

Генетика, 2022, T. 58, № 7, стр. 815-821
Мутационный анализ гена F11 у пациентов с дефицитом FXI в России
О. С. Пшеничникова 1, *, В. Л. Сурин 1, Н. В. Сац 1, Е. В. Яковлева 1, О. С. Димитриева 1, Н. И. Зозуля 1
1 Национальный медицинский исследовательский центр гематологии Министерства здравоохранения
Российской Федерации
125167 Москва, Россия
* E-mail: pshenichnikovaolesya@gmail.com
Поступила в редакцию 01.12.2021
После доработки 21.01.2022
Принята к публикации 03.02.2022
- EDN: RQSMDE
- DOI: 10.31857/S001667582207013X
Аннотация
Фактор FXI является важным участником каскада свертывания крови и кодируется геном F11, мутации в котором приводят к крайне редкому (1 : 1 000 000 человек) аутосомному заболеванию – дефициту FXI, известному также как гемофилия С. Наиболее часто дефицит FXI диагностируется у евреев Ашкенази с тремя преобладающими мутациями. Целью данного исследования было первичное описание мутационного спектра гена F11 в российской популяции. В ходе исследования секвенировали все функционально важные области гена F11 для 11 неродственных пациентов с диагнозом гемофилия С. Всего было выявлено 10 различных генных нарушений, из них пять миссенс-мутаций, одна нонсенс-мутация, три frameshift-делециии одна inframe-делеция, все они достаточно равномерно распределялись по всему гену. В нашей выборке из распространенных в мире генетических нарушений встретилась лишь мутация II типа p.Glu135Ter, характерная для евреев Ашкенази. Два ранее не описанных варианта (c.1768del и p.His53Tyr) были охарактеризованы нами как вероятно патогенные. По лабораторным показателям наблюдалась картина, характерная для неполного доминирования, – значительное снижение уровня активности FXI и удлинение активированного частичного тромбопластинового времени (АЧТВ) в случае повреждения двух копий гена и небольшие отклонения от нормы при нарушении одной копии. Таким образом, мы описали нарушения, встречающиеся в гене F11 в российской популяции у пациентов с дефицитом FXI. Полученные данные свидетельствуют о высокой степени гетерогенности спектра мутаций, вызывающих гемофилию С в России.
FXI представляет собой зимоген протеазы FXIa, играющей важную роль в активации FIX в каскаде свертывания крови. Этот гликопротеин синтезируется в гепатоцитах и находится в плазме крови в комплексе с высокомолекулярным кининогеном. FXI представляет собой гомодимер из двух соединенных бисульфидным мостиком субъединиц, имеющих массу 80 кДа и включающих четыре apple-домена и один каталитический домен (сериновая протеаза). Полноразмерную субъединицу (625 аминокислотных остатков) кодирует ген F11, который локализован на длинном плече 4-й хромосомы (4q35.2), имеет общую длину 24 тпн и состоит из 15 экзонов. Первый экзон является некодирующим, второй экзон кодирует сигнальный пептид (17 аминокислотных остатков) [1–3].
Мутации в гене F11 приводят к крайне редкому аутосомному заболеванию с неполным доминированием – дефициту FXI, известному также как гемофилия С, дефицит плазменного предшественника тромбопластина, или синдром Розенталя (OMIM 612416). В настоящее время в базах данных (http://www.factorxi.org/, http://www.hgmd. cf.ac.uk/) описано 272 патогенных варианта в гене F11, из которых значительную часть составляют миссенс-мутации (154) [3].
В отличие от гемофилии А и В дефицит FXI встречается с примерно равной частотой у мужчин и женщин и характеризуется более мягким течением, кровотечения чаще всего развиваются в результате травм или хирургического вмешательства, спонтанные кровотечения редки даже при тяжелой форме болезни. Клиническая картина может быть различной и зависит от того, сколько копий гена повреждено. Тяжелая форма (FXI < 20%) развивается у гомозиготных носителей мутации или у компаундов (носителей двух разных патогенных вариантов на разных хромосомах). У гетерозиготных носителей одного генного дефекта болезнь носит более легкий характер, активность фактора при этом находится в пределах 20–70%. Предполагают дефицит FXI при продолжительных кровотечениях при увеличении активированного частичного тромбопластинового времени (АЧТВ) [4].
