

Геохимия, 2021, T. 66, № 11, стр. 1067-1074
О некоторых базовых положениях общей теории эволюции вселенной на первых этапах зарождения жизни
Л. А. Грибов a, *, В. И. Баранов a, И. В. Михайлов a
a Институт геохимии и аналитической химии им. В.И. Вернадского РАН
119991 Москва, ул. Косыгина, 19, Россия
* E-mail: l_gribov@mail.ru
Поступила в редакцию 04.02.2021
После доработки 21.03.2021
Принята к публикации 29.03.2021
Аннотация
Рассмотрены некоторые общие положения физической теории строения и свойств молекул и химических превращений, которые приводят к выводам, согласующимся с наблюдаемыми особенностями процессов эволюции молекулярного мира. Показано, что высокая плотность энергетических уровней, достижимая только в больших и сложных молекулярных объектах, резко увеличивает вероятность реакций и структурных перестроек и приводит к заметному снижению энергии активации. Приведен пример построения математической модели многоступенчатого процесса, включающего в себя внутримолекулярные переходы, сопровождающиеся поглощением или излучением электромагнитных волн, и реакционные превращения – мономолекулярные (структурная изомеризация), синтез и разложение.
ВВЕДЕНИЕ
Настоящая работа выполнена в рамках широкого круга исследований, направленных на объяснение и описание эволюционных процессов и их свойств (Галимов, 2008а, 2013), и является прямым продолжением опубликованных ранее в журнале Геохимия статей (Грибов, 2007; Грибов, Михайлов, 2010; Грибов и др., 2012; Баранов и др., 2013; Грибов, Баранов, 2014; Баранов и др., 2016; Баранов и др., 2016а; Грибов и др., 2018; Baranov, Gribov, 2014). Следует особо отметить, что существенной причиной возросшего интереса к изучению различных аспектов возможности появления и существования наблюдаемых в течение миллиардов лет устойчивых процессов превращения части материи во все более высокоорганизованные формы стало появление концепции устойчивого упорядочения академика РАН Эрика Михайловича Галимова (Галимов, 2001, 2008; Galimov, 2004).
В наших прежних работах показано, что описание важнейших экспериментальных фактов эволюционного развития Вселенной на первых стадиях образования молекулярного мира возможно с помощью математических моделей, в основе которых лежит понятие о причинно-следственных связях, явным образом прослеживающихся в процессах эволюции. Важнейшей особенностью таких моделей, представимых в виде цепочек импликаций разной сложности с количественными характеристиками в виде вероятностей превращений, является их опора на фундаментальный физический закон – второе начало термодинамики. То обстоятельство, что отправляясь от общего положения физики – монотонного возрастания энтропии, – удается прийти к тем же закономерностям, что и наблюдаемые особенности эволюционных процессов окружающего мира, свидетельствует не только о корректности предложенного подхода, но и о том, что законы эволюции не являются случайными, а вполне согласуются с базовыми физическими утверждениями.
Еще одним важнейшим результатом наших исследований является ответ на принципиальный вопрос о вероятностях образования тех или иных молекулярных объектов из сгустка атомов, локализованных в малой области пространства (Баранов и др., 2013). Проблема заключается в том, что молекулы имеют вполне определенные структуры, и такое расположение атомов, в результате которого могут сразу образоваться, например, плоские молекулы бензола, представляется очень маловероятным. Вопрос снимается, если учесть, что уже сравнительно малые молекулы (а в открытом космосе сейчас обнаружены и весьма крупные (Tielens, 2013; Endres et al., 2016; Mcguire et al., 2018)) имеют большое число структурных изомеров. Появление любого такого изомера уже не случайно, а закономерно, и может привести в результате мономолекулярной реакции к появлению структур, сильно отличающихся от начальной. Также было показано, что на ранних стадиях эволюции молекулярного мира образование ациклических (протяженных) структур, является более вероятным, чем циклических (свернутых, объемных) (Баранов и др., 2016).
