

Геохимия, 2022, T. 67, № 1, стр. 69-83
Причины образования кислых дренажных вод в отвалах сульфидсодержащих пород
В. А. Алексеев *
Институт геохимии и аналитической химии им. В.И. Вернадского РАН
119991 Москва, ул. Косыгина, 19, Россия
* E-mail: alekseyev-v@geokhi.ru
Поступила в редакцию 23.09.2020
После доработки 17.03.2021
Принята к публикации 22.03.2021
- EDN: FVJWUJ
- DOI: 10.31857/S0016752522010022
Аннотация
Выполнен критический анализ экспериментальных и расчетных исследований по актуальной экологической проблеме образования кислых дренажных вод в отвалах сульфидсодержащих пород. Упор сделан на оценку причин изменения скорости окисления пирита (r), самого распространенного сульфида. Зависимости r от температуры, рН, содержаний О2 и Fe3+ в растворе оказались чуть ли не единственными, выраженными в виде уравнений. Медленная стадия окисления Fe2+ в кислых растворах не может контролировать r в природных условиях, т.к. она ускоряется железоокисляющими бактериями. Величина r изменяется также в зависимости от вида и содержания изоморфных примесей в пирите, уменьшается под влиянием некоторых лигандов, других сульфидов (гальванический эффект), а также с течением времени. Влияние времени обычно объясняется образованием поверхностного протектирующего слоя продуктов реакции, но может иметь и другое объяснение, а именно, растворение (исчезновение) поверхностных дефектов. В зависимости от содержания и активности минералов, производящих и нейтрализующих кислоту, выветривание отвалов может протекать с образованием кислых или близнейтральных растворов. Тот или иной сценарий прогнозируется с помощью статических и кинетических тестов, которые имеют существенные недостатки, связанные с различиями лабораторных и полевых условий. Привлечение для этой цели математического моделирования перспективно, но пока ограничено вследствие упрощения моделей и погрешностей расчетов. Тем не менее, моделирование убедительно показало, что величина r в отвалах определяется скоростью доставки О2, которая в свою очередь зависит от размера пор, степени заполнения пор водой, градиентов температуры и давления. Для предотвращения и рекультивации кислых дренажных вод используют материалы, которые изолируют отдельные зерна сульфидов (микроинкапсуляция) или весь отвал от проникноверия О2, щелочные материалы, нейтрализующие кислоту, бактерициды, понижающие активность железоокисляющих бактерий, а также биореакторы, где под действием сульфат-редуцирующих бактерий происходит осаждение сульфидов металлов.
ВВЕДЕНИЕ
При добыче полезных ископаемых вблизи шахт и карьеров образуются отвалы вскрышных пород, содержащие сульфиды. Выветривание сульфидных минералов с участием кислорода и воды приводит к образованию кислых вод, обогащенных нормируемыми элементами (Замана, Чечель, 2014; Рыбникова, Рыбников, 2019; Bowell et al., 1999; Evangelou, Zhang, 1995; Nordstrom et al., 2015). Этот процесс называется кислым шахтным дренажом (КШД) или кислым дренажом породы (КДП). В зарубежной литературе аналогами являются термины acid mine drainage (AMD) или acid rock drainage (ARD). Разгрузка кислых вод в водоносные горизонты, реки и озера создает угрозу здоровью людей, животных, растений и может приводить к нарушению экологического баланса. Выполнены экспериментальные исследования механизмов и скоростей окислительного растворения (далее просто окисления) сульфидов, в первую очередь наиболее распространенного пирита (Chandra, Gerson, 2010; Wang et al., 2019; Williamson, Rimstidt, 1994). В результате обнаружены зависимости и параметры, влияющие на скорость окисления пирита (r). Некоторые из этих зависимостей, выраженные в виде уравнений, используются при математическом моделировании процесса окисления сульфидов в отвалах и хвостах (Gerke et al., 1998; Molson et al., 2005; Romano et al., 2003; Wunderly et al., 1996). При этом возникают дополнительные зависимости и параметры, влияющие на r. Целью настоящей работы является критический анализ экспериментальных и расчетных исследований проблемы образования КДП. Акцент сделан на влияющие процессы, их эффективность, механизмы и параметры, а также на нерешенные вопросы, связанные с достоверностью прогнозов. В конечном итоге хотелось бы прояснить вопрос: какие существуют узкие места, которые мешают расчетным методам адекватно предсказывать эволюцию КДП? Важной частью исследований КДП является также разработка способов уменьшения скоростей окисления сульфидов в отвалах (Naidu et al., 2019; Pozo-Antonio et al., 2014; Skousen et al., 2019). Большое число существующих публикаций по КДП трудно совместить с ограниченным объемом журнальной статьи. Поэтому каждый параметр или фактор, влияющий на r, иллюстрирован здесь ограниченным числом ссылок, которые, тем не менее, показывают характерную степень влияния.