Принято оценивать частоту встречаемости этой болезни 1 : 1 000 000 человек, при этом недавние исследования, основанные на анализе большого массива данных полноэкзомного и полногеномного секвенирования, показали, что частота носительства мутаций в гене F11 в мировой популяции примерно в 13 раз выше, чем считалось ранее [5]. Наиболее часто дефицит FXI диагностируется у евреев Ашкенази (частота встречаемости гомозигот и компаундов 1 : 450 человек, гетерозиготного носительства 1 : 11 человек) с тремя преобладающими патогенными вариантами, обусловленными эффектом основателя: мутация II типа (p.Glu135Ter), III типа (p.Phe301Leu) и более редкая мутация I типа (c.1716 + 1G>A) [6–9]. Среди других мажорных вариантов, для которых также показан эффект основателя, наиболее известны p.Gln106Ter, p.Cys56Arg и p.Cys146Ter, преобладающие среди французов Нанта и Баскских земель и британцев соответственно. Также были описаны варианты с эффектом основателя в Южной Корее – p.Gln281Ter и p.Gln244Ter [10]. Анализ данных полноэкзомного секвенирования позволил выявить еще несколько мажорных, характерных для определенных этносов, вариантов: p.Phe241Leu у африканцев, p.Gln281Ter и p.(Leu442Cysfs*8) в Восточной Азии и p.Ala430Thr у латиноамериканцев [5]. Однако в большей части популяций мутационный спектр гена F11 значительно разнообразнее [10–17]. Целью данного исследования было первичное описание мутационного спектра гена F11 в российской популяции.
МАТЕРИАЛЫ И МЕТОДЫ
В исследование были включены 11 неродственных пациентов с первичным диагнозом дефицит FXI разной степени тяжести, а также шесть близких родственников одного из них после получения информированного согласия. ДНК выделяли из ядерных клеток периферической крови после селективного лизиса эритроцитов 0.8%-ным раствором хлорида аммония по стандартной методике, включающей обработку додецилсульфатом натрия (0.5%) и протеиназой К (200 мкг/мл) с последующей фенол-хлороформной экстракцией.
Для проведения молекулярно-генетического анализа все функционально важные участки гена F11 амплифицировали в виде девяти фрагментов длиной от 286 до 1047 пар нуклеотидов (пн). Исследуемые фрагменты включали кодирующие экзоны 2–15 и экзон-интронные сочленения. Амплификацию проводили в системе PCR Master Mix (ThermoScientific, США) с праймерами, разработанными на основании нуклеотидной последовательности NC_000004.12 (табл. 1) и синтезированными в ЗАО “Синтол” (Москва, Россия) с 0.01–0.02 мкг геномной ДНК в условиях 94°C – 1 мин, 62°C – 1 мин, 72°C – 3 мин, 30 циклов. Визуализацию продуктов ПЦР проводили с помощью регистрации в УФ-свете после разделения электрофорезом в 6%-ном полиакриламидном геле (ПААГ) и прокрашивания бромистым этидием.
Таблица 1.
Праймеры, использованные в работе
| Название | Нуклеотидная последовательность | Экзоны | Размер фрагмента, пн |
|---|---|---|---|
| F11D1 | ctactcccctctctccctat | Экзон 2 | 286 |
| F11R1 | acctcaattcctaccctgct | ||
| F11D2 | tagagctacttgccttgcct | Экзон 3 | 381 |
| F11R2 | tgcgtaaacaccatcacttct | ||
| F11D3 | tttggctttctgtgtgctgact | Экзон 4 | 322 |
| F11R3 | ggagggcaggcatgtatgta | ||
| F11D4 | agttgaaaggatgagtcagga | Экзон 5 | 294 |
| F11R4 | tctggcataaagttgatggca | ||
| F11D5 | catgcccagccattcagcct | Экзоны 6–7 | 824 |
| F11R5 | gcaacttttatcctatactgaca | ||
| F11D6 | catgagctgactttactttctc | Экзоны 8–10 | 722 |
| F11R6 | cctgaatgttctcccttctgt | ||
| F11D7 | gacagccttgtagtaccaca | Экзон 11 | 567 |
| F11R7 | tgtacctgcacctgttctga | ||
| F11D8 | gccacacacttcacaatgtc | Экзоны 12–13 | 989 |
| F11R8 | cttcagctacatgacttgaca | ||
| F11D9 | gtttatgtgtattgtgtatggt | Экзоны 14–15 | 1047 |
| F11R9 | agcgtgttactgtggaggaa |
ПЦР-фрагменты для секвенирования очищали на колонках Wizard (Promega, США). Секвенирование по Сэнгеру проводили в ЦКП “Геном” ИМБ РАН с помощью набора реактивов BigDye Terminator v3.1 (Applied Biosystems, США) с последующим анализом продуктов реакции на автоматическом секвенаторе ABI PRISM 3100Avant (Applied Biosystems).
