

Генетика, 2022, T. 58, № 2, стр. 219-231
Алгоритм фильтрации наборов данных ДНК-штрихкодирования природных сообществ
А. Ю. Краснопеев 1, *, Ю. С. Букин 1, **, С. А. Потапов 1, О. И. Белых 1
1 Лимнологический институт Сибирского отделения Российской академии наук
664033 Иркутск, Россия
* E-mail: andrewkrasnopeev@gmail.com
** E-mail: bukinyura@mail.ru
Поступила в редакцию 21.04.2021
После доработки 11.08.2021
Принята к публикации 28.09.2021
- EDN: PKATTP
- DOI: 10.31857/S0016675822020084
Аннотация
Определение таксономического разнообразия сообществ является важным этапом многих экологических и мониторинговых исследований экосистем. В основном для таких работ используют методы ампликонной метагеномики (метабаркодинга). В особенности широко метагеномный подход применяется при анализе сообществ микроорганизмов. Представленность определенного таксона в метагеномной пробе при этом определяется количеством (покрытием) расшифрованных нуклеотидных последовательностей. Несмотря на большие размеры выборок расшифрованных нуклеотидных последовательностей, получаемых на платформах секвенирования нового поколения, открытым остается вопрос о статистической достоверности оцениваемых показателей представленности таксонов. Главным образом это касается минорной компоненты исследуемых сообществ. Для решения задачи по оценке статистической репрезентативности выборок расшифрованных последовательностей ДНК для метабаркодинговых исследований нами разработан алгоритм, основанный на использовании бутстреп-метода. Применение данного алгоритма позволяет оценить доверительные интервалы, абсолютные и относительные ошибки показателей представленности таксонов в сообществах. Исходный код, примеры работы и рабочие файлы доступны по адресу: https://github.com/Krasnopeev/otu_error_filtering.
Определение таксономического разнообразия сообществ является важным этапом многих экологических и мониторинговых исследований экосистем. В основном для таких работ применяют методы “shotgun” метагеномики (метод дробовика – секвенирования случайных фрагментов) и секвенирования “ампликонов” (ПЦР-продуктов, полученных в результате проведения полимеразной цепной реакции) – ДНК-штрихкодирование [1, 2] с применением технологий секвенирования нового поколения (NGS, Next Generation Sequencing). В задачи такого анализа входят идентификация таксонов организмов, генетический материал которых находился в смеси ДНК, и определение показателей представленности таксонов – количества или доли прочтений, приходящихся на каждый таксон в исследуемом образце.
В “ампликонной” метагеномике (ДНК-штрихкодировании) на этапе пробоподготовки производится ПЦР-амплификация какого-либо широко используемого в филогении и ДНК-штрихкодировании генетического маркера, например, вариабельного фрагмента гена 16S или 18S рРНК, фрагмента митохондриального гена COX1, ITS1 или ITS2 (internal transcribed spacers) – внутренних спейсеров 1 и 2 ядерного рибосомного кластера генов соответственно. Фрагменты ДНК из смеси продуктов амплификации расшифровываются с помощью методов высокопроизводительного секвенирования. Анализ выборок расшифрованных последовательностей выполняется в два этапа. Первый этап предполагает кластеризацию последовательностей с применением ДНК k-мерного анализа [9, 10] или кластеризацию по генетическим расстояниям, рассчитанным после процедуры выравнивания [11]. На втором этапе последовательности из кластеров сравниваются с референсными базами данных [12–14]. Во многих случаях исследователи оперируют не идентифицированными до вида группами последовательностей, а идентификаторами кластеров ОТЕ – оперативными таксономическими единицами, выделенными на генетических расстояниях уровня вида. Например для бактериальных сообществ при использовании маркерного гена 16S рРНК порог кластеризации на уровне вида оставляет 3% нуклеотидных замен [9, 11].
Итоговый результат таксономической идентификации расшифрованных фрагментов ДНК при анализе серии образцов в рамках одного исследования – таблица, столбцы которой характеризуют отдельные образцы, а строки содержат информацию о количестве последовательностей (показатели представленности), приходящихся на идентифицированные таксоны. Полученные таблицы представленности таксонов являются отправной точкой при анализе и сравнении сообществ различными методами вычислительной экологии: расчет индексов α-разнообразия, например, индексы Chao1 [15] и ACE [16] (Abundance Coverage Estimator), которые позволяют оценить реальное (максимальное) количество таксонов в образце, и β-разнообразия – различные виды кластерного анализа (UPGMA, UniFrac), метрические и неметрические статистики, оценка влияния различных биотических и абиотических факторов на состав и структуру сообщества методами многомерного шкалирования (MDS, PCA) и т.д. Все эти подходы широко применяют при анализе бактериальных сообществ [17, 18], например при исследованиях сезонной динамики численности разнообразных групп микроорганизмов [19].
Несмотря на большие размеры выборок расшифрованных нуклеотидных последовательностей, получаемых на платформах секвенирования нового поколения, открытым остается вопрос о статистической достоверности оцениваемых показателей представленности таксонов. В основном это касается минорной компоненты исследуемых сообществ. Для того чтобы избежать ложных выводов при использовании статистик вычислительной экологии, исследователь должен оценить статистическую ошибку определения показателей представленности таксонов в разных метагеномных образцах, связанную с ограничением размера выборки расшифрованных последовательностей ДНК. Задача сводится к расчетам доверительных интервалов в абсолютных и относительных величинах для показателей представленности каждого таксона в пробе, т.е. таксоны, показатели представленности которых определены с большой относительной ошибкой, должны удаляться из анализа перед использованием методов вычисли-тельной экологии.
Одним из наиболее простых методов, который может быть использован для определения абсолютных и относительных ошибок показателей представленности таксонов в пробах при работе с данными секвенирования по методу дробовика и ампликонной метагеномики, является непараметрический бутстреп-анализ [20]. Принцип бутстреп-семплирования заключается в создании случайной выборки методом Монте-Карло для оценки различных статистик (дисперсия, доверительные интервалы, систематические ошибки и т.д.) анализируемой модели.
Бутстреп-метод широко применяется в вычислительной экологии. Так, например используют бутстреп для определения доверительных интервалов для индекса Шеннона – меры α-разнообразия сообщества [21].
Цель нашего исследования – разработка универсального алгоритма для расчета абсолютных и относительных ошибок показателей представленности таксонов в выборке данных, полученных при анализе серии проб с помощью методов метагеномики. Анализ предполагает использование непараметрического бутстрепа с реализацией в свободно распространяемой среде программирования R. Одной из задач алгоритма является автоматизированная фильтрация набора данных, связанная с удалением таксонов, показатели представленности которых оценены с большой относительной ошибкой. Исходный код алгоритма, примеры работы и рабочие файлы доступны по адресу: https://github.com/Krasnopeev/otu_error_filtering.
МАТЕРИАЛЫ И МЕТОДЫ
Описание алгоритма
В качестве входных данных для работы используется таблица представленности таксонов, полученная в ходе анализа результатов секвенирования ДНК. Она представляет собой матрицу, в которой по вертикали расположены наименования проб, по горизонтали – таксоны. Соответственно в каждой ячейке отмечается количество последовательностей, приходящихся на данный таксон в образце (показатель представленности таксона в пробе). Для удобства обозначим таксон в массиве данных как ОТЕ – оперативная таксономическая единица. Всего в рамках анализа исследуется n проб (образцов), в которых в совокупности выделено k ОТЕ. Индекс пробы обозначим i, i принимает значения от 1 до n. Индекс ОТЕ обозначим j, j принимает значении от 1 до k. ОТЕij – количество расшифрованных последовательностей, приходящихся на таксон с индексом j в i пробе (показатель представленности таксона в пробе).
На первом этапе анализа для каждой пробы i формируется символьный вектор Si следующего содержания:

Количеств элементов Ni в векторе Si задается формулой:
На следующем этапе каждый вектор, как описано в работе [21], подвергается бутстреп-семплированию с применением непараметрического бутстрепа с возвратом. Всего проводится h циклов бутстреп-семплирований. В результате для пробы i после каждой итерации получается вектор (Si)l – бутстреп реплика набора с индексом l (l принимает значения от 1 до h).
На основе векторов (Si)l формируется бутстреп-выборка значений показателей представленности (ОТЕij)l каждого таксона в каждой пробе. Значения (ОТЕij)l определяются как количество записей элемента “ОТЕj” в векторе (Si)l. Для того, чтобы найти доверительный интервал уровня вероятности P согласно процедуре бутстреп-анализа [21], значения в векторах (ОТЕij)l сортируются в порядке возрастания. В отсортированном векторе элемент с индексом h(1 – P)/2 будет XMINij левой границей доверительного интервала, а элемент с индексом h(P + 1)/2 XMAXij – правой границей доверительного интервала. Далее, рассчитывается Dij – относительная ошибка определения показателя представленности ОТЕij по формуле:
По умолчанию в программной реализации алгоритма количество бутстреп-реплик h = 1000, ширина доверительного интервала задается уровнем вероятности P = 0.95. Но допустимы изменения этих параметров пользователем.
Далее, производится фильтрация – удаление ОТЕ, относительная ошибка определения которых превышает значение порога в 0.2 – минимальный порог достоверности данных, приемлемый для биологических исследований [22], заданный по умолчанию в алгоритме. Такие ОТЕ исключаются из дальнейшего анализа (табл. 1). Параметр порога относительной ошибки может меняться пользователем алгоритма. Фильтрация – удаление ОТЕ по уровню ошибки может быть отключена, тогда будет рассчитываться только относительная и абсолютная ошибка определения показателей представленности ОТЕ в пробах.
Таблица 1.
Пример таблицы данных с указанием величины относительной ошибки представленности ОТЕ в пробе, полученной в результате работы предложенного алгоритма
| S8 | S10 | S11 | S12 | … | Sn | |
|---|---|---|---|---|---|---|
| OTE1 | 0.034* | 1 | 0.015* | 0.074* | … | 0.05* |
| OTE2 | 0.045* | 0.55 | 0.22 | 0.886 | … | 0.94 |
| OTE3 | 0.0564* | 0.32 | 0.01* | 0.13* | … | 1 |
| OTE4 | 1 | 1 | 1 | 0.02* | … | 1 |
| OTE5 | 0.167* | 1 | 0.178* | 1 | … | 0.01* |
| … | … | … | … | … | … | 0.04* |
| OTEi | 0.87 | 0.01* | 0.21 | 0.44 | … | 0.03* |
Соответственно, оставшаяся часть ОТЕ с относительной ошибкой меньше либо равной 0.2 является достоверно представленной. После этой процедуры значения ошибки и количество последовательностей достоверно оцененных ОТЕ переносятся в новую таблицу. В итоге, полученный список таблиц объединяется в одну общую матрицу, имеющую вид оригинальной таблицы представленности, но содержащую в себе только те ОТЕ, которые были оценены с заданным размером ошибки. Отсутствующие значения, которые отброшены в ходе работы алгоритма, заменяются нулями в соответствующих ячейках новой таблицы. Блок-схема работы алгоритма фильтрации данных представлена на рис. 1.
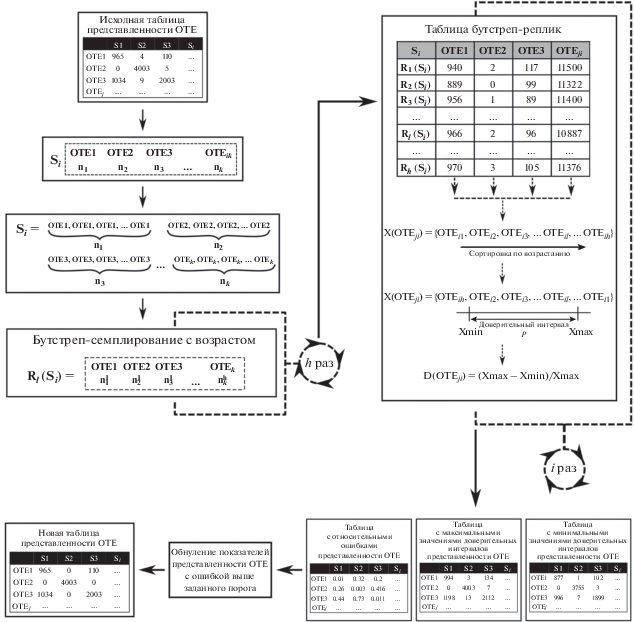
Рис. 1.
Блок-схема работы алгоритма. S – идентификатор пробы. Значения показателей представленности в ячейках показаны для примера. R – бутстреп-реплика конкретной пробы. Исходная таблица представленности разбивается на отдельные таблицы соответственно каждой пробе и преобразуется в список. Каждый элемент списка преобразуется в вектор повторяющихся идентификаторов ОТЕj в зависимости от числа последовательностей, принадлежащих этой ОТЕj в рассматриваемой пробе. Далее производится бутстреп-семплирование (генерация случайной выборки) с возвратом для каждого вектора (т.е. пробы) в количестве h штук (значение h устанавливается пользователем, по умолчанию h = 1000). Реплики векторов трансформируются в таблицы представленности соответственно пробам. После этого рассчитываются относительные ошибки представленности ОТЕ в образце и генерируется новая таблица, содержащая в себе достоверные, согласно алгоритму, ОТЕ, а также отдельные таблицы с минимальными и максимальными значениями доверительных интервалов, заданных пользователем.
Для работы с программой пользователю необходимо загрузить в рабочую среду R свою таблицу представленности таксонов. Входная функция, выполняющая первичную обработку данных, это getSub (data, value = 0.95, cutoff = 1, subsample = = FALSE, subsize = NULL) с параметрами по умолчанию, где data – таблица представленности таксонов (ОТЕ), value – размер выборки от общего числа (по умолчанию 0.95–95% доминирующий пул всех ОТЕ), cutoff – значение нижней границы численности анализируемых ОТЕ (по умолчанию – 2, т.е. удаляем из набора все ОТЕ с суммарной численностью 2 и менее), subsample – логический оператор, позволяет указать нужно ли выполнять семплирование образцов с размером выборки subsize (на усмотрение пользователя).
Далее происходит генерация векторов, имитирующих сообщества в пробе с помощью функции getOtuSample (data, trimmedby = 0), где data – это образец-столбец из таблицы представленности, а trimmedby (по умолчанию равно 0) – дополнительный вариант фильтрации уже отдельного образца по аналогии с cutoff из функции getSub.
Полученный список векторов передается в функцию, которая выполняет непосредственно бутстреп-семплирование – getReplica (data, n = 1000, scaling = “int”), где data – вектор, полученный с помощью функции getOtuSample, n – число бутстреп-семплирований (по умолчанию 1000), scaling – преобразование полученных количественных значений в доли представленности – “percent”, приведение к натуральному логарифму по основанию 10 – “log”, оригинальные количественные показатели представленности – “int” (по умолчанию).
Последним шагом является расчет относительных ошибок представленности ОТЕ с помощью функции repError (data, rate = 0.2, filterError = = TRUE, p = 0.95), где data – набор реплик конкретного образца, rate – максимальный размер ошибки представленности (задается на усмотрение пользователя), рассчитанной по формуле (3), filterError – логический оператор, отвечающий за фильтрацию ОТЕ по значению rate (по умолчанию TRUE), p – доверительный интервал уровня вероятности (по умолчанию 0.95).
В результате работы последней функции пользователь получит список таблиц: (i) таблица представленности ОТЕ, скорректированная с учетом значения rate и переменной filterError, (ii) таблица с показателями представленности ОТЕ, соответствующих верхнему пределу доверительного интервала, и (iii) таблица с показателями представленности ОТЕ нижнего предела доверительного интервала уровня вероятности. Для удобства пользователя значения в таблицах (ii) и (iii) конвертированы в относительные доли представленности.
Пример пошаговой работы алгоритма можно также найти в файле README.md в репозитории по адресу https://github.com/Krasnopeev/otu_error_filtering.
Тестирование алгоритма
В качестве тестовых данных использовали результаты секвенирования с помощью ДНК-штрихкодирования 28 образцов воды (табл. 2), отобранных в озере Байкал в течение 2017–2018 гг. Образцы получены на разных станциях и глубинах во всех трех котловинах озера как в подледный период (март), так и в период открытой воды (июнь–сентябрь). Исследуемые пробы различались по составу и количественным показателям развития фито- и бактериопланктона, в том числе по доминирующим группам первичных продуцентов. Кроме того, места отбора проб характеризовались различным уровнем антропогенной нагрузки [23–25]. Можно ожидать, что исследованные образцы будут достоверно различаться по составу таксонов и видовому богатству прокариотических микроорганизмов.
Таблица 2.
Характеристика образцов тестового набора
| Название пробы | Глубина, м | Станции отбора | Дата отбора 2018 г. |
|---|---|---|---|
| S8 | 0–5 | 7 км от п. Листвянка | Март |
| S10 | 20–25 | 7 км от п. Листвянка | Март |
| S11 | 50–100 | 7 км от п. Листвянка | Март |
| S12 | 0–5 | Ц. р. п. Листвянка–п. Танхой | Июнь |
| S13 | 10–15 | Ц. р. п. Листвянка–п. Танхой | Июнь |
| S14 | 20–25 | Ц. р. п. Листвянка–п. Танхой | Июнь |
| S15 | 50–100 | Ц. р. п. Листвянка–п. Танхой | Июнь |
| S16 | 0–5 | Ц. р. м. Ухан–м. Тонкий | Июнь |
| S17 | 10–15 | Ц. р. м. Ухан–м. Тонкий | Июнь |
| S18 | 20–25 | Ц. р. м. Ухан–м. Тонкий | Июнь |
| S19 | 50–100 | Ц. р. м. Ухан–м. Тонкий | Июнь |
| S20 | 0–5 | Пролив Малое Море | Июнь |
| S22 | 0–5 | Баргузинский залив | Июнь |
| S23 | 0–5 | Ц. р. м. Елохин–п. Давша | Июнь |
| S24 | 10–15 | Ц. р. м. Елохин–п. Давша | Июнь |
| S25 | 20–25 | Ц. р. м. Елохин–п. Давша | Июнь |
| S26 | 50–100 | Ц. р. м. Елохин–п. Давша | Июнь |
| S27 | 0–50 | Ц. р. п. Листвянка–п. Танхой | Сентябрь |
| S28 | 0–50 | Ц. р. м. Ухан–м. Тонкий | Сентябрь |
| S29 | 0–50 | Ц. р. м. Елохин–п. Давша | Сентябрь |
| S30 | 0 | Пролив Малое Море | Август |
| S31 | 0 | Пролив Малое Море | Август |
| S32 | 0 | Пролив Малое Море | Август |
| S35 | 0–5 | Ц. р. п. Листвянка–п. Танхой | Август |
| S36 | 10–15 | Ц. р. п. Листвянка–п. Танхой | Август |
| S37 | 25–50 | Ц. р. п. Листвянка–п. Танхой | Август |
| S38 | 0 | м. Лиственичный, около пос. Листвянка | Июнь |
| S40 | 0 | м. Елохин | Июнь |
Пробы воды фильтровали через поликарбонатные фильтры с диаметром пор 0.2 мкм (Sartorius, Германия). Суммарную ДНК выделяли методом ферментативного лизиса с использованием лизоцима (Roche, Швейцария), протеиназы К (Thermo Scientific, США) и додецилсульфата натрия (VWR Life Science AMRESCO, США) с последующей экстракцией фенолом (Медиген, Россия) хлороформом (Экос-1, Россия) [26]. Библиотеки ампликонов фрагмента V3–V4-гена 16S рРНК получены с помощью пары универсальных праймеров 343F (5′-CTCCTACGGRRSGCAGCAG-3′) и 806R (5′-GGACTACNVGGGTWTCTAAT-3′) [27] на платформе Illumina MiSeq (Illumina, США) в Евроген (Москва, Россия) с использованием протокола парно-концевых прочтений 2 × 250 пн. Полученные библиотеки ампликонов депонированы в базу данных NCBI GenBank с доступом по номеру PRJNA637453.
Первичную обработку данных секвенирования проводили с использованием программного пакета Trimmomatic [28] с параметрами MINLEN:200. Для удаления праймеров и адаптеров использовали cutadapt v1.14 [29]. Полученные в результате первичной обработки парные последовательности сшиты с помощью FLASH [30] с параметрами по умолчанию. Удаление химер, прочтений короче 400 и длиннее 435 пн, а также кластеризацию последовательностей в ОТЕ проводили при помощи программного пакета mothur v. 1.40.5 (https://mothur.org/) с порогом кластеризации в 97% гомологии согласно стандартному протоколу для наборов данных, полученных с секвенаторов Illumina [31]. Выходные данные оформлены в виде таблицы представленности ОТЕ. Для каждого образца рассчитывали индексы α-разнообразия Шеннона, Симпсона и Chao1. Показаны результаты корреляционного анализа матриц расстояний по метрике Брея–Кёртиса [32] и Евклидовых расстояний. Корреляционный анализ между матрицами расстояний выполнен с использованием непараметрического критерия Спирмена. Алгоритм реализован при помощи средств языка программирования R [33, 34].
РЕЗУЛЬТАТЫ
Всего для 28 тестовых проб (образцов) получено 2 109 405 парно-концевых прочтений. В среднем на каждый образец приходилось по 75 336 последовательностей (от 20 155 до 195 476 прочтений). Средняя длина фрагмента составила 418 пн.
До проведения процедуры фильтрации предложенным алгоритмом количество ОТЕ составляло от 824 до 5338 (рис. 2,а). Значения индекса Шеннона варьировали в диапазоне 3.18–4.54. Индекс Симпсона для исходного набора данных составлял от 0.83 до 0.97. Индекс Chao1 принимал значения от 634 до 3262 (табл. 3).
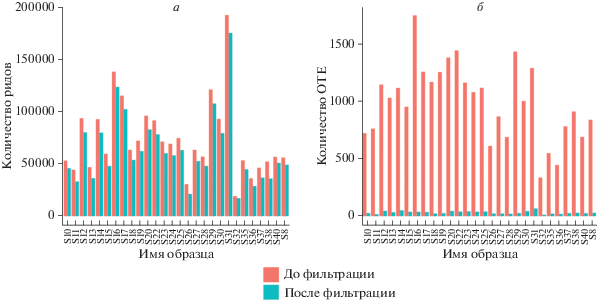
Таблица 3.
Сравнительная таблица наборов данных до и после фильтрации. Simpson – индекс Симпсона, Shannon – индекс Шеннона, Chao1 – индекс Chao1, N ОТЕ – количество ОТЕ, N прочтений – количество прочтений в образце
| Название пробы | До фильтрации | После фильтрации | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N прочтений | N ОТЕ | Shannon | Simpson | Chao1 | N прочтений | N ОТЕ | Shannon | Simpson | доля потерь прочтений | Chao1 | |
| S8 | 57 355 | 2246 | 3.55 | 0.92 | 1914 | 49 098 | 29 | 2.66 | 0.89 | 14.4 | 29 |
| S10 | 54 086 | 1780 | 3.58 | 0.93 | 1547 | 45 769 | 27 | 2.7 | 0.9 | 15.4 | 27 |
| S11 | 45 230 | 1675 | 3.71 | 0.92 | 1666 | 33 095 | 17 | 2.24 | 0.86 | 26.8 | 17 |
| S12 | 95 652 | 3261 | 4.18 | 0.96 | 2732 | 80 117 | 46 | 3.31 | 0.95 | 16.2 | 46 |
| S13 | 49 298 | 3529 | 4.54 | 0.97 | 2547 | 36 243 | 34 | 3.16 | 0.94 | 26.5 | 34 |
| S14 | 94 799 | 3339 | 4.28 | 0.97 | 2558 | 79 801 | 51 | 3.43 | 0.95 | 15.8 | 51 |
| S15 | 61 176 | 2578 | 4.3 | 0.96 | 2133 | 47 860 | 38 | 3.2 | 0.94 | 21.8 | 38 |
| S16 | 141 690 | 5338 | 3.28 | 0.83 | 3262 | 123 465 | 38 | 2.34 | 0.77 | 12.9 | 38 |
| S17 | 117 288 | 3426 | 3.22 | 0.83 | 2455 | 102 160 | 36 | 2.34 | 0.77 | 12.9 | 36 |
| S18 | 65 364 | 3174 | 3.37 | 0.85 | 2937 | 53 721 | 23 | 2.19 | 0.78 | 17.8 | 23 |
| S19 | 74 259 | 3481 | 3.27 | 0.83 | 2752 | 62 143 | 26 | 2.14 | 0.75 | 16.3 | 26 |
| S20 | 99 253 | 4630 | 4.1 | 0.95 | 3090 | 82 802 | 45 | 3.1 | 0.93 | 16.6 | 45 |
| S22 | 94 560 | 4514 | 3.96 | 0.93 | 3194 | 78 175 | 40 | 2.88 | 0.89 | 17.3 | 40 |
| S23 | 74 318 | 4244 | 4.37 | 0.97 | 2871 | 60 195 | 42 | 3.3 | 0.95 | 19 | 42 |
| S24 | 72 027 | 3983 | 4.26 | 0.96 | 2228 | 58 050 | 39 | 3.15 | 0.93 | 19.4 | 39 |
| S25 | 77 453 | 4008 | 4.27 | 0.96 | 2550 | 63 102 | 41 | 3.23 | 0.94 | 18.5 | 41 |
| S26 | 30 990 | 1056 | 4.18 | 0.96 | 1032 | 21 246 | 23 | 2.84 | 0.92 | 31.4 | 23 |
| S27 | 64 182 | 1667 | 3.68 | 0.94 | 1529 | 52 583 | 23 | 2.66 | 0.91 | 18.1 | 23 |
| S28 | 57 482 | 1339 | 3.47 | 0.92 | 1235 | 47 990 | 21 | 2.54 | 0.89 | 16.5 | 21 |
| S29 | 124 128 | 4503 | 3.49 | 0.91 | 2385 | 107 617 | 27 | 2.57 | 0.88 | 13.3 | 27 |
| S30 | 94 676 | 2762 | 4.11 | 0.95 | 1765 | 79 355 | 44 | 3.23 | 0.93 | 16.2 | 44 |
| S31 | 195 476 | 4599 | 3.94 | 0.94 | 1914 | 175 031 | 67 | 3.28 | 0.92 | 10.5 | 67 |
| S32 | 20 155 | 1339 | 3.18 | 0.91 | 634 | 17 232 | 12 | 2.24 | 0.87 | 14.5 | 12 |
| S35 | 53 855 | 1109 | 3.25 | 0.89 | 1179 | 44 775 | 22 | 2.33 | 0.84 | 16.9 | 22 |
| S36 | 36 494 | 824 | 3.59 | 0.93 | 958 | 28 715 | 20 | 2.59 | 0.89 | 21.3 | 20 |
| S37 | 47 400 | 1932 | 4.07 | 0.96 | 1789 | 36 749 | 27 | 2.93 | 0.93 | 22.5 | 27 |
| S38 | 53 134 | 1888 | 4.48 | 0.96 | 1381 | 35 979 | 29 | 2.92 | 0.92 | 32.3 | 29 |
| S40 | 57 625 | 1685 | 3.29 | 0.91 | 1476 | 50 859 | 26 | 2.58 | 0.89 | 11.7 | 26 |
С помощью разработанного алгоритма в наборе данных идентифицированы ОТЕ, ошибка определения представленности которых была меньше 0.2. Для наборов, состоявших из достоверно представленных ОТЕ, рассчитаны новые значения индексов Шеннона и Симпсона для сравнения с исходными значениями. Несмотря на значительный объем библиотек ампликонов, после использования предложенного алгоритма оценки количество ОТЕ сократилось в несколько раз относительно изначального значения в среднем – с 2854 до 33 ОТЕ (рис. 2). Значения индексов α‑разнообразия Шеннона и Симпсона в большинстве проб до и после фильтрации с помощью алгоритма изменились незначительно (табл. 3) (в среднем значения индексов сократились на 5–10%). Это говорит о том, что после фильтрации наборов данных информация о сложности устройства сообществ практически не была потеряна. Количество потерянных прочтений (рис. 2) при удалении ОТЕ с большими ошибками определения показателей представленности в большинстве случаев составило небольшую долю от общего числа исходных расшифрованных последовательностей в пробе. В небольшом количестве проб (рис. 2, табл. 3) доля потери прочтений в результате фильтрации достигала 30%, однако и в них значения индексов α-разнообразия Шеннона и Симпсона изменились в незначительных пределах. Можно сделать вывод, что отфильтрованные последовательности и ОТЕ сами по себе не вносили достаточно весомый вклад в α-разнообразие исследованных проб (табл. 3).
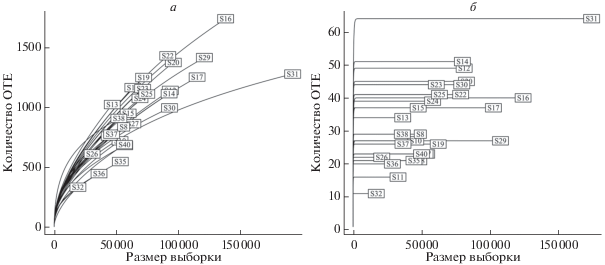
Ожидаемое резкое изменение характера кривых насыщения также наблюдали в отфильтрованном (рис. 3,б) наборе данных по сравнению с исходным (рис. 3,а). Такое сильное изменение показателей величин насыщения, очевидно, было вызвано значительным уменьшением количества ОТЕ в отфильтрованном наборе, что, в целом, упрощает дальнейший анализ данных для исследователя.
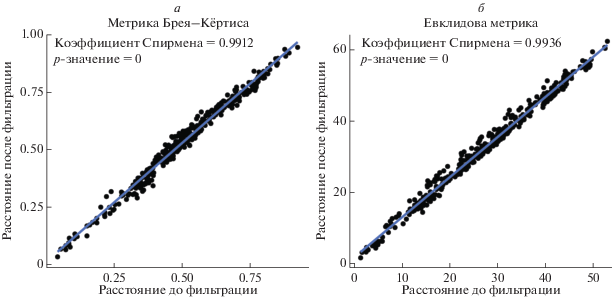
Для сравнения проб по таксономическому составу и уровню представленности различных ОТЕ были рассчитаны матрицы попарных расстояний Брея–Кёртиса и Евклидовых расстояний. Матрицы рассчитаны для наборов данных до фильтрации и после фильтрации – удаления недостоверно представленных ОТЕ (ОТЕ, для которых уровень относительной ошибки определения показателей представленности в пробах был больше заданного по умолчанию порога в 0.2), согласно предложенному в работе алгоритму оценки. Для сравнения матриц расстояний проведен корреляционных анализ, который показал, что прослеживалась строгая линейная зависимость между матрицами расстояний до и после фильтрации (рис. 4). Это было подтверждено высоким значением коэффициента Спирмена 0.9912 (p-value = 0) для матриц по метрике Брея–Кёртиса и 0.9936 (p-value = 0) для матриц Евклидовых расстояний.
Рис. 4.
Корреляционный анализ между матрицами дистанций анализируемых образцов до и после фильтрации. а – сравнение матриц по метрике Брея–Кёртиса, б – сравнение матриц дистанций по Евклидовым расстояниям.
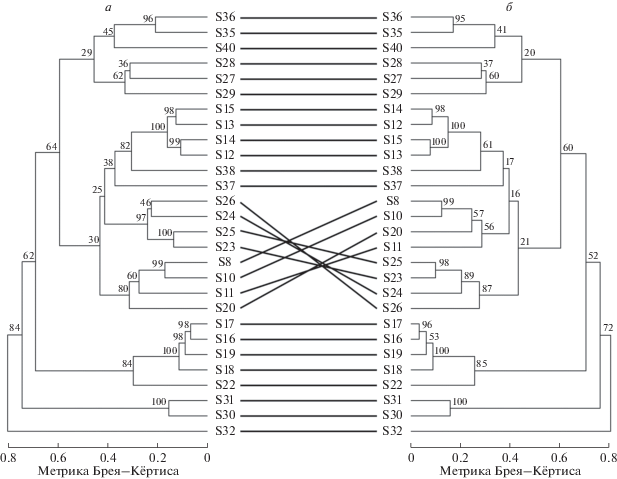
Также при сравнении UPGMA-дендрограм кластеризации образцов на основе матриц расстояний Брея–Кёртиса до и после применения алгоритма фильтрации (рис. 5) показано, что топология дендрограм в обоих случаях сохранялась для большей части образцов. Наблюдалось изменение топологии некоторых ветвей, которые изначально имели меньшие значения бутстреп-поддержек, однако внутри узла топология сохраняла свою структуру. Такое распределение показывает, что вклад, вносимый отфильтрованной частью данных, был незначителен.
Рис. 5.
Сравнение UPGMA-дендрограм на основе дистанций Брея–Кёртиса до (а) и после (б) использования предложенного алгоритма. Цифрами на ветвях обозначены значения рассчитанных бутстреп-поддержек.
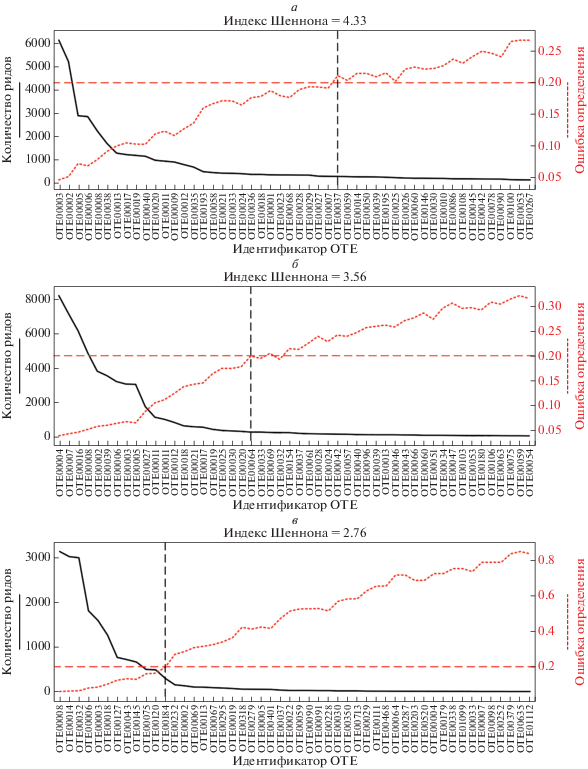
При сравнении проб с различными индексами α-разнообразия была обнаружена интересная зависимость между величиной индекса Шеннона и количеством достоверно определенных ОТЕ в образцах. На рис. 6 показаны графики, отражающие тенденцию к уменьшению достоверно определенных ОТЕ с уменьшением индекса Шеннона. Для примера мы выделили пробы с максимальным, средним и минимальным индексом Шеннона соответственно. В каждой из этих проб был выбран пул доминирующих ОТЕ в количестве 50 штук. Следует отметить, что с падением показателей индекса Шеннона в пробе также увеличивалось количество ОТЕ, определенных с ошибкой выше порога в 0.2. Такое быстрое увеличение значения ошибки могло быть вызвано недостаточной глубиной прочтения. Согласно графикам (рис. 6,а–в), при высоком значении индекса Шеннона доля достоверно определенных ОТЕ была гораздо выше, чем при низком значении, что позволило нам использовать в анализе гораздо большую выборку ОТЕ из этого набора.
Рис. 6.
Графики отношения между количеством и представленностью ОТЕ (черная линия) от величины ошибки их определения (красная линия). Проба с наибольшим показателем индекса Шеннона – а, проба со средним показателем индекса Шеннона – б, проба с наименьшим показателем индекса Шеннона – в. ОТЕ ранжированы в порядке убывания представленности в сообществе.
ОБСУЖДЕНИЕ
Предложенный в работе алгоритм позволяет решать несколько важных задач, связанных с анализом данных исследований методом ДНК-штрихкодирования. Информация об уровне ошибок определения показателей представленности видов в сообществах, анализируемых с помощью ДНК-штрихкодирования, требуется при проведении мониторинговых и скрининговых работ [35, 36]. В таких исследованиях часто ставится задача определить, меняется ли представленность каких-либо таксонов в сообществе. Сделать выводы о таких изменениях можно только тогда, когда доверительные интервалы показателей представленности вида в двух сравниваемых пробах не пересекаются. Предлагаемый в работе алгоритм помогает решить задачу такого сопоставления, представляя исследователю данные о доверительных интервалах определения показателей представленности всех таксонов в сравниваемых пробах.
Часто для сравнения сообществ, информация о составе которых получена методом ДНК-штрихкодирования, применяют методы многомерной статистики (кластерный анализ на основе различных метрик расстояний, метод главных компонент – PCA, многомерное шкалирование и т.п.) [37]. С помощью таких методов сопоставляют различные пробы на основе показателей представленности входящих в них видов. Разрешающие способности и точности методов многомерной статистики падают, если в массивах информации присутствуют неточные – “зашумленные данные” (данные о видах, показатели представленности которых определены с большими ошибками). Применение нашего алгоритма позволяет отфильтровать такие данные, сделав массив более пригодным для многомерного статистического анализа.
В работах по ДНК-штрихкодированию различных сообществ [38, 39] для определения глубины прочтения ампликонов часто применяют методы, позволяющие оценить количество недоучтенных (потерянных видов) из-за недостаточно количества расшифрованных последовательностей ДНК. К таким методам относятся расчеты индексов Chao1 [15], ACE [16] и др. Если доля недоучтенных видов по сравнению с оцененным разнообразием сообщества мала, то массив данных пригоден для анализа. Предложенный в работе подход, в отличие от индексов ACE, Chao1, может определить достаточность размера выборки расшифрованных последовательностей ДНК для оценки показателей представленности каждого вида в отдельности. Если для каких-либо видов (таксонов) ошибка оценки показателей представленности высока и не позволяет сделать правильных выводов, то исследователь может принять решение о дополнительном секвенировании пробы, даже если индексы ACE, Chao1 говорят о достаточности размера выборки.
Таким образом, предложенный нами алгоритм фильтрации наборов данных может существенно облегчить работу специалистов-микробиологов и ученых, занимающихся экологической статистикой и сравнительным анализом различных сообществ. Разработанный подход позволяет отфильтровать так называемый шум из данных, который может влиять на результаты как многомерной статистики, так и при учете индексов видового разнообразия.
Авторы выражают благодарность сотрудникам Иркутского Суперкомпьютерного Центра СО РАН за предоставление доступа к вычислительным ресурсам кластера “Академик В.М. Матросов”. Работа выполнена в рамках темы госзадания ЛИН СО РАН № 0279-2021-0015 (разработка алгоритма и анализ данных), а также при финансовой поддержке проектов РФФИ № 18-34-00513 мол_а (пробоподготовка и выделение ДНК) и РФФИ № 18-54-05005 Арм_а (секвенирование).
Настоящая статья не содержит каких-либо исследований с использованием в качестве объекта животных.
Настоящая статья не содержит каких-либо исследований с участием в качестве объекта людей.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Zakharenko A.S., Galachyants Y.P., Morozov I.V. et al. Bacterial communities in areas of oil and methane seeps in pelagic of Lake Baikal // Microbial Ecology. 2019. V. 78. № 2. P. 269–285. https://doi.org/10.1007/s00248-018-1299-5
Potapov S.A., Tikhonova I.V., Krasnopeev A.Yu. et al. Metagenomic analysis of virioplankton from the pelagic zone of Lake Baikal // Viruses. 2019. I. 11. P. 1–15. https://doi.org/10.3390/v11110991
Wood D.E., Salzberg S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments // Genome Biol. 2014. V. 15. P. 46. https://doi.org/10.1186/gb-2014-15-3-r46
Meyer F., Paarmann D., D’Souza M. et al. The metagenomics RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes // BMC Bioinformatics. 2008. V. 9. P. 386. https://doi.org/10.1186/1471-2105-9-386
Buchfink B., Xie C., Huson D. Fast and sensitive protein alignment using DIAMOND // Nat. Methods. 2015. V. 12. P. 59–60. https://doi.org/10.1038/nmeth.3176
Roux S., Faubladier M., Mahul A. et al. Metavir: A web server dedicated to virome analysis // Bioinformatics. 2011. V. 27. I. 21. P. 3074–3075. https://doi.org/10.1093/bioinformatics/btr519
Kang D.D., Li F., Kirton E. et al. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies // PeerJ. 2019. V. 7. P. e7359. https://doi.org/10.7717/peerj.7359
Wu Y.-W., Simmons B., Singer S. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets // Bioinformatics. 2016. V. 32. I. 4. P. 605–607. https://doi.org/10.1093/bioinformatics/btv638
Edgar R. UPARSE: Highly accurate OTU sequences from microbial amplicon reads // Nat. Methods. 2013. V. 10. P. 996–998. https://doi.org/10.1038/nmeth.2604
Callahan B., McMurdie P., Rosen M. et al. DADA2: High-resolution sample inference from Illumina amplicon data // Nat. Methods. 2016. V. 13. P. 581–583. https://doi.org/10.1038/nmeth.3869
Schloss P., Westcott S., Ryabin T. et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities // Applied and Envir. Microbiology. 2009. V. 75. P. 7537–7541. https://doi.org/10.1128/AEM.01541-09
Quast C., Pruesse E., Yilmaz P. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools // Nucl. Ac. Res. 2013. V. 41. I. D1. P. 590–596. https://doi.org/10.1093/nar/gks1219
Wang Q., Garrity G., Tiedje J., Cole J. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy // Applied and Envir. Microbiology. 2007. V. 73. P. 5261–5267. https://doi.org/10.1128/AEM.00062-07
DeSantis T., Hugenholtz P., Larsen N. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB // Applied and Envir. Microbiology. 2006. V. 72. P. 5069–5072. https://doi.org/10.1128/AEM.03006-05
Chao A. Non-parametric estimation of the number of classes in a population // Scand. J. Stat. 1984. V. 11. № 4. P. 265–270. URL: http://www.jstor.org/stable/4615964 (дата обращения: 15.09.2021).
Chao A., Lee S.M. Estimating the number of classes via sample coverage // J. Amer. Stat. Association. 1992. V. 87. I. 417. P. 210–217. http://chao.stat.nthu.edu.tw/ wordpress/paper/1992_JASA_87_P210.pdf (дата обращения: 15.09.2021).
Sorokin D.Y., Berben T., Melton E.D. et al. Microbial diversity and biogeochemical cycling in soda lakes // Extremophiles. 2014. V. 18. P. 791–809. https://doi.org/10.1007/s00792-014-0670-9
Feng C., Jia J., Wang C. et al. Phytoplankton and bacterial community structure in two chinese lakes of different trophic status // Microorganisms. 2019. V. 7. P. 621. https://doi.org/10.3390/microorganisms7120621
Eiler A., Heinrich F., Bertilsson S. Coherent dynamics and association networks among lake bacterioplankton taxa // ISME. 2012. V. 6. P. 330–342. https://doi.org/10.1038/ismej.2011.113
Efron B. Bootstrap methods: Another look at the jackknife // The Annals of Statistics. 1979. V. 7. P. 1–26.
Pla L. Bootstrap confidence intervals for the Shannon biodiversity index: A simulation study // JABES. 2004. V. 9. I. 42. P. 42–56. https://doi.org/10.1198/1085711043136
Рокицкий П.Ф. Биологическая статистика. Минск: “Вышейш. шк.”, 1973. 320 с.
Mincheva E.V., Peretolchina T.E., Bukin Yu.S. et al. The composition of the communities of green algae and microeukaryotes at the sites of Lake Baikal contrasting in anthropogenic pressure // Limnol. and Freshwater Biol. 2020. I. 4. P. 729–730. https://doi.org/10.31951/2658-3518-2020-A-4-729
Мальник В.В., Штыкова Ю.Р., Сутурин А.Н., Тимошкин О.А. Влияние населенных пунктов на санитарно-микробиологическое состояние малых притоков и прибрежных вод на примере залива Лиственничный (Южный Байкал) // География и природные ресурсы. 2019. № 4. С. 84–92. https://doi.org/10.21782/GIPR0206-1619-2019-4(84-92)
Timoshkin O.A., Klump J.V., Rudstam L.G. et al. Recent research on Lake Baikal: The 2018 Conference, freshwater ecosystems-key problems // J. of Great Lakes Res. 2020. V. 46. I. 1. P. 1–3. https://doi.org/10.1016/j.jglr.2019.12.004
Maniatis T., Fritsch E.F., Sambrook J. Molecular Cloning. The Laboratory Manual. N.Y.: Cold Spring Harbor Laboratory, 1982. P. 545. https://doi.org/10.1016/0307-4412(83)90068-7
Caporaso J., Lauber C., Walters W. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and Metagenomic sequencing platform // ISME J. 2012. I. 6. P. 1621–1624. https://doi.org/10.1038/ismej.2012.8
Bolger A.M., Lohse M., Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data // Bioinformatics. 2014. V. 30. P. 2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads // EMBnet J. 2011. I. 17. P. 10–12. https://doi.org/10.14806/ej.17.1.200
Magoč T., Salzberg S. FLASH: Fast length adjustment of short reads to improve genome assemblies // Bioinformatics. 2011. V. 27. I. 21. P. 2957–2963. https://doi.org/10.1093/bioinformatics/btr507
Schloss P. MiSeq SOP: Mothur. 2015. URL: http://www.mothur.org/wiki/MiSeq_SOP (дата обращения: 15.09.2021).
Bray J.R., Curtis J.T. An ordination of the upland forest communities of southern Wisconsin // Ecol. Monographs. 1957. V. 27. I. 4. P. 325–349. https://doi.org/10.2307/1942268
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical // Computing. 2019. URL: http://www.R-project.org/ (дата обращения: 15.09.2021).
Wickham H. The Split-Apply-Combine strategy for data analysis // J. Stat. Software. 2011. V. 40. I. 1. P. 1–29. https://doi.org/10.18637/jss.v040.i01
Galan M., Razzauti M., Bard E. et al. 16S rRNA amplicon sequencing for epidemiological surveys of bacteria in wildlife // mSystems. 2016. V. 1. I. 4. e00032-16. https://doi.org/10.1128/mSystems.00032-16
Yoshitake K., Kimura G., Sakami T. et al. Development of a time-series shotgun metagenomics database for monitoring microbial communities at the Pacific coast of Japan // Sci. Rep. 2021. V. 11. https://doi.org/10.1038/s41598-021-91615-3
Rudi K., Zimonja M., Trosvik P., Næs T. Use of multivariate statistics for 16S rRNA gene analysis of microbial communities // Int. J. Food Microbiol. 2007. V. 120. № 2. P. 95–99. https://doi.org/10.1016/j.ijfoodmicro.2007.06.004
Sinclair L., Osman O.A., Bertilsson S., Eiler A. Microbial community composition and diversity via 16S rRNA gene amplicons: evaluating the illumina platform // PLoS One. 2015. V. 10. e0116955. https://doi.org/10.1371/journal.pone.0116955
Bukin Y., Galachyants Y., Morozov I. et al. The effect of 16S rRNA region choice on bacterial community metabarcoding results // Sci. Data. 2019. I. 6. https://doi.org/10.1038/sdata.2019.7
Дополнительные материалы отсутствуют.