КИНЕТИКА И МЕХАНИЗМЫ ОКИСЛЕНИЯ ПИРИТА
Наиболее распространенным сульфидным минералом в земной коре (и, соответственно, в шахтных отвалах) является пирит (FeS2), поэтому основное внимание при исследовании проблемы КДП уделяется этому минералу. В присутствии кислорода (О2) пирит растворяется в воде по реакциям (Chandra, Gerson, 2010; Evangelou, Zhang, 1995; Singer, Stumm, 1970):
(1)
${\text{Fe}}{{{\text{S}}}_{{\text{2}}}} + 3.5{{{\text{O}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {\text{F}}{{{\text{e}}}^{{2 + }}} + 2{\text{SO}}_{4}^{{2 - }} + 2{{{\text{H}}}^{ + }},$(2)
${\text{F}}{{{\text{e}}}^{{2 + }}} + 0.25{{{\text{O}}}_{2}} + {{{\text{H}}}^{ + }} \to {\text{F}}{{{\text{e}}}^{{3 + }}} + 0.5{{{\text{H}}}_{{\text{2}}}}{\text{O,}}$(3)
${\text{F}}{{{\text{e}}}^{{3 + }}} + 3{{{\text{H}}}_{{\text{2}}}}{\text{O}} = {\text{Fe}}{{\left( {{\text{OH}}} \right)}_{{\text{3}}}} + 3{{{\text{H}}}^{ + }}.$По реакции (1) пиритная сера S(-I) окисляется до сульфата, а железо переходит в раствор, не меняя валентность. Растворенное железо (Fe2+) окисляется кислородом по реакции (2) до Fe3+, который выпадает в осадок в виде гидроксида Fe(OH)3 по реакции (3). В кислых растворах, где растворимость Fe(OH)3 довольно высока, в растворе по реакции (2) накапливается трехвалентное железо Fe3+, которое становится основным окислителем пирита, т.е. вместо реакции (1) протекает реакция (4):
(4)
$\begin{gathered} {\text{Fe}}{{{\text{S}}}_{{\text{2}}}} + 14{\text{F}}{{{\text{e}}}^{{3 + }}} + 8{{{\text{H}}}_{{\text{2}}}}{\text{O}} \to \\ \to 15{\text{F}}{{{\text{e}}}^{{2 + }}} + 2{\text{SO}}_{4}^{{2 - }} + 16{{{\text{H}}}^{ + }}. \\ \end{gathered} $Реакции (2) и (4) описывают цикл, в котором Fe2+ сначала окисляется до Fe3+ кислородом, а затем Fe3+ восстанавливается до Fe2+ пиритом. Этот цикл повторяется многократно на протяжении всего процесса окисления пирита (Singer, Stumm, 1970). Реакции (1), (2) и (4) протекают вдали от равновесия, т.е. при отсутствии заметной скорости обратной реакции, а реакция (3) является обратимой реакцией растворения-осаждения. Максимальная степень окисления серы (VI) в виде сульфата по реакциям (1) и (4) характерна лишь для кислых растворов. С повышением рН увеличивается доля менее окисленных форм серы: тиосульфатов, политионатов, сульфитов, элементарной серы (Chandra, Gerson, 2010). Вместе с Fe(OH)3 иногда образуется гидрооксисульфат FeOHSO4 гетит FeOOH (Todd et al., 2003), швертманит Fe8O8(OH)6SO4 и ярозит KFe3(SO4)2(OH)6 (Nordstrom et al., 2015). Тем не менее, для вычисления скорости окисления пирита обычно используют общую реакцию, которая представляет собой сумму реакций (1)–(3) или (4), (2) и (3):
(5)
$\begin{gathered} {\text{Fe}}{{{\text{S}}}_{2}} + 3.75{{{\text{O}}}_{2}} + 3.5{{{\text{H}}}_{{\text{2}}}}{\text{O}} \to \\ \to 4{{{\text{H}}}^{ + }} + 2{\text{SO}}_{4}^{{2 - }} + {\text{Fe}}{{\left( {{\text{OH}}} \right)}_{{\text{3}}}}. \\ \end{gathered} $Масса пирита, окислившегося в экспериментах (в изолированных или проточных реакторах), обычно измеряется по содержанию сульфата в растворе с учетом стехиометрии реакции (5). Нормирование этой массы к единице времени и площади поверхности дает скорость окисления пирита. Мольное отношение S/Fe в пирите несколько отклоняется от 2 вследствие присутствия изоморфных примесей Ag, As, Au, Bi, Co, Ni, Pb, Sb, Zn и других элементов (Abraitis et al., 2004). Колебания их содержания вызывает изменение электрической проводимости пиритов на 4 порядка вплоть до смены типа электрической проводимости, что влияет на скорость его окисления (Chandra, Gerson, 2010). Высокие содержания примесей обычно увеличивают скорость окисления пирита, но расхождения в скоростях окисления разных образцов пирита обычно не превышают одного порядка (Rimstidt, Vaughan, 2003; Williamson, Rimstidt, 1994).