Патогенные варианты выявляли путем сравнения нуклеотидной последовательности гена F11 пациента с референсными последовательностями ДНК и мРНК (NC_000004.12 и NM_000128.4 соответственно). Названия вариантам присваивали согласно рекомендациям HGVS (Human Genome Variation Society, http://varnomen.hgvs.org/). Выявленные варианты проверяли по базам данных (http://www.factorxi.org/, http://www.hgmd.cf.ac.uk/, https://www.ncbi.nlm.nih.gov/clinvar/), ранее не описанные варианты проверяли на патогенность с помощью калькулятора патогенности МГНЦ (http://calc.generesearch.ru/)и алгоритмов, предсказывающих патогенность: MutationTaster [18], PolyPhen-2 v2.2.2 [19], PROVEAN v1.1.5 [20], SIFT v6.2.1 [21], FATHMM (v2.3) [22].
Статистические расчеты проводили в программной среде R.
РЕЗУЛЬТАТЫ
У всех исследованных пациентов с первичным диагнозом дефицит FXI были выявлены патогенные варианты (табл. 2, рис. 1). Всего выявлено 10 различных генных нарушений, из них пять миссенс-мутаций, одна нонсенс-мутация, три делеции со сдвигом рамки считывания (frameshift) и одна inframe-делеция. Один найденный вариант (делеция c.1768del) ранее описан не был, он представляет собой делецию нуклеотида G, приводящую к сдвигу рамки считывания и образованию стоп-кодона в следующей позиции (p.(Val590Ter)). Согласно калькулятору патогенности это вероятно патогенный вариант (критерии PM2, PVS3). Еще один вариант – миссенс-мутация p.His53Tyr (c.157 C>T) не была описана в базах данных по мутациям, а также в базе ClinVar, однако была отмечена в базе gnomAD как встреченная однажды в гетерозиготной форме (1 : 251 458 аллелей, частота 0.000004) и зарегистрирована в базе dbSNP как rs55956266. Компьютерными алгоритмами она определяется как вероятно патогенная (MutationTaster: disease causing p = 0.89; Polyphen2: HumDiv/HumVar probably damaging p = 0.99; SIFT: Damaging, score = = 0.001; Provean: Deleterious, score = –2.57; fathmm: Damaging, score = –2.48).
Таблица 2.
Список выявленных нарушений в гене F11, их статус и локализация, а также лабораторные характеристики пациентов
| Пациент | Пол | FXI, % (норма 70–120) | АЧТВ, с (норма 29–38) | Патогенный вариант | Тип | Статус варианта | Экзон | Домен | Ссылка |
|---|---|---|---|---|---|---|---|---|---|
| C1-1 | Ж | 0.7 | 89.8 | p.Glu135Ter (c.403G>T) | Нонсенс | Гомозигота | 5 | Apple 2 | [6] |
| C2 | М | 62 | 39 | p.Glu135Ter (c.403G>T) | Нонсенс | Гетерозигота | 5 | Apple 2 | [6] |
| C3 | Ж | 1.1 | 165.6 | c.1768del (p.(Val590Ter)) | Делеция, frameshift | Гомозигота | 15 | Сериновая протеаза | New |
| C4 | М | 1.8 | 92.3 | c.961_962del (p.(Cys321Hisfs*37)) | Делеция, frameshift | Гомозигота | 9 | Apple 4 | [10] |
| C5 | М | 49.9 | 48.2 | c.73_86del (p.(Lys26Ter)) | Делеция, frameshift | Гетерозигота | 3 | Apple 1 | [11] |
| C6 | М | 44.3 | 47.5 | c.644_649del (p.(Ile215_Asp216del)) | Делеция, inframe | Гетерозигота | 7 | Apple 3 | [23] |
| C7 | Ж | 35.2 | 42.5 | p.Ala43Thr (c.127G>A) | Миссенс | Гетерозигота | 3 | Apple 1 | [24] |
| HM25 | Ж | 32.4 | 60.4 | p.Arg326His (c.977G>A) | Миссенс | Гетерозигота | 9 | Apple 4 | [15] |
| C8 | Ж | 1.3 | 86.8 | p.His53Tyr (c.157C>T) | Миссенс | Гетерозигота | 3 | Apple 1 | New |
| p.Thr60Pro (c.178A>C) | Миссенс | Гетерозигота | 3 | Apple 1 | [25] | ||||
| C9 | Ж | 37.9 | 39.4 | p.Arg443Cys (c.1327C>T) | Миссенс | Гетерозигота | 12 | Сериновая протеаза | [10] |
| C10 | Ж | 1.1 | 114.8 | c.1768del (p.(Val590Ter)) | Делеция, frameshift | Гомозигота | 15 | Сериновая протеаза | New |
Рис. 1.