В настоящей работе рассмотрен важнейший вопрос о причинах, приводящих к малым энергиям активации многих биохимических реакций, и показано, что некоторые разработанные в физической теории строения и свойств молекул положения могут быть с успехом применены для заполнения промежутков в логике общей эволюционной теории развития Вселенной.
НЕКОТОРЫЕ ОБЩИЕ ПОЛОЖЕНИЯ ФИЗИЧЕСКОЙ ТЕОРИИ СТРОЕНИЯ И СВОЙСТВ МОЛЕКУЛ
Все процессы в микромире подчиняются законам не классической, а квантовой теории. Последняя базируется на представлении о дискретных уровнях энергии. В свою очередь, такие дискретные энергетические состояния возникают тогда, когда на понимаемое в широком смысле движение частиц накладываются определенные ограничения. В теории вводится понятие потенциальной ямы и считается, что устойчивый молекулярный объект находится внутри ямы. Принципиально важно, что количество уровней (возможных состояний, в которых может находиться система) в яме зависит от ее глубины, ширины и размерности (числа степеней свободы).
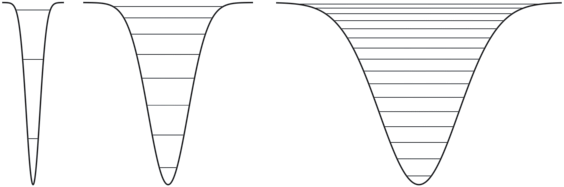
Увеличение плотности уровней в ямах одинаковой глубины, имеющих разную ширину, иллюстрирует рис. 1. Все расчеты, результаты которых приводятся здесь и далее, выполнены в среде MATLAB (https://www.mathworks.com), в том числе с помощью пакета программ Chebfun (http:// www.chebfun.org).
Рис. 1.
Иллюстрация возрастания плотности уровней при увеличении ширины потенциальных ям: показаны ямы, имеющие одинаковую глубину, но разную ширину, и соответствующие возможные энергетические состояния.
Число уровней (состояний системы) увеличивается также с ростом числа степеней свободы молекулярного объекта, т.е. при его усложнении. На рис. 2 показано как меняется плотность уровней гармонического осциллятора в одном и том же энергетическом промежутке при увеличении размерности осциллятора.
Рис. 2.
Иллюстрация возрастания плотности уровней при увеличении размерности: показаны энергетические уровни гармонического осциллятора, имеющего одну (1), две (2) и шесть (3) степеней свободы.

Глубина ямы для молекул определяется энергией электронно-ядерных взаимодействий, которая возрастает при увеличении числа атомов. Получается, что чем сложнее молекулярный объект, тем более плотно (на меньших расстояниях друг от друга) расположены энергетические уровни в ямах. Это очень важный фактор, определяющий, как увидим ниже, увеличение вероятности превращений (химических реакций) с участием крупных молекул по сравнению с малыми.
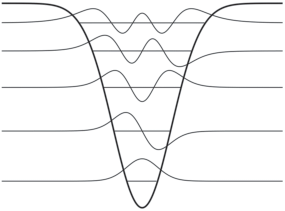
Каждому уровню энергии (состоянию системы) отвечает своя волновая функция. Квадрат волновой функции определяет вероятность нахождения частицы в единичном объеме в окрестности некоторой точки пространства, и его величина быстро падает до нуля за пределами ямы, что хорошо видно на рис. 3.
Рис. 3.
Энергетические уровни и волновые функции для (гауссовой) потенциальной ямы. Нулевые значения волновых функций совмещены с энергиями соответствующих состояний.