На основании экспериментальных исследований выведены разные уравнения скорости, суммированные в обзорах (Chandra, Gerson, 2010; Wang et al., 2019). Их анализ показал, что для практического применения лучше подходят следующие уравнения, полученные в результате критического обобщения разных кинетических данных для 25°С (Williamson, Rimstidt, 1994):
(7)
$r = {{10}^{{ - 6.07}}}m_{{{\text{F}}{{{\text{e}}}^{{3 + }}}}}^{{0.93}}m_{{{\text{F}}{{{\text{e}}}^{{2 + }}}}}^{{ - 0.4}},$Рис. 1.
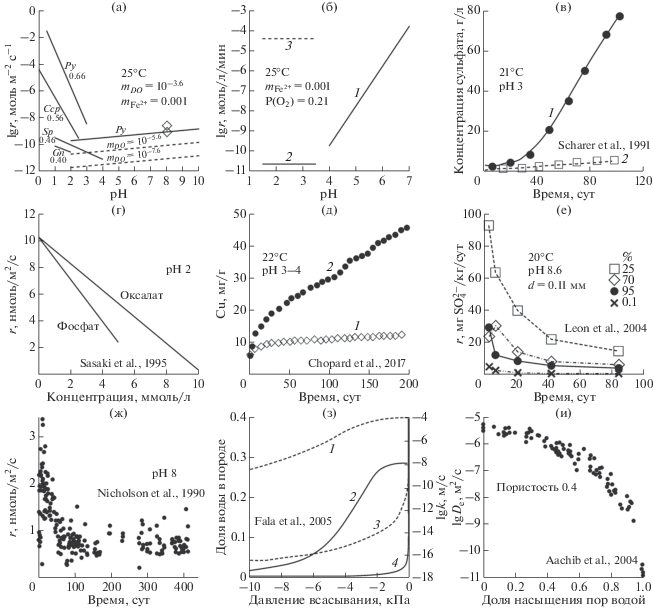
Зависимости, влияющие на образование кислого дренажа пород: (а) Скорости растворения (r) пирита Py (уравнения (6) и (7)), халькопирита Ccp (Kimbal et al., 2010), сфалерита Sp (Acero et al., 2007a) и галенита Gn (Acero et al., 2007b) в растворах с разным рН. Числа показывают потенциал покоя сульфидов (Chandra, Gerson, 2010). ${{m}_{{{\text{F}}{{{\text{e}}}^{{{\text{3 + }}}}}}}}$ определялась растворимостью ферригидрита (Yu et al., 2002). Пунктирные линии соответствуют r при уменьшении концентрации растворенного кислорода (DO) на 2 и 4 порядка. Значки при рН 8 показывают значения r для условий, когда в порах между зернами пирита был не только раствор, но и воздух (Nicholson et al., 1990). (б) Скорости окисления Fe2+ в растворе кислородом в зависимости от рН при отсутствии микроорганизмов (1 и 2) по уравнениям (9) и (10) (2) и в их присутствии (3) по данным (Nordstrom, 1985) и др. (в) Изменение концентрации растворенного сульфата во времени при окислении пирита в присутствии бактерий (1) и в их отсутствии (2). (г) Скорость окисления пирита в зависимости от концентрации фосфата и оксалата. (д) Масса меди, перешедшая со временем в раствор из халькопирита (1) и из смеси халькопирита с пиритом (2). (е) Эволюция во времени скорости перехода в раствор сульфата при окислении дробленого пирита в условиях разного содержания воздуха в порах. (ж) Изменение во времени скорости окисления дробленого пирита в условиях, когда в порах присутствует и раствор, и воздух. (з) Доля воды (2, 4) и гидравлическая проводимость k (1, 3) для песка (1, 2) и гравия (3, 4) в зависимости от всасывающего давления. (и) Эффективный коэффициент диффузии кислорода (De) в зависимости от доли насыщения пор водой.