Распределение выявленных патогенных вариантов в гене F11 и их соотношение с белковой структурой. В скобках указано число пациентов с данным вариантом, красным обозначены ранее не описанные патогенные варианты.
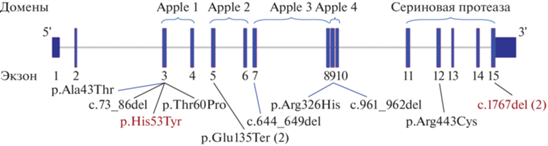
Только у двух пациентов выявлено наиболее распространенное генное нарушение – мутация II типа p.Glu135Ter, характерная для евреев Ашкенази. У четырех пациентов патогенный вариант был в гомозиготной форме, у остальных в гетерозиготной форме. У одного пациента было выявлено два патогенных варианта в гетерозиготной форме.
У пациентов с двумя поврежденными копиями гена F11 (гомозиготные варианты и компаунд из двух гетерозиготных вариантов) активность FXI была статистически значимо меньше (среднее: 1.2%, min–max: 0.7–1.8%, t-test: p = 0.0002, Cohen’s d = 1.377 (large)), а АЧТВ – больше (среднее: 109.9 с, min–max: 86.8–165.6 с, t-test: p = 0.0112, Cohen’s d = 2.62 (large)) по сравнению с пациентами, у которых была нарушена лишь одна копия гена (FXI среднее: 43.6%, min–max: 39–62%, АЧТВ среднее: 46.1 с, min–max: 39–60.4 с).
Для одной пациентки с гомозиготной мутацией II типа p.Glu135Ter был доступен биологический материал и лабораторные данные от родственников – матери, сестры и четырех детей. В связи с этим для них было определено соотношение фенотипа и мутационного статуса (табл. 3). Только сестра пробанда не являлась носительницей патогенного варианта, остальные члены семьи были гетерозиготными носителями. При этом у двух дочерей и сына пациентки наблюдалось характерное для гетерозиготных носителей снижение активности FXI и удлинение АЧТВ. Интересно, что у матери пробанда и показатели активности фактора, и АЧТВ были в рамках нормы, а у одной из дочерей при нормальной активности фактора АЧТВ увеличено.
Таблица 3.
Мутационный статус семьи пациентки с патогенным вариантом p.Glu135Ter (c.403G>T) и его соответствие показателям активности FXI и АЧТВ
| Пациент | Родство | FXI, % (норма 70–120%) | АЧТВ, с (норма 29–38 с) | Статус варианта |
|---|---|---|---|---|
| C1-1 | Пробанд | 0.7 | 89.8 | Гомозигота |
| C1-2 | Мать пробанда | 95 | 31.9 | Гетерозигота |
| C1-3 | Сестра пробанда | 147.1 | 30.1 | Норма |
| C1-4 | Дочь пробанда | 60.8 | 43 | Гетерозигота |
| C1-5 | Дочь пробанда | 49.1 | 40.2 | Гетерозигота |
| C1-6 | Сын пробанда | 53.8 | 40.1 | Гетерозигота |
| C1-7 | Дочь пробанда | 98.4 | 41.1 | Гетерозигота |
ОБСУЖДЕНИЕ
В данном исследовании мы выявили 10 различных патогенных вариантов гена F11 у российских пациентов с дефицитом FXI. Это первая работа, посвященная изучению мутационного спектра гена F11, в нашей стране. В нашей выборке из распространенных в мире генетических нарушений встретилась лишь мутация II типа p.Glu135Ter, характерная для евреев Ашкенази [6], у нас она была выявлена только у двух пациентов. По типам генетических нарушений в спектре преобладали миссенс-мутации (50%), что характерно для данного гена [3, 5, 10–17]. Два ранее не описанных варианта (c.1768del и p.His53Tyr) были охарактеризованы нами как вероятно патогенные.
Для некоторых наследственных заболеваний системы свертывания крови характерно наличие мажорных мутаций, которые позволяют разработать более быстрый и дешевый алгоритм молекулярно-генетической диагностики. В одном молекулярно-генетическом исследовании гемофилии С, проведенном в Великобритании на представительной выборке пациентов, было предложено начинать диагностику с трех ампликонов, включающих в себя экзоны 5, 15, 8–10 с наиболее часто встречающимися мутациями [11], тем не менее в нашей популяции такой алгоритм выглядит нецелесообразным. Все выявленные нами нарушения достаточно равномерно распределялись по всему гену F11 (рис. 1), затрагивая все белковые домены, и разнообразие встреченных вариантов было высоким. Единственным исключением может быть уникальная для нашей популяции микроделеция c.1767del, найденная дважды в гомозиготном состоянии, но ее реальная частота встречаемости может быть оценена только после существенного увеличения выборки обследованных пациентов. Этот вариант в обоих случаях был выявлен с полиморфизмом c.485+66T>C в гетерозиготном состоянии, что указывает на низкую вероятность того, что пробанды с данной микроделецией являются родственниками.
У пациентов наблюдалась картина, характерная для неполного доминирования заболевания, – значительные изменения лабораторных показателей коагулограммы (активность FXI и АЧТВ) в случае повреждения двух копий гена и небольшие отклонения от нормы при нарушении одной копии.
Таким образом, мы описали генетические нарушения, встречающиеся в гене F11 в российской популяции у пациентов с дефицитом FXI. Полученные данные свидетельствуют о высокой степени гетерогенности спектра мутаций, вызывающих гемофилию С в России.
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики.
От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Emsley J., McEwan P.A., Gailani D. Structure and function of factor XI // Blood. 2010. V. 115. № 13. P. 2569–2577. https://doi.org/10.1182/blood-2009-09-199182
Calderara D.B., Zermatten M.G., Aliotta A., Alberio L. How to capture the bleeding phenotype in FXI-deficient patients // Hamostaseologie. 2020. V. 40. № 4. P. 491–499. https://doi.org/10.1055/a-1227-8122
Harris V.A., Lin W., Perkins S.J. Analysis of 272 genetic variants in the upgraded interactive FXI web database reveals new insights on FXI deficiency // TH Open. 2021. https://doi.org/10.1055/a-1683-8605
Seligsohn U. Factor XI deficiency in humans // J. Thromb. Haemost. 2009. V. 7. Suppl. 1. P. 84–87. https://doi.org/10.1111/j.1538-7836.2009.03395
Asselta R., Paraboschi E.M., Rimoldi V. et al. Exploring the global landscape of genetic variation in coagulation factor XI deficiency // Blood. 2017. V. 130. № 4. P. e1–e6. https://doi.org/10.1182/blood-2017-04-780148
Asakai R., Chung D.W., Ratnoff O.D., Davie E.W. Factor XI (plasma thromboplastin antecedent) deficiency in Ashkenazi Jews is a bleeding disorder that can result from three types of point mutations // PNAS. 1989. V. 86. № 20. P. 7667–7671. https://doi.org/10.1073/pnas.86.20.7667
Peretz H., Mulai A., Usher S. et al. The two common mutations causing factor XI deficiency in Jews stem from distinct founders: one of ancient Middle Eastern origin and another of more recent European origin // Blood. 1997. V. 90. № 7. P. 2654–2659.
Peretz H., Salomon O., Mor-Cohen R. et al. Type I mutation in the F11 gene is a third ancestral mutation which causes factor XI deficiency in Ashkenazi Jews // J. Thromb. Haemost. 2013. V. 11. № 4. P. 724–730. https://doi.org/10.1111/jth.12137
Shpilberg O., Peretz H., Zivelin A. et al. One of the two common mutations causing factor XI deficiency in Ashkenazi Jews (type II) is also prevalent in Iraqi Jews, who represent the ancient gene pool of Jews // Blood. 1995. V. 85. № 2. P. 429–432.
Kim J., Song J., Lyu C.J. et al. Population-specific spectrum of the F11 mutations in Koreans: evidence for a founder effect // Clin. Genet. 2012. V. 82. № 2. P. 180–186. https://doi.org/10.1111/j.1399-0004.2011.01732.x
Mitchell M., Mountford R., Butler R. et al. Spectrum of factor XI (F11) mutations in the UK population–116 index cases and 140 mutations // Hum. Mutat. 2006. V. 27. № 8. P. 829. https://doi.org/10.1002/humu.9439
Zucker M., Zivelin A., Landau M. et al. Characterization of seven novel mutations causing factor XI deficiency // Haematologica. 2007. V. 92. № 10. P. 1375–1380. https://doi.org/10.3324/haematol.11526
Gueguen P., Chauvin A., Quemener-Redon S. et al. Revisiting the molecular epidemiology of factor XI deficiency: nine new mutations and an original large 4qTer deletion in western Brittany (France) // Thromb. Haemost. 2012. V. 107. № 1. P. 44–50. https://doi.org/10.1160/TH11-06-0415
Liu H., Wang H.F., Tang L. et al. Genetic analysis in Factor XI deficient patients from central China: Identification of one novel and seven recurrent mutations // Gene. 2015. V. 561. № 1. P. 101–106. https://doi.org/10.1016/j.gene.2015.02.021
Shao Y., Cao Y., Lu Y. et al. Clinical manifestations and mutation spectrum of 57 subjects with congenital factor XI deficiency in China // Blood Cells, Mol. Dis. 2016. V. 58. P. 29–34. https://doi.org/10.1016/j.bcmd.2016.01.004
Esteban J., de la Morena-Barrio M.E., Salloum-Asfar S. et al. High incidence of FXI deficiency in a Spanish town caused by 11 different mutations and the first duplication of F11: Results from the Yecla study // Haemophilia. 2017. V. 23. № 6. P. e488–e496. https://doi.org/10.1111/hae.13356
Colakoglu S., Bayhan T., Tavil B. et al. Molecular genetic analysis of the F11 gene in 14 Turkish patients with factor XI deficiency: identification of novel and recurrent mutations and their inheritance within families // Blood Transfus. 2018. V. 16. № 1. P. 105–113. https://doi.org/10.2450/2016.0098-16
Schwarz J.M., Cooper D.N., Schuelke M., Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age // Nat. Methods. 2014. V. 11. № 4. P. 361–362. https://doi.org/10.1038/nmeth.2890
Adzhubei I.A., Schmidt S., Peshkin L. et al. A method and server for predicting damaging missense mutations // Nat. Methods. 2010. V. 7. № 4. P. 248–249. https://doi.org/10.1038/nmeth0410-248
Choi Y., Sims G.E., Murphy S. et al. Predicting the functional effect of amino acid substitutions and indels // PLoS One. 2012. V. 7. № 10. P. e46688. https://doi.org/10.1371/journal.pone.0046688
Sim N.L., Kumar P., Hu J. et al. SIFT web server: Predicting effects of amino acid substitutions on proteins // Nucl. Acids Res. 2012. V. 40. P. W452–W457. https://doi.org/10.1093/nar/gks539
Shihab H.A., Gough J., Cooper D.N. et al. Predicting the functional consequences of cancer-associated amino acid substitutions // Bioinformatics. 2013. V. 29. № 12. P. 1504–1510. https://doi.org/10.1093/bioinformatics/btt182
Zadra G., Asselta R., Malcovati M. et al. Molecular genetic analysis of severe coagulation factor XI deficiency in six Italian patients // Haematologica. 2004. V. 89. № 11. P. 1332–1340.
Dai L., Rangarajan S., Mitchell M. Three dominant-negative mutations in factor XI-deficient patients // Haemophilia. 2011. V. 17. № 5. P. e919–e922. https://doi.org/10.1111/j.1365-2516.2011.02519.x
Mulder R., Wiewel-Verschueren S., Meijer K., Mulder A.B. Identification of a novel factor XI gene mutational event in a Dutch Caucasian family with inherited factor XI deficiency // Thromb. Haemost. 2013. V. 109. № 6. P. 1183–1185. https://doi.org/10.1160/TH12-12-0933
Дополнительные материалы отсутствуют.