syndrome-associated calmodulin missense variant, E141G // Circ. Cardiovasc. Genet. 2016. V. 9. № 2. P. 136–146. https://doi.org/10.1161/CIRCGENETICS.115.001323
Chin D., Winkler K.E., Means A.R. Characterization of substrate phosphorylation and use of calmodulin mutants to address implications from the enzyme crystal structure of calmodulin-dependent protein kinase I // J. Biol. Chem. 1997. V. 272. № 50. P. 31235–31240. https://doi.org/10.1074/JBC.272.50.31235
Junge H.J., Rhee J.S., Jahn O. et al. Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity // Cell. 2004. V. 118. № 3. P. 389–401. https://doi.org/10.1016/J.CELL.2004.06.029
Crotti L., Spazzolini C., Tester D.J. et al. Calmodulin mutations and life-threatening cardiac arrhythmias: Insights from the International Calmodulinopathy Registry // Eur. Heart J. 2019. V. 40. № 35. P. 2964–2975. https://doi.org/10.1093/EURHEARTJ/EHZ311
Marsman R.F., Barc J., Beekman L. et al. A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence // J. Am. Coll. Cardiol. 2014. V. 63. № 3. P. 259–266. https://doi.org/10.1016/J.JACC.2013.07.091
Mohler P.J., Schott J.J., Gramolini A.O. et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death // Nature. 2003. V. 421. № 6923. P. 634–639. https://doi.org/10.1038/NATURE01335
Mohler P.J., Splawski I., Napolitano C. et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function // Proc. Natl Acad. Sci. USA. 2004. V. 101. № 24. P. 9137–9142. https://doi.org/10.1073/PNAS.0402546101
Mohler P.J., Le Scouarnec S., Denjoy I. et al. Defining the cellular phenotype of “ankyrin-B syndrome” variants: Human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes // Circulation. 2007. V. 115. № 4. P. 432–441. https://doi.org/10.1161/CIRCULATIONAHA.106.656512
Westenskow P., Splawski I., Timothy K.W. et al. Compound mutations: A common cause of severe long-QT syndrome // Circulation. 2004. V. 109. № 15. P. 1834–1841. https://doi.org/10.1161/01.CIR.0000125524.34234.13
Adler A., Novelli V., Amin A.S. et al. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome // Circulation. 2020. V. 141. № 6. P. 418. https://doi.org/10.1161/CIRCULATIONAHA.119.043132
Лебедев Д.С., Михайлов Е.Н., Неминущий Н.М. и др. Желудочковые нарушения ритма. Желудочковые тахикардии и внезапная сердечная смерть. Клинические рекомендации // Рос. кардиол. журн. 2020. Т. 26. № 7. https://doi.org/10.15829/1560-4071-2021-4600
Mazzanti A., Maragna R., Faragli A. et al. Gene-specific therapy with mexiletine reduces arrhythmic events in patients with long QT syndrome type 3 // J. Am. Coll. Cardiol. 2016. V. 67. № 9. P. 1053–1058. https://doi.org/10.1016/J.JACC.2015.12.033
Bos J.M., Crotti L., Rohatgi R.K. et al. Mexiletine shortens the QT interval in patients with potassium channel-mediated type 2 long QT syndrome // Circ. Arrhythm. Electrophysiol. 2019. V. 12. № 5. https://doi.org/10.1161/CIRCEP.118.007280
Etheridge S.P., Compton S.J., Tristani-Firouzi M. et al. A new oral therapy for long QT syndrome: Long-term oral potassium improves repolarization in patients with HERG mutations // J. Am. Coll. Cardiol. 2003. V. 42. № 10. P. 1777–1782. https://doi.org/10.1016/J.JACC.2003.07.006
Crotti L., Celano G., Dagradi F. et al. Congenital long QT syndrome // Orphanet J. Rare Dis. 2008. V. 3. № 1. P. 1–16. https://doi.org/10.1186/1750-1172-3-18/FIGURES/6
Priori S.G., Wilde A.A., Horie M. et al. HRS/EHRA/ APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes // J. Arrhythmia. 2014. V. 30. № 1. P. 1–28. https://doi.org/10.1016/J.JOA.2013.07.002
Дополнительные материалы отсутствуют.