Чтобы учесть влияние температуры, в правую часть уравнений (6) и (7) следует добавить сомножитель (Palandri, Kharaka, 2004):
(8)
$\exp \left[ {\frac{{ - {{E}_{a}}}}{R}\left( {\frac{1}{T} - \frac{1}{{298.15}}} \right)} \right],$Скорость окисления Fe2+ кислородом в растворе по реакции (2) при 25°С описывается следующими уравнениями (Singer, Stumm, 1970; Stumm, Lee, 1961):
(9)
$ - d{{m}_{{{\text{F}}{{{\text{e}}}^{{2 + }}}}}}{\text{/}}dt = k{{m}_{{{\text{F}}{{{\text{e}}}^{{2 + }}}}}}m_{{{\text{O}}{{{\text{H}}}^{ - }}}}^{2}{{P}_{{{{{\text{O}}}_{2}}}}},$(10)
$ - d{{m}_{{{\text{F}}{{{\text{e}}}^{{2 + }}}}}}{\text{/}}dt = k'{{m}_{{{\text{F}}{{{\text{e}}}^{{2 + }}}}}}{{P}_{{{{{\text{O}}}_{2}}}}},$Некоторые ацидофильные бактерии являются сильными катализаторами реакции (2), т.к. используют ее энергию для своей жизнедеятельности. Наиболее распространенными и изученными среди них являются Acidithiobacillus (ранее Thiobacillus) ferrooxidans. Эти бактерии активны при рН 1.5–5 и 5–55°С с максимальной активностью при рН 3 и 30°С (Jaynes et al., 1984). В этих условиях скорость реакции (2) растет пропорционально массе бактерий (Pesic et al., 1989) и может повышаться более чем в 106 раз по сравнению с небактериальным окислением (линия 3 на рис. 1б). Это так называемое непрямое метаболическое окисление пирита, суть которого заключается в регенерации Fe3+, основного оксиданта пирита (Evangelou, Zhang, 1995). В работе (Singer, Stumm, 1970) сделан вывод, что стадией, контролирующей окисление пирита в кислых растворах, является медленная реакция (2), которая поставляет окислитель Fe3+ для основной реакции (4). Реакции (1) в этом сценарии отводилась лишь роль инициатора процесса (появление Fe2+ в растворе). При этом, однако, был не замечен тот факт, что ускорение реакции (2) бактериями лишает ее контролирующей роли, которая переходит к реакции (4) (Williamson et al., 2006). Реакция (4) под действием бактерий тоже ускоряется, но в значительно меньшей степени. Например, скорость появления сульфата в растворе в результате окисления пирита при рН 3 и 21°С в присутствии бактерий была в ∼20 раз выше, чем при их отсутствии (рис. 1в), но при снижении температуры до 6°С эти скорости сравнивались (Scharer et al., 1991). Добавление железоокисляющих бактерий (107–108 клеток в 1 см3) увеличивало скорость окисления халькопирита при рН 2 лишь в 2 раза (Халезов, 2009). В экспериментах по окислению сульфидов с бактериями рН раствора был на две единицы ниже, а содержания сульфатов и тяжелых металлов до 16 раз больше, чем в экспериментах без бактерий (Blackmore et al., 2018). В близнейтральных условиях железоокисляющие бактерии также способны сохранять свою активность, поселяясь в пористом слое вторичных минералов на сульфиде и создавая благоприятную для себя кислую среду на микроуровне путем интенсификации окисления сульфида (Dockrey et al., 2014). Это прямое метаболическое окисление пирита характеризуется непосредственным контактом сульфида и бактерий, в результате чего на поверхности пирита образуются округлые ямки травления, по форме и размеру совпадающие с бактериями (Mielke et al., 2003). Сложность в оценке эффективности влияния бактерий заключается в измерении концентрации живых клеток.
Лиганды Cl– и ${\text{SO}}_{4}^{{2 - }}$ уменьшают скорость окисления пирита лишь в небольшой степени (Williamson, Rimstidt, 1994), а ${\text{PO}}_{4}^{{3 - }}$ и ${{{\text{C}}}_{2}}{\text{O}}_{4}^{{2 - }}$ значительно сильнее (рис. 1г). Сдерживающее влияние фосфата и оксалата объясняется низким стандартным окислительно-восстановительным потенциалом доминантных комплексов Fe3+, которые действуют как оксиданты, а также образованием коллоидных и полиядерных частиц Fe(III), которые осаждаются на поверхности пирита (Sasaki et al., 1995). Карбонатные растворы при рН 8–11 увеличивают скорость окисления пирита до 2-х раз по сравнению с растворами NaOH за счет более легкого переноса электронов от карбонатных комплексов Fe(II) к О2, увеличения растворимости железа и создания буферированной щелочной среды (Caldeira et