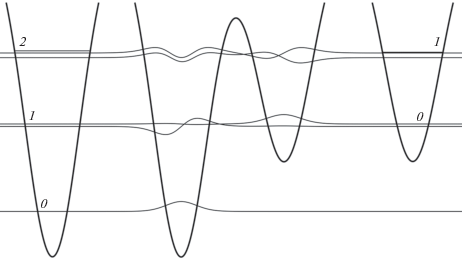
В результате химической реакции одни устойчивые объекты (исходные молекулы) превращаются в другие, тоже устойчивые. В теории такой процесс в простейшем случае мономолекулярной реакции описывается как переход из одной потенциальной ямы в другую. В том случае, когда ямы расположены достаточно близко друг к другу, могут возникнуть уровни с волновыми функциями, отличными от нуля в пределах не одной ямы, а сразу двух. Такая ситуация показана на рис. 4. В центре рисунка изображена двухминимумная потенциальная яма, образующаяся при сближении показанных по краям потенциалов, имеющих две пары состояний с очень близкими (но не равными) энергиями. Первую пару составляют состояние 1 левой ямы и состояние 0 правой ямы, вторую – состояние 2 левой ямы и состояние 1 правой ямы. Хорошо видно, что, если для первой пары состояний каждая их двух волновых функций локализована только в пределах своей ямы, то для второй пары состояний, энергии которых значительно ближе к барьеру, наблюдается размазывание обеих волновых функций по всей области двухминимумного потенциала. Возникающий дублет (резонансный уровень) определяет возможность химического (структурного) превращения – перехода из одной ямы в другую.
Рис. 4.
Волновые функции для двухминимумной потенциальной ямы (в центре). Нулевые значения волновых функций совмещены с энергиями соответствующих состояний. Для сравнения по краям показаны те же потенциальные ямы, находящиеся на большом расстоянии друг от друга и уровни энергии в них. Состояния 1 левой ямы и 0 правой, а также 2 левой ямы и 1 правой образуют пары с близкими энергиями.
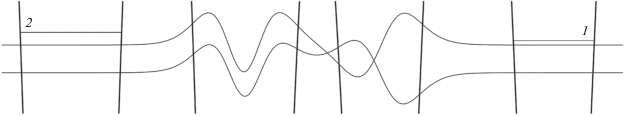
На рис. 4 волновые функции, нулевые значения которых совмещены с энергиями соответствующих состояний, намеренно изображены не только внутри ям, но и далеко за их пределами. Это с одной стороны сделано для иллюстрации близости энергий состояний в упоминавшихся выше парах уровней правой и левой ям, а с другой является попыткой показать уменьшение энергии уровней в двухминимумном потенциале по сравнению с отдельными ямами. Такое уменьшение особенно выражено для второй пары состояний и хорошо видно на рис. 5, который является увеличенным участком рис. 4.
Рис. 5.
Участок рисунка 4, отвечающий области энергий состояний второй пары (уровень 2 левой ямы и уровень 1 правой), волновые функции которых отличны от нуля в обеих частях двухминимумного потенциала (в центре). Справа и слева для сравнения показано положение уровней энергии в левой и правой яме по отдельности.
Характеристики потенциалов, показанных на рис. 4 и 5, специально подобраны таким образом, чтобы появлялись пары уровней имеющих очень близкие энергии. Очевидно, что в ямах с небольшим числом уровней достаточная для возникновения резонанса близость энергий состояний является маловероятной. Именно поэтому такую важную роль играет большая плотность уровней в сложных молекулах, которая резко повышает вероятность резонанса. Одновременно уменьшаются и энергия уровня, и энергия активации реакции. Отсюда следует, что для начала того, что принято называть жизнью, эволюция молекулярного мира должна от стадии простых малых молекул перейти в стадию крупных.
В общей физической теории химических реакций показано, что в полном соответствии с экспериментом реакции с участием крупных молекул возможны только тогда, когда имеются так называемые реакционные центры (Баранов и др., 2013; Грибов, Баранов, 2012а; Баранов и др., 2016а). Это малые участки сложных молекулярных объектов, где и происходят перестановки атомов, приводящие к превращению исходных структур в продукты. Реакционные центры с большей вероятностью образуются на концах протяженных (ациклических), т.е. вытянутых в одном направлении молекул (участков молекул), чем в объемных (циклических). А, как уже говорилось во Введении, согласно теории ациклические молекулы образуются на ранних стадиях с большей вероятностью, чем циклические (объемные) (Баранов и др., 2016).
Все биохимические реакции происходят в жидкой конденсированной среде. Для протекания реакции требуется, чтобы, во-первых, до реакции реагирующие частицы были подвижными и в результате хаотического движения они могли встретиться друг с другом, а во-вторых, число реагирующих частиц в объеме было достаточно велико.
На основании всего вышесказанного можно сделать следующие выводы. Во-первых, реакции происходят в ограниченной области пространства в конденсированной среде (жидкости). Во-вторых, в реакциях должны участвовать крупные молекулы протяженной структуры. В-третьих, реакционные центры в таких молекулах должны быть расположены на концах структур. Все это точно соответствует эксперименту. Можно, поэтому, заключить, что для объяснения особенностей биохимических реакций не надо вводить какие-то новые химические силы.
МОДЕЛИРОВАНИЕ МНОГОСТУПЕНЧАТЫХ РЕАКЦИЙ
В предыдущем разделе показано, что общие положения физической теории химических реакций приводят к заключениям общего характера, согласующимся с наблюдаемыми закономерностями эволюции молекулярного мира. Рассмотрим теперь, как современное состояние теории позволяет изучать особенности развития процессов (в том числе и геохимических) любой сложности во времени на примере построения системы кинетических уравнений для описания многоступенчатого превращения, схема которого показана на рис. 6.
Рис. 6.
Схема многоступенчатого процесса, включающего в себя внутримолекулярные переходы, структурные перестройки и химические реакции. Цифрами 1–11 обозначены номера уровней, которые соответствуют последовательности превращений и могут относиться к разным молекулярным объектам. Пунктирными линиями показано расширение схемы превращений для моделирования реакции третьего порядка.
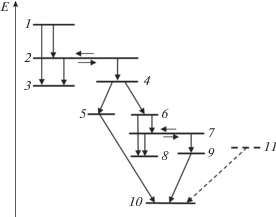
Будем считать, что в результате внешнего воздействия на ансамбль исходных молекул (состояния 1, 2 и 3) заселяется возбужденное состояние 1. Величину этой заселенности удобно принять равной единице. Так как уровень 2 резонансный (общий для двух ям), то в процессе релаксации становятся возможными переходы на уровень 4 второй молекулы – структурного изомера исходной. Этот изомер является неустойчивым (переходная структура) и распадается на две части – одной соответствует состояние 5, другой – состояния 6, 7 и 8. Для последней снова возможен изомерный переход, так как уровень 7 резонансный. Уровни 8 и 9 являются основными состояниями структурных изомеров, имеющих меньшее число атомов, чем первая (исходная) и вторая (неустойчивая) молекулы. Появляющиеся в ходе многоступенчатого процесса структуры с уровнями 5 и 9 способны вступать в реакцию присоединения с образованием молекулы 10 с тем же числом атомов, что и исходная, т.е. структура 10 является изомером первой молекулы (описываемой уровнями 1, 2 и 3). Таким образом, последовательность превращений, показанная сплошными линиями, включает в себя внутримолекулярные переходы между уровнями энергии, две реакции структурной изомеризации (два резонансных уровня), одну реакцию разложения и одну реакцию присоединения. Конечными состояниями являются состояния 3, 8, 10 и одно из состояний 5 или 9 (в зависимости от соотношения вероятностей превращений).
В системе дифференциальных уравнений для описания развития многоступенчатого процесса во времени должны быть учтены все четыре перечисленных выше типа превращений.
Реакция разложения исходного состояния 4 (Ван-дер-Ваальсова комплекса, способного к распаду с вероятностью α), в результате которой появляются компоненты 5 и 6, описывается системой трех уравнений
Вероятность реакции присоединения β определяется вероятностью одновременного нахождения структур 5 и 9 в одном и том же малом объеме V. Последняя равна произведению вероятностей нахождения объеме V частиц 5 и 9 по отдельности: n5n9/V 2. Тогда процесс появления новой молекулы 10 можно охарактеризовать также системой трех уравнений
Для внутримолекулярного перехода между двумя нерезонансными уровнями, например, 1 и 3 с заселенностями n1 и n3, характеризующегося вероятностью p13, изменение заселенностей состояний записывается в виде
Для внутримолекулярных переходов между двумя уровнями, один из которых резонансный, например, 1 → 2, 2 → 3 или 2 → 4 следует учитывать, что резонансный уровень является не просто общим для двух ям (в многоминимумных потенциалах все уровни энергии общие), но и имеет волновую функцию, локализованную (отличную от нуля) в области обеих ям (пример такой функции показан на рис. 5 предыдущего раздела). В результате получается дублет с близкими энергиями, функции которого имеют вид, с хорошей степенью точности отвечающий выражению (φi ± φj)/√2, где φi и φj − участки функций, локализованные в левой и правой ямах. При такой форме волновых функций переход из состояния 1 с функцией, локализованной в пространстве только левой ямы, на уровень дублета (состояние 2) создает условия дальнейшего развития процесса в двух направлениях – на нижележащие уровни 3 и 4 с функциями, локализованными только в левой и правой ямах соответственно. Переход 2 → 4 отвечает появлению структурной формы (в нашем случае неустойчивой), являющейся продуктом химической реакции, необходимым условием которой можно считать существование состояния с “размазанной” волновой функцией.
Расстояние между уровнями энергии дублета мало, и вместо дублета можно ввести один дважды вырожденный уровень с энергией E = (Ei + Ej)/2. Для квадрата волновой функции резонансного уровня можно записать
и говорить о безызлучательном переходе из левой ямы в правую (Грибов, Баранов, 2006; Грибов и др., 2019).Это означает, что вероятности переходов на уровни дублета (1 → 2) и с них (2 → 3 и 2 → 4) будут зависеть от величины частоты квантовых биений ω и определяться квадратами матричных элементов дипольного момента, умноженными на осциллирующие функции cos2ωt или sin2ωt. Выбор конкретного множителя определяется начальными условиями, т.е. направлением химического превращения (Грибов, Баранов, 2006). Для переходов 1 → 2 и 2 → 3 между уровнями энергии исходного вещества (донора) вероятность перехода умножается на cos2ωt, а для перехода 2 → 4 между уровнями продукта реакции (акцептора) – на sin2ωt:
Все вышесказанное позволяет записать систему дифференциальных уравнений первого порядка, описывающую сложный многоступенчатый процесс превращений, в виде
Важно отметить, в общей физической теории химических реакций разработаны методы и алгоритмы расчета вероятностей переходов (pij) и частот квантовых биений (ω2 и ω7), являющихся параметрами системы (Грибов и др., 2019). Оценки величин других параметров (вероятности α и β) также могут быть получены средствами теории.
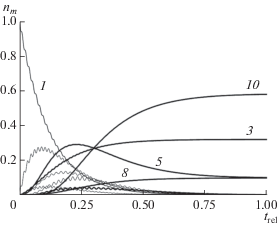
Результат решения системы уравнений будет зависеть от начальных условий. Как уже говорилось выше, в нашем случае естественно принять n1(0) = 1, n2,…,10(0) = 0. Полученные временные зависимости заселенностей уровней энергии молекул показаны на рис. 7. Видно, что сохранится определенное число исходных молекул (состояние 3), появятся ее изомерные формы (состояние 10) и одинаковое число еще двух меньших по размеру молекул (состояния 5 и 8). Использован некоторый временной интервал от 0 до tmax, а все результаты представлены для безразмерных величин trel = t/tmax.
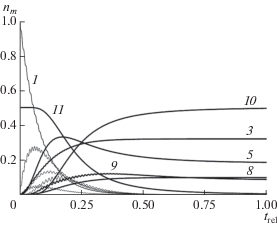
Схему переходов между уровнями энергии, показанную на рис. 6 сплошными линиями, можно распространить на случай, когда изначально присутствуют два типа молекул, причем молекулы второго типа реагируют с продуктами превращений первого соединения. Тогда могут появиться реакции третьего порядка. Дополнительные уровни и превращения для такого процесса показаны на рис. 6 штриховыми линиями. Соответствующая система уравнений по сравнению с записанной выше изменится незначительно – появится одно новое уравнение dn11/dt = −β n5n9n11, а в правых частях уравнений для заселенностей n5, n9 и n10 слагаемое β n5n9 следует заменить на β n5n9n11. Результат решения расширенной системы с начальными условиями n1(0) = 1, n2,…,10(0) = 0 и n11(0) = 0.5 показан на рис. 8.
ЗАКЛЮЧЕНИЕ
Показано, что общие положения физической теории строения и свойств молекул и химических превращений приводят к выводам, которые согласуются с наблюдаемыми особенностями процессов эволюции молекулярного мира. Высокая плотность энергетических уровней, достижимая только в больших и сложных молекулярных объектах, резко увеличивает вероятность реакций и структурных перестроек и приводит к заметному снижению энергии активации. Отсюда следует, что то, что принято называть жизнью, не могло возникнуть на ранних стадиях эволюции молекулярного мира.
Кроме размера и сложности молекулярных объектов для многих биохимических реакций наблюдаются и другие характерные особенности. Во-первых, такие реакции происходят в ограниченной области пространства в конденсированной среде. Во-вторых, молекулы, участвующие в таких реакциях, обычно имеют в своей структуре протяженные участки. В-третьих, реакционные центры в таких молекулах обычно расположены на концах протяженных участков. Все это хорошо согласуется с выводами теории.
Приведен пример моделирования сложной цепочки превращений, в которой происходят как процессы внутримолекулярные (они приводят к поглощению или излучению электромагнитных волн), так и реакционные – мономолекулярные (изомеризация), синтеза и разложения. Показанная возможность построения математической модели сложного процесса, с помощью которой можно описывать, в том числе и процессы эволюции молекулярного мира на ранних стадиях развития, принципиально важна. Целью любой естественной науки является не только понимание, но, и это главное, прогноз, который невозможен без использования моделей достаточно хорошо описывающих явления в природе. Давно известно, что любая отрасль науки достигает совершенства тогда, когда она начинает пользоваться математикой. Геохимия исключением не является, и перспектива ее развития определяется не только накоплением экспериментальных фактов, но и переходом к построению математических моделей.
Список литературы
Баранов В.И., Грибов Л.А., Эляшберг М.Е. (2013) Структурная изомеризация и эволюция молекулярного мира на ранних стадиях образования Вселенной. Геохимия (3), 256-261.
Baranov V.I., Gribov L.A., Elyashberg M.E. (2013) Structural isomerization and the evolution of the molecular world during the early universe evolution. Geochem. Int. 51(3), 231-236.
Баранов В.И., Грибов Л.А., Михайлов И.В. (2016) Исследование процессов развития молекулярного мира на ранних стадиях его образования на основе изучения поведения энтропии колебаний молекул. Геохимия (11), 963-969.
Baranov V.I., Gribov L.A., Mikhailov I.V. (2016) Processes of the development of the molecular world at early stages of its formation: Evidence from the investigation of the behavior of the vibrational entropy of molecules. Geochem. Int. 54(11), 929-935.
Баранов В.И., Грибов Л.А., Дементьев В.А., Михайлов И.В. (2016а) Некоторые общие закономерности формирования сложных молекулярных объектов на ранних стадиях образования биосферы как следствие физических свойств конденсированных сред. Геохимия (11), 1046-1054.
Baranov V.I., Gribov L.A., Dement’ev V.A., Mikhailov I.V. (2016a) Some general relations of the development of complex molecular objects at early stages of biosphere formation as a consequence of the physical properties of condensed medium. Geochem. Int. 54(11), 1011-1018.
Галимов Э.М. (2001) Феномен жизни: между равновесием и нелинейностью. Происхождение и принципы эволюции. М.: URSS, 256 с.
Галимов Э.М. (2008) Концепция устойчивого упорядочения и АТФ-зависимый механизм происхождения жизни. “Проблемы зарождения и эволюции биосферы” (Под ред. Галимова Э.М.). М.: Книжный дом “ЛИБРОКОМ”, 23-31.
Галимов Э.М. (2008а) Сб. “Проблемы зарождения и эволюции биосферы” (Под ред. Галимова Э.М.). М.: Книжный дом “ЛИБРОКОМ”, 552 с.
Галимов Э.М. (2013) Сб. “Проблемы зарождения и эволюции биосферы: Допланетная стадия развития Солнечной системы. Реконструкция химических и геологических условий на ранней Земле. Теоретические и экспериментальные исследования предбиологических химических систем. События и факторы” (Под ред. Галимова Э.М.). М.: КРАСАНД, 640 с.
Грибов Л.А., Баранов В.И. (2006) Теория и методы расчета молекулярных процессов: спектры, химические превращения и молекулярная логика. М.: КомКнига, 480 с.
Грибов Л.А. (2007) О некоторых закономерностях формирования органического вещества на ранних стадиях геохимической эволюции. Геохимия (1), 89-93.
Gribov L.A. (2007) Some principles in the formation of organic matter during the early stages of chemical evolution. Geochem. Int. 45(1), 80-83.
Грибов Л.А., Михайлов И.В. (2010) Стрела эволюции в процессе формирования биосферы. Геохимия (6), 646-650.
Gribov L.A., Mikhailov I.V. (2010) Evolution arrow in the origin of the biosphere. Geochem. Int. 48(6), 606-610.
Грибов Л.А., Баранов В.И., Михайлов И.В. (2012) Стрела времени на ранних стадиях эволюции биосферы. Детерминизм и множественность. Геохимия (5), 435-452.
Gribov L.A., Baranov V.I., Mikhailov I.V. (2012) The arrow of time at the early stages of biosphere evolution. Determinism and pluralism. Geochem. Int. 50(5), 393-408.
Грибов Л.А., Баранов В.И. (2012а) От молекул к жизни. М.: КРАСАНД, 208 с.
Грибов Л.А., Баранов В.И. (2014) Химическое пространство и пространство биосферы. Геохимия (9), 854-858.
Gribov L.A., Baranov V.I. (2014) Chemical space and biospheric space. Geochem. Int. 52(9), 783-787.
Грибов Л.А., Баранов В.И., Михайлов И.В. (2018) Закономерности эволюции биосферы и энтропия. Геохимия (9), 837-847.
Gribov L.A., Baranov V.I., Mikhailov I.V. (2018) Evolution of the Biosphere and Entropy. Geochem. Int. 56(9), 871-880.
Грибов Л.А., Баранов В.И., Михайлов И.В. (2019) Физическая теория химических реакций. М.: Русайнс, 368 с.
Baranov V.I., Gribov L.A. (2014) Early Stage of the Evolution of the Universe: Molecular Medium and the Emergence of Properties of Functioning of Living Systems. Geochem. Int. 52(13), 1103-1145.
Galimov E.M. (2004) Phenomenon of life: between equilibrium and non-linearity. Origin Life Evol. Biosph. 6, 599-613.
Endres C., Schlemmer S., Schilke P., Stutzki J., Müller H. (2016) The Cologne Database for Molecular Spectroscopy, CDMS, in the Virtual Atomic and Molecular Data Centre, VAMDC. J. Mol. Spectrosc. 327, 95-104.
Mcguire B., Burkhardt A., Kalenskii S., Shingledecker C., Remijan A., Herbst E., McCarthy M. (2018) Detection of the aromatic molecule benzonitrile (c-C6H5CN) in the interstellar medium. Science 359(6372). 202-205.
Tielens A.G.G.M. (2013) The molecular universe. Rev. Mod. Phys. 85, 1021-1081.
Дополнительные материалы отсутствуют.