

Генетика, 2022, T. 58, № 7, стр. 798-807
Микроматричный анализ транскриптома мононуклеаров периферической крови у больных раком легкого
В. И. Минина 1, 2, *, В. Г. Дружинин 1, А. В. Ларионов 1, Е. Д. Баранова 1, В. Ю. Буслаев 2, Л. В. Мацкова 3, М. Л. Баканова 2
1 Кемеровский государственный университет
650065 Кемерово, Россия
2 Федеральный исследовательский центр угля и углехимии Сибирского отделения
Российской академии наук
650065 Кемерово, Россия
3 Балтийский федеральный университет им. И. Канта
236000 Калининград, Россия
* E-mail: vminina@mail.ru
Поступила в редакцию 25.11.2021
После доработки 14.02.2022
Принята к публикации 22.02.2022
- EDN: UIAKCY
- DOI: 10.31857/S0016675822070128
Аннотация
Рак легкого является наиболее распространенным онкологическим заболеванием у мужчин во всем мире. Для поиска новых биологических маркеров этой патологии с помощью технологии микроматричного анализа SurePrint G3 Human Gene Expression был исследован транскриптом мононуклеаров крови пациентов и здоровых доноров (жители Кемеровской обл., Россия). В результате было выявлено 288 дифференциально экспрессирующихся генов, 108 генов с повышенной и 180 с пониженной экспрессией. Функциональный анализ обогащения с использованием ресурса WebGestalt и различных баз (Gene Ontology, KEGG и Reactome) указал на изменение экспрессии генов, вовлеченных в процессы иммунологического ответа, синтеза белка, контроля клеточного цикла и апоптоза. Анализ белок-белковых взаимодействий с применением алгоритма STRING позволил выявить функциональные кластеры продуктов генов с разным уровнем их экспрессии.
Рак легкого (РЛ) занимает лидирующие места по показателям заболеваемости и смертности населения от онкологических патологий во всем мире [1]. Ежегодно Всемирная организация здравоохранения (ВОЗ) регистрирует около 2.1 миллионов диагнозов и 1.75 миллионов летальных исходов. Выявление на поздних стадиях, активное метастазирование, многообразие гистологических форм – отличительная особенность данной патологии [2]. Курение, действие канцерогенов окружающей или производственной среды рассматривается в качестве ведущих факторов риска РЛ [3]. Действие полициклических ароматических углеводородов (ПАУ), тяжелых металлов, ионизирующего излучения индуцирует окислительный стресс, приводящий к накоплению повреждений ДНК [4, 5] и росту заболеваемости.
Развитие постгеномных технологий ускорило возможности поиска новых биологических маркеров. Секвенирование РНК и микроматричный анализ относятся к основным подходам для изучения экспрессии генов (и их продуктов), вовлеченных во множественные сигнальные пути. Предполагают, что секвенирование РНК позволяет выявить потенциальные генетические маркеры развития РЛ [6]. Технология микроматричного анализа SurePrint G3 Human Gene Expression недавно использовалась для изучения иммунного микроокружения немелкоклеточного РЛ и поиска новых молекулярных детерминант мелкоклеточного РЛ [7, 8]. Данная технология позволяет описать паттерны экспрессии, ассоциированные с опухолеобразованием для наилучшего понимания клеточных процессов, связанных с РЛ.
Аннотация транскриптома напрямую связана с использованием крупных репозиториев. К ним относятся Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) и Reactome. База данных GO представляет собой биоинформатический ресурс, который позволяет проводить анализ различных групп генов и их продуктов. В настоящее время были разработаны программы для работы с данными GO [9]. База данных GO является ключевым ресурсом для исследований функциональной геномики при интерпретации результатов секвенирования РНК [10]. KEGG содержит данные о метаболоме, а также о геномных и протеомных последовательностях. Данный ресурс был эффективно использован в исследовании вклада экзосомальных РНК в развитие аденокарциномы легкого [11]. Reactome содержит информацию о важных биологических сигнальных путях у различных организмов, включая человека. Методы машинного обучения с использованием информации о генах, полученной в Reactome, позволили выявить новые молекулярные маркеры мелкоклеточного РЛ [12]. Таким образом, использование ресурсов нескольких биоинформатических баз данных способно дать ценную информацию и наиболее полно охарактеризовать результаты транскриптомных исследований.
Цель данного исследования – микроматричный анализ транскриптома мононуклеаров периферической крови у больных РЛ.
МАТЕРИАЛЫ И МЕТОДЫ
Материалы
Данное исследование было выполнено в лаборатории цитогенетики Кемеровского государственного университета (Кемерово, Россия) и в Томском национальном исследовательском медицинском центре (Томск, Россия). Материалом послужили образцы периферической крови пациентов Кемеровского областного онкологического диспансера, которые впервые обратились для постановки диагноза и лечения (диагноз выставлялся позднее после всех видов инструментальных обследований специалистами диспансера). Сбор материала был проведен в период январь–сентябрь 2019 г. (до начала пандемии covid-19). В качестве группы сравнения использовались образцы крови здоровых мужчин близкого возраста, проживающих в той же местности, работников Кемеровского государственного университета. Забор крови производился в стерильные вакутейнеры с ЭДТА в асептических условиях. Образцы немедленно доставлялись в лабораторию, где проводили выделение мононуклеаров крови (в градиенте плотности фиколла) и выделение РНК. Все участники эксперимента (семь больных немелкоклеточным РЛ и восемь здоровых мужчин, жители Кемеровской обл., средний возраст 59 лет) были осведомлены о цели, методах исследования и оформляли информированное согласие.
Выделение РНК и пробоподготовка
Стабилизацию РНК в образцах проводили с помощью RNAlater/RNAStabilization Reagent (Qiagen, Германия). Выделение тотальной РНК из мононуклеаров периферической крови выполняли с использованием реагента TRIzol (Invitrogen, США) согласно рекомендациям производителя. Для очистки РНК использовался коммерчески доступный набор “RNeasy Mini Kit” (Qiagen, Германия). Концентрация РНК и показатели стабильности РНК (RIN-values) верифицировалась с применением электрофоретической системы “Agilent 2100 Bioanalyzer” (Agilent Technologies, США). Реагент “RNA Spike-In” добавлялся для калибровки РНК. РНК далее транскрибировалась в кДНК с использованием набора “T7 Primer Mix” (Merck, Германия). Далее “Transcription Master Mix” использовался для транскрибирования кДНК в копии молекул кРНК.
Определение генной экспрессии
В данном исследовании экспрессия генов была изучена с помощью “SurePrint G3 Human Gene Expression 8 × 60K Microarray Kit” (Agilent Technologies, США). Микрочип содержит пробы для 26 803 уникальных генов и 30 606 длинных некодирующих РНК. Количественный анализ экспрессии выполнялся согласно протоколу производителя (One-color microarray-based gene expression analysis, v. 6.9.1, Agilent Technologies, США). Микрочипы были просканированы с использованием “SureScan Microarray Scanner” (Agilent Technologies, США). Программное обеспечение “Feature Extraction v. 12.0” (Agilent Technologies, США) использовалось для оценки интенсивности флуоресцентного сигнала.
Процессинг полученных данных и статистический анализ
Ресурсы Gene Ontology, KEGG и Reactome использовались для аннотации результатов транскриптомного анализа. Функциональный анализ обогащения выполнялся с помощью функций программы WebGestalt. Алгоритм STRING использовался для оценки белок-белковых взаимодействий продуктов дифференциально экспрессирующихся генов. Достоверный уровень белок-белковых взаимодействий принимался при условии коэффициента score > 0.4. Поправка FDR на множественные сравнения использовалась для установления значимости различий в экспрессии генов между двумя группами сравнения. В качестве пороговых значений были использованы p < 0.05 и отличия в экспрессии между группами более чем в 1.5 раза (lgFC > |0.6|) и более чем в 2 раза (lgFC > > |1.0|). Выбор данных значений обусловлен тем, что наиболее часто используемым порогом отличий между группами образцов по уровням экспрессии генов являются отличия в 2 и более раз [13]. Более мягкий порог отличий в 1.5 раза используется для исследований, направленных на анализ обогащения отдельных функциональных групп среди дифференциально экспрессирующихся генов [14, 15].
РЕЗУЛЬТАТЫ
В результате транскриптомного анализа было установлено, что больные РЛ и здоровые индивиды отличаются между собой по экспрессии большого числа генов (108 генов с повышенной и 180 с пониженной экспрессией). Несмотря на то, что при использовании поправки на множественность сравнений Бенджамини–Хохберга (false discovery rate – FDR) между группами индивидов не было обнаружено статистически значимых дифференциально экспрессирующихся генов (ДЭГ), возможно провести анализ наборов генов с наибольшими отличиями в экспрессии между группами. В опытной группе было выявлено повышение экспрессии более чем в 1.5 раза для 108 проб на микрочипе, а также снижение экспрессии более чем в 1.5 раза для 180 проб на микрочипе (табл. 1). Часть проб на микрочипе относится к различным некодирующим транскриптам, гипотетическим транскриптам, транскриптам псевдогенов и другим слабо аннотированным частям генома. Поэтому для дальнейшего анализа использовалась только информация, полученная с проб для транскриптов известных генов человека (на основе базы данных Entrez Gene – https://www.ncbi.nlm.nih.gov/gene/). Число таких ДЭГ с повышенной экспрессией в опытной группе составило 87 (более чем в 1.5 раза) и девять генов (более чем в 2 раза). Сниженную экспрессию в опытной группе демонстрировали 175 и 50 генов (более чем в 1.5 и 2 раза соответственно) (табл. 1).
Таблица 1.
Число дифференциально экспрессирующихся генов (ДЭГ) у больных раком легкого
| Гены с повышенной экспрессией | Гены со сниженной экспрессией | Все ДЭГ | ||
|---|---|---|---|---|
| LgFC > |0.6| | Число проб на чипе | 108 | 180 | 288 |
| Число уникальных транскриптов | 108 | 180 | 288 | |
| Число уникальных генов (Entrez) | 87 | 175 | 262 | |
| LgFC > |1.0| | Число проб на чипе | 10 | 35 | 45 |
| Число уникальных транскриптов | 10 | 35 | 45 | |
| Число уникальных генов (Entrez) | 9 | 50 | 59 | |
Функциональный анализ экспрессии генов
Функциональный анализ дифференциально экспрессирующихся генов с использованием баз данных Gene Ontology (GO) указало на обогащение групп генов, вовлеченных в процессы иммунологического ответа, синтеза белка и контроля клеточного цикла. У пациентов с РЛ было отмечено понижение экспрессии генов врожденного и специфического иммунного ответа. Среди генов с пониженной экспрессией (в 61 раз) выявлены гены, кодирующие компоненты телец Кахаля (GO:1904869: CCT6A, TCP1, DKC1), факторы инициации синтеза белка и транспорта молекул к эндоплазматическому ретикулуму (GO:0006614, GO:0006613, GO:0045047, GO:0072599, GO:0006413, GO:0070972) имели наибольшую степень обогащения в группе пациентов. Транскрипты с повышенной экспрессией были представлены факторами иммунного ответа (GO:0006955), хемокиновыми рецепторами, специфическими киназами и другими сигнальными молекулами, связанными с реакциями иммунитета. Исходя из полученных результатов, было также отмечено, что в группе больных РЛ факторы апоптоза характеризуются повышенным уровнем синтеза (GO:0042981). Гены, которые имели как повышенную, так и пониженную экспрессию, были задействованы в регуляции клеточного цикла и иммунологическом ответе (GO:0001775; GO:0006955).
Функциональный анализ на основе данных KEGG позволил определить понижение в группе больных РЛ экспрессии факторов, которые были ассоциированы с иммунологическим ответом, функциональной активностью фагосом и рибосом, а также дифференциацией остеокластов. Наибольшим уровнем обогащения характеризовались факторы резистентности к туберкулезу. Повышенная экспрессия была характерна для факторов, связанных с развитием острых иммунных заболеваний. Центральная роль была определена для компонентов сигнального пути Т-клеточных рецепторов (hsa04660). Компоненты, задействованные в иммунологическом ответе и синтезе белка, были также определены как низко экспрессирующиеся в результате функционального анализа с использованием базы данных Reactome. Данные гены кодируют факторы, которые задействованы во всех этапах синтеза белка, формировании рибосомального комплекса, а также активации компонентов иммунитета. В отличие от баз данных GO и KEGG группы генов с повышенной экспрессией не были обнаружены в ходе анализа.
Белок-белковые взаимодействия
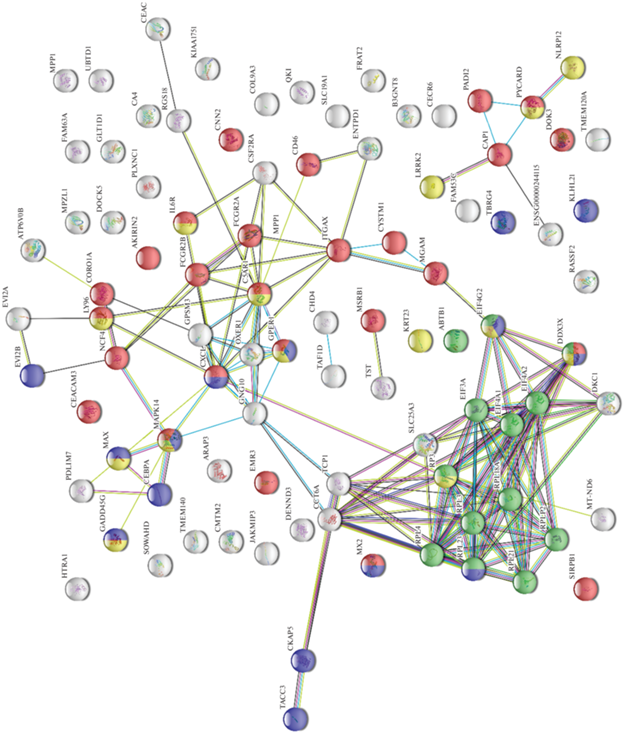
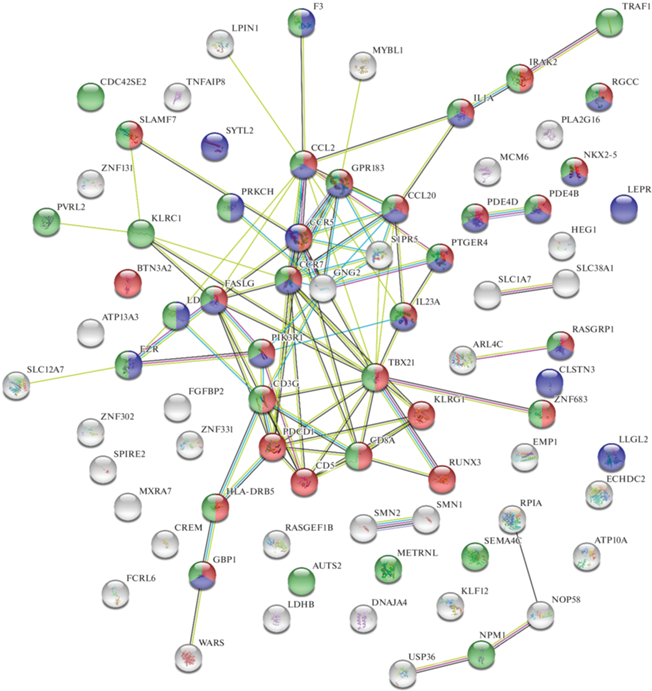
Группы дифференциально экспрессирующихся генов были проанализированы с помощью алгоритма STRING и репозитория GO [16]. Были построены сети, отражающие особенности взаимодействий между продуктами этих генов. Сильный и значимый уровень взаимодействий был определен при значении показателя score parameter > 0.4. В ходе анализа были идентифицированы кластеры генов с разным уровнем экспрессии. Используя ресурсы GO было установлено, что гены с пониженной экспрессией были представлены факторами синтеза белка (GO:0006412), иммунологического ответа (GO:0006955), регуляции клеточного цикла (GO:0051726) и клеточной смерти (апоптоза) (GO:0008219) (рис. 1). Функциональные кластеры продуктов генов с повышенной экспрессией были также задействованы в иммунологических реакциях (GO:0002376), локализации молекул в клетке (GO:0032879) и ответа клеток на стимулы (GO:0048583) (рис. 2).
ОБСУЖДЕНИЕ
На основе проведенного транскриптомного анализа впервые в России были установлены изменения паттернов генной экспрессии в мононуклеарах периферической крови у больных РЛ. Группа генов иммунного ответа была определена как наиболее обогащенная среди ДЭГ. Рецептор к интерлейкину-6 IL6R (1q21.3) является компонентом сигнального пути IL-6/IL6R, который вносит существенный вклад в опухолеобразование. Особенности экспрессии данного цитокинового рецептора рассматриваются в контексте развития некоторых гистопатологических типов РЛ [22]. Экспрессия данного фактора негативно коррелирует с показателями выживаемости при развитии аденокарциномы легкого и положительно при плоскоклеточном раке. Хемокины, продуцируемые макрофагами, выполняют роль регуляторов реакций врожденного иммунитета. Ген CXCL8 (14q13.3) кодирует цитокин ИЛ-8, участвующий в активации Т-лимфоцитов. У лабораторных мышей (линия Lewis) был отмечен противораковый эффект ингибирования CXCL8-CXCR1/2 [23]. Однако повышение экспрессии CXCL8 вносит вклад в повышение показателей выживаемости. Рецептор C5AR1 r (CD88) (19q13.32) участвует в развитии провоспалительных реакиций и их контроле. Повышение экспрессии мРНК C5AR1 может быть ассоциировано с развитием метастаз при тяжелом течении немелкоклеточного РЛ [24]. Рецепторы семейства G-белков вовлечены в процессы развития врожденного и специфического иммунитета. Один из них кодируется геном GPER (7p22.3), имеет способность к связыванию с эстрадиолом и может способствовать проявлению гормональных эффектов. Данный фактор имеет повышенную экспрессию в составе тканей легочных неоплазм. Роль GPER в развитии и РЛ может быть главным образом связана с регуляцией NOTCH1 [25].
Цитоплазматический белок NLRP12 (19q13.42) участвует в активации провоспалительных факторов. Уровень мРНК в легких лабораторных мышей может подавляться при воздействии взвешенных твердых частиц (PM2.5) [26]. Белки, связанные с аденилатциклазой (CAP’s), задействованы в циклическим сигнальном пути AМФ и в контроле иммунных реакций. При использовании клеточных линий РЛ обнаружили повышение экспрессии гена CAP1 при развитии немелкоклеточного РЛ, что способствует усилению клеточной пролиферации. Повышенная экспрессия CAP1 также может быть связана с развитием метастазов [27, 28].
Гены рибосомальных белков относятся к числу “генов домашнего хозяйства”. В ходе данного исследования было выявлено изменение уровня их экспрессии в мононуклеарах периферической крови у больных РЛ. Фактор инициации синтеза белка EIFA42 (3q27.3) обеспечивает присоединение мРНК к рибосомальному комплексу и является высококонсервативным геном. Влияние его экспрессии было определено для особенностей разных гистопатологических типов РЛ и определенной степени прогрессии опухолеобразования [29]. Отмечено, что потеря экспрессии EIFA42 понижает выживаемость у пациентов с немелкоклеточным РЛ. Рибосомальный белок RPL4 (15q22.31) выполняет важную функцию при развитии клеточного роста и пролиферации через участие в MDM-p53 [30]. Рибосомальный белок RPL4 (15q22.31) выполняет важные функции при реализации сигнального пути RPL23-MDM-p53, обладающего противораковым потенциалом [31]. Фактор eIF3a (10q26.11) работает в составе мультипротеинового комплекса при инициации трансляции. Его экспрессия может быть связана с резистентностью к химиотерапии на основе платины при лечении РЛ [32]. Аналогичные особенности были отмечены при исследовании циркулирующих молекул РНК, ассоциированных с eIF3a [33].
Факторы, регулирующие клеточный цикл и апоптоз, играют важнейшую роль в инициации канцерогенеза. Некоторые из них были также идентифицированы в ходе транскриптомного анализа. LRRK2 (12q12), относящийся к ферментам-киназам, имеет сильную ассоциацию с развитием нейродегенеративных и аутоиммунных заболеваний, включая болезни Паркинсона и Крона. Однако последние исследования выявили ассоциацию гена LRRK2 с РЛ [34]. Пациенты с низкой экспрессией фактора LRRK2 имели сниженные показатели выживаемости. Лабораторные мыши, нокаутные по гену LRRK2, демонстрировали повышенный рост опухолей, подтверждая потенциальную роль данного гена в прогрессировании РЛ. Некоторые транскрипционные факторы модулируют экспрессию генов, участвующих в реакциях клеточного цикла. Ген CEBPA (19q13.11) кодирует белок CEBPA (CAAT/enhancer binding protein alpha) – ключевой регулятор эмбриогенеза, дифференциации клеток крови и некоторых метаболических путей. Он принадлежит к семейству bZIP-подобных транскрипционных факторов и может присоединяться к определенным энхансерам. В опухолях немелкоклеточного РЛ лабораторных мышей было обнаружено нарушение регуляции экспрессии данного гена [35].
Транскрипционный фактор MAX (14q23.3) также может рассматриваться в качестве важного регулятора клеточного цикла. Уровень синтеза его мРНК (наряду с факторами MTURN и HLA-B) был определен как повышенный в составе тромбоцитов на ранних стадиях развития РЛ [36]. GADD45-подобные белки имеют множественную функциональную активность и участвуют во многих процессах (репарация ДНК, старение и генотоксический стресс), влияющих на онкогенез [37]. Данное суперсемейство включает GADD45G (9q22.2), который также проявляет множественную активность.
С применением ресурса STRING в ходе исследования были получены данные о взаимодействиях продуктов ДЭГ. В данном исследовании была построена белковая сеть из факторов с низким и повышенным уровнем синтеза. Был идентифицирован кластер, состоящий из факторов синтеза белка (RPL4, RPL23, PL21, RPLP2, EIF3A, EIF4A1, EIF4A2). Эти белки имели связь с факторами SLC25A3 и DOX3X, ассоциированными с такими важными процессами как митохондриальный транспорт и контроль клеточных сигнальных путей. Кластер факторов иммунологического ответа и клеточного апоптоза включал в себя: CXCL, GPSM3, FCGR2B, IL6R, GNG10, CXER1, GPER1, C5AR1, FCGR2A, C5F2RA, ITGAX, а также белки CAP1, PADI2, PYCARD, NLRP12. Белки GADD45G, CEBPA, MAX, MAPK14, PDLM7 составили кластер клеточного цикла и апоптоза.
Результаты экспериментальных исследований профилей экспрессии генов при РЛ весьма разнородны и представлены в таких базах как Gene Expression Omnibus (GEO), Genotype-Tissue Expression (GTEx) project, the Cancer Genome Atlas (TCGA), A molecular cell atlas of the human lung from single cell RNA sequencing и других. Так, был охарактеризован транскриптом опухолевых и нормальных тканей легкого [38]. С использованием TWAS-подхода (transcriptome-wide association study), объединившего результаты GWAS (genome-wide association studies) и eQTL (expression quantitative trait loci), выявлены гены: IREB2 и AQP3, изменение экспрессии которых значимо увеличивает риск РЛ [39]. Китайскими авторами недавно были описаны пять новых генов (DCAF16, CBL, ATR, GYPE и PARD3), которые были значимо ассоциированы с риском РЛ в различных моделях (cross-tissue и lung tissue models) [40]. Fanlu Meng c коллегами, выполнили мета-анализ результатов изучения экспрессии генов, основываясь на данных, размещенных в базе GEO, результатах функционального анализа обогащения (на основе данных KEGG и GO), изучения белок-белковых взаимодействий и анализа связывания транскрипционных факторов при аденокарциноме легкого [41]. Было выявлено повышение экспрессии 207 и снижение экспрессии 367 генов в опухолевых тканях легкого. Дальнейший анализ позволил выявить ряд таргетных генов, таких как AURKA, CCNB1, KIF11, CCNA2, TOP2A, CENPF, KIF2C, TPX2, HMMR, MAD2L1 и генов транскрипционных факторов (SP1, CTNNB1, MYC, CEBPA и NFKB1), оказывающих наиболее значимое влияние на патогенез РЛ. Они вовлечены в управление такими биологическими процессами как пролиферация, иммунный ответ, миграция лейкоцитов, клеточная адгезия, ангиогенез функционирование микротрубочек, контроль топологии ДНК, клональный рост клеток, дифференцировка и апоптоз.
У пациентов с аденокарциномой легкого также проводилось секвенирование РНК одиночных клеток с помощью HiSeq 4000 (Illumina, San Diego, California, USA) для оценки клеточных популяций в составе микроокружения опухоли [42]. Было обнаружено, что гетерогенный клеточный состав микроокружения опухоли был ассоциирован со степенью дифференцировки клеток карциномы. Были выявлены два различных паттерна микроокружения опухоли (CP2E и N3MC), отличающихся по составу клеток и прогнозу. Таким образом, профилирование РНК одиночных клеток предоставляет дополнительную прогностическую информацию, основанную на микроокружении, что может помочь предсказать ответ на терапию и выявить возможные популяции клеток-мишеней [42].
Полномасштабный анализ транскриптома цельной крови ранее выполнялся с помощью чипов WG6 (Illumina), которые сканировались на Illumina BeadStation 500× [43]. Полученные результаты свидетельствуют о том, что профили экспрессии генов РНК-стабилизированных образцов цельной крови (использовались пробирки PAXgene) могут быть использованы для детекции рака легкого у курильщиков. Перечень генов, использовавшихся для построения классификации был обогащен генами, контролирующими иммунный ответ. Авторы постулируют, что классификатор основан на транскрипции генов, контролирующих иммунный ответ именно в клетках крови, а не под влиянием транскриптов редких опухолевых клеток, иногда обнаруживаемых в крови раковых пациентов (хотя эту возможность нельзя исключать) [43].
M. Rotunno с соавт. выполняли анализ транскриптома крови у больных раком легкого с помощью чипов Affymetrix GeneChip® HG-U133A v2.0 [44]. Авторы сфокусировались на анализе случаев больных аденокарциномой легкого на первой стадии и выявили гены, экспрессия которых в клетках крови статистически значимо отличается от здоровых индивидов. Было описано восемь ключевых генов (RUNX3, TGFBR3, TRGC2, TRGV9, TARP, ACP1, VCAN и TSTA3), вовлеченных в регуляцию процессов репарации ДНК, пролиферации, миграции, адгезии клеток, ангиогенеза и функционирования иммунной системы. Интересно то, что данные особенности были описаны у пациентов с I-ой стадией развития РЛ. Поэтому, по мнению авторов, особенности экспрессии данных генов в клетках крови (при условии верификации) могут оказаться полезны в качестве ранних маркеров риска рака легкого [44].
Отдельные типы клеток крови, например мононуклеары широко используются для исследования патологий, связанных со злокачественной трансформацией. Например, линия таких клеток использовалась для поиска потенциальных биомаркеров гепатоцеллюлярного ракa [45]. В настоящем исследовании впервые с помощью чипа SurePrint G3 Human Gene Expression (Agilent Technologies) было выполнено изучение транскриптома мононуклеаров крови у пациентов с РЛ и выявлено обогащение групп генов, вовлеченных в процессы иммунного ответа, синтеза белка и пролиферации. Это согласуется с выводами работ, выполненных ранее непосредственно на опухолевых клетках тканей легкого [46, 47], клетках эпителия дыхательных путей [48] и клетках цельной крови пациентов с РЛ [43, 44].
Масштабные изменения, которые наблюдаются в мононуклеарах крови, отражают глобальные перестройки экспрессии генов, происходящие в организме при формировании солидных опухолей. Результаты проведенных исследований указывают на то, что профиль экспрессии генов в клетках крови можно рассматривать как промежуточный молекулярный фенотип между генотипом и фенотипом пациента [48], потенциально значимый в качестве биомаркера РЛ. Безусловно полученные результаты нуждаются в верификации другими методами (например количественной ПЦР) на больших по объему выборках. Однако полученные данные могут быть полезны для выбора направлений будущих исследований.
ЗАКЛЮЧЕНИЕ
С использованием технологии микроматричного анализа транскриптома и биоинформатического анализа была изучена экспрессия генов в мононуклеарах периферической крови больных РЛ и здоровых доноров. В опытной группе была изменена экспрессия генов иммунного ответа и генов, продукты которых участвуют в регуляции клеточного цикла. На фоне подобных изменений возможно накопление большого количества повреждений ДНК в клетках и инициация канцерогенеза. Кроме того, нарушенные реакции иммунного ответа могут способствовать прогрессированию злокачественной трансформации в тканях. Предстоит оценить перспективность применения выявленных молекулярных сигнатур в системе ранней диагностики и профилактики РЛ.
Исследование поддержано грантом РНФ № 18-14-00022.
От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Bray F., Ferlay J., Soerjomataram I. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries // CA: A Cancer J. for Clinicians. 2018. V. 68. № 6. P. 394–424. https://doi.org/10.3322/caac.21492
Zappa C., Mousa S.A. Non-small cell lung cancer: Current treatment and future advances // Translational Lung Cancer Res. 2016. V. 5. № 3. P. 288–300. https://doi.org/10.21037/tclr.2016.06.07
Yang X., Man J., Chen H. et al. Temporal trends of the lung cancer mortality attributable to smoking from 1990 to 2017: A global, regional and national analysis // Lung Cancer. 2021. V. 152. P. 49–57. https://doi.org/10.1016/j.lungcan.2020.12.007
Moorthy B., Chu C., Carlin D.J. Polycyclic aromatic hydrocarbons: from metabolism to lung cancer // Toxicol Sci. 2015. V. 145. № 1. P. 5–15. https://doi.org/10.1093/toxsci/kfv040
Ewa B., Danuta M.-Š. Polycyclic aromatic hydrocarbons and PAH-related DNA adducts // J. Applied Genetics. 2017. V. 58. № 3. P. 321–330. https://doi.org/10.1007/s13353-016-0380-3
Han S.-S., Kim W.J., Hong Y. et al. RNA sequencing identifies novel markers of non-small cell lung cancer // Lung Cancer. 2014. V. 84. № 3. P. 229–235. https://doi.org/10.1016./j.lungcan.2014.03.018
Öjlert Å.K., Halvorsen A.R., Nebdal D. et al. The immune microenvironment in non-small cell lung cancer is predictive of prognosis after surgery // Mol. Oncology. 2019. V. 13. № 5. P. 1166–1179. https://doi.org/10.1002/1878-0261.12475
Misono S., Mizuno K., Suetsugu T. et al. Molecular signature of small cell lung cancer after treatment failure: The MCM complex as therapeutic target // Cancers. 2021. V. 13. № 6. P. 187–203. https://doi.org/10.3390/cancers13061187
Pomaznoy M., Ha B., Peters B. GOnet: A tool for interactive gene ontology analysis // BMC Bioinformatics. 2019. V. 19. № 1. P. 470–478. https://doi.org/10.1186/s12859-018-2533-3
Zhu J., Zhao Q., Katsevich E., Sabatti C. Exploratory gene ontology analysis with interactive visualization // Sci. Rep. 2019. V. 9. № 1. P. 7793–7801. https://doi.org/10.1038/s41598-019-42178-x
Liu X-Q., Tufman A., Kiefl R. et al. Identification of lung adenocarcinoma-specific exosome RNAs in peripheral blood by RNA-Seq analysis // European Review for Med. and Pharmacol. Sci. 2020. V. 24. № 4. P. 1877–1886. https://doi.org/10.26355/eurrev_202002_20366
Juarez-Flores A., Zamudio G.S., José M.V. Novel gene signatures for stage classification of the squamous cell carcinoma of the lung // Sci. Rep. 2021. V. 11. № 1. P. 4835–4845. https://doi.org/10.1038/s41598-021-83668-1
Gov E., Arga K.Y. Differential co-expression analysis reveals a novel prognostic gene module in ovarian cancer // Sci. Rep. 2017. V. 7. № 1. P. 4996. https://doi.org/10.1038/s41598-017-05298-w
Fajarda O., Duarte-Pereira S., Silva R.M., Oliveira J.L. Merging microarray studies to identify a common gene expression signature to several structural heart diseases // BioData Min. 2020. V. 13. № 8. https://doi.org/10.1186/s13040-020-00217-8
Yosef N., Shalek A.K., Gaublomme J.T. et al. Dynamic regulatory network controlling TH17 cell differentiation // Nature. 2013. V. 496. № 7446. P. 461–468. https://doi.org/10.1038/nature11981
Szklarczyk D., Gable A.L., Lyon D. et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets // Nucl. Ac. Res. 2019. V. 47. № 1. P. 607–613. https://doi.org/10.1093/nar/gky1131
Chae Y.K., Arya A., Iams W. et al. Immune checkpoint pathways in non-small cell lung cancer // Ann. of Translational Med. 2018. V. 6. № 5. P. 88–108. https://doi.org/10.21037/atm.2017.09.30
Seidel J.A., Otsuka A., Kabashima K. Anti-PD-1 and Anti-CTLA-4 therapies in cancer: Mechanisms of action, efficacy, and limitations // Frontiers in Oncology. 2018. V. 8. P. 86–100. https://doi.org/10.3389/fonc.2018.00086
Paz-Elizur T., Leitner-Dagan Y., Meyer K.B. et al. DNA repair biomarker for lung cancer risk and its correlation with airway cells gene expression // JNCI Cancer Spectrum. 2020. V. 4. № 1. P. 1–10. https://doi.org/10.1093/jncics/pkz067
Nan Y.-L., Hu Y.-L., Liu Z.-K. et al. Relationships between cell cycle pathway gene polymorphisms and risk of hepatocellular carcinoma // World J. of Gastroenterology. 2016. V. 22. № 24. P. 5558–5567. https://doi.org/10.3748/wjg.v22.i24.5558
McKay J.D., Hung R.J., Han Y. et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes // Nature Genetics. 2017. V. 49. № 7. P. 1126–1132. https://doi.org/10.1038/ng.3892
Xu B., Chen Q., Yue C. et al. Prognostic value of IL6R mRNA in lung adenocarcinoma and squamous cell carcinoma // Oncology Letters. 2018. V. 16. № 3. P. 2935–2948. https://doi.org/10.3892/ol.2018.9044
Hsu S.-Y., Yu H.-Y., Lee W.-C. et al. A novel CXCL8 analog is effective in inhibiting the growth via cell cycle arrest and attenuating invasion of lewis lung carcinoma // OncoTargets and Therapy. 2019. V. 12. P. 7611–7621. https://doi.org/10.2147/OTT.S215824
Ajona D., Zandueta C., Corrales L. et al. Blockade of the complement C5a/C5aR1 axis impairs lung cancer bone metastasis by CXCL16-mediated effects // Am. J. of Respiratory and Critical Care Med. 2018. V. 197. № 9. P. 1164–1176. https://doi.org/10.1164/rccm.201703-0660OC
Shen Y., Li C., Zhou L., Huang J. G protein-coupled oestrogen receptor promotes cell growth of non-small cell lung cancer cells via YAP1/QKI/circNOTCH1/ m6A methylated NOTCH1 signalling // J. Cellular and Molecular Medicine. 2021. V. 25. № 1. P. 284–296. https://doi.org/10.1111/jcmm.15997
Jin Y., Wu W., Zhang W. et al. Involvement of EGF receptor signaling and NLRP12 inflammasome in fine particulate matter-induced lung inflammation in mice: EGFR and NLRP12 Involved in PM2.5-induced lung inflammation in mice // Environmental Toxicology. 2017. V. 32. № 4. P. 1121–1134. https://doi.org/10.1002/tox.22308
Tan M., Song X., Zhang G. et al. Overexpression of adenylate cyclase-associated protein 1 is associated with metastasis of lung cancer // Oncol. Reports. 2013. V. 30. № 4. P. 1639–1644. https://doi.org/10.3892/or.2013.2607
Kolegova E.S., Kakurina G.V., Kondakova I.V. et al. Adenylate cyclase-associated protein 1 and cofilin in progression of non-small cell lung cancer // Bull. of Exp. Biol. and Med. 2019. V. 167. № 3. P. 393–395. https://doi.org/10.1007/s10517-019-04534-9
Shaoyan X., Juanjuan Y., Yalan T. et al. Downregulation of EIF4A2 in non–small-cell lung cancer associates with poor prognosis // Clin. Lung Cancer. 2013. V. 14. № 6. P. 658–665. https://doi.org/10.1016/j.cllc.2013.04.011
He X., Li Y., Dai M.-S., Sun X.-X. Ribosomal protein L4 is a novel regulator of the MDM2-p53 loop // Oncotarget. 2016. V. 7. № 13. P. 16217–16226. https://doi.org/10.18632/oncotarget.7479
Wang J., Zhang Z., Li F. et al. Triptolide interrupts rRNA synthesis and induces the RPL23-MDM2-p53 pathway to repress lung cancer cells // Oncol. Reports. 2020. V. 43. № 6. P. 1863–1874. https://doi.org/10.3892/or.2020.7569
Yin J.-Y., Shen J., Dong Z.-Z. et al. Effect of eIF3a on response of lung cancer patients to platinum-based chemotherapy by regulating DNA repair // Clin. Cancer Res. 2011. V. 17. № 13. P. 4600–4609. https://doi.org/10.1158/1078-0432.CCR-10-2591
Huang M., Yuan F., Gao Y. et al. Circular RNA screening from EIF3a in lung cancer // Cancer Med. 2019. V. 8. № 9. P. 4159–4168. https://doi.org/10.1002/cam4.2338
Lebovitz C., Wretham N., Osooly M. et al. Loss of Parkinson’s susceptibility gene LRRK2 promotes carcinogen-induced lung tumorigenesis // Sci. Rep. 2021. V. 11. № 1. P. 2097–3011. https://doi.org/10.1038/s41598-021-81639-0
Sato A., Yamada N., Ogawa Y., Ikegami M. CCAAT/enhancer-binding protein-α suppresses lung tumor development in mice through the p38α MAP kinase pathway // PLoS One. 2013. V. 8. № 2. e57013–e57022. https://doi.org/10.1371/journal.pone.0057013
Liu L., Song X., Li X. et al. A three-platelet mRNA set: MAX, MTURN and HLA-B as biomarker for lung cancer // J. of Cancer Res. and Clin. Oncol. 2019. V. 145. № 11. P. 2713–2723. https://doi.org/10.1007/s00432-019-03032-9
Tamura R.E., de Vasconcellos J.F., Sarkar D. et al. GADD45 proteins: Central players in tumorigenesis // Curr. Mol. Med. 2012. V. 12. № 5. P. 634–651. https://doi.org/10.2174/156652412800619978
Bang M.S., Kang K., Lee J. et al. Transcriptome analysis of non-small cell lung cancer and genetically matched adjacent normal tissues identifies novel prognostic marker genes // Genes & Genomics. 2017. V. 39. № 3. P. 277–284. https://doi.org/10.1007/s13258-016-0492-5
Bossé Y., Li Z., Xia J. et al. Transcriptome-wide association study reveals candidate causal genes for lung cancer // Int. J. Cancer. 2020. V. 146. № 7. P. 1862–1878. https://doi.org/10.1002/ijc.32771
Zhu M., Fan J., Zhang C. et al. A cross-tissue transcriptome-wide association study identifies novel susceptibility genes for lung cancer in Chinese populations // Hum. Mol. Genet. 2021. V. 30. № 17. P. 1666–1676. https://doi.org/10.1093/hmg/ddab119
Meng F., Zhang L., Ren Y., Ma Q. Transcriptome analysis reveals key signature genes involved in the oncogenesis of lung cancer // Cancer Biomark. 2020. V. 29. № 4. P. 475–482. https://doi.org/10.3233/CBM-200110
Bischoff P., Trinks A., Obermayer B. et al. Single-cell RNA sequencing reveals distinct tumor microenvironmental patterns in lung adenocarcinoma // Oncogene. 2020. V. 40(50). P. 6748–6798. https://doi.org/10.1038/s41388-021-02054-3
Zander T., Hofmann A., Staratschek-Jox A. et al. Blood-based gene expression signatures in non-small cell lung cancer // Cancer Res. 2011. V. 17. № 10. P. 3360–3367. https://doi.org/10.1158/1078-0432.CCR-10-0533
Rotunno M., Hu N., Su H. A gene expression signature from peripheral whole blood for stage I lung adenocarcinoma // Cancer Prev. Res. (Phila). 2011. V. 4. № 10. P. 1599–1608. https://doi.org/10.1158/1940-6207
Kunadirek P., Ariyachet C., Sriphoosanap S. et al. Identification of BHLHE40 expression in peripheral blood mononuclear cells as a novel biomarker for diagnosis and prognosis of hepatocellular carcinoma // Sci. Rep. 2021. V. 11. № 1. P. 11201–11213. https://doi.org/10.1038/s41598-021-90515-w
Xiong Y., Feng Y, Qiao T., Han Y. Identifying prognostic biomarkers of non-small cell lung cancer by transcriptome analysis // Cancer Biomark. 2020. V. 27. № 2. P. 243–250. https://doi.org/10.3233/CBM-190222
Billatos E., Vick J.L., Lenburg M.E., Spira A.E. The airway transcriptome as a biomarker for early lung cancer detection // Clin. Cancer Res. 2018. V. 24. № 13. P. 2984–2992. https://doi.org/10.1158/1078-0432.CCR-16-3187
Lopatkina M.E., Lebedev I.N. Transcriptome analysis as a tool for investigation of pathogenesis of chromosomal diseases // Russian J. Genetics. 2020. V. 56. Issue 5. P. 548–561. https://doi.org/10.1134/S1022795420050099
Дополнительные материалы
- скачать ESM.docx
- Таблица 1. - Таблица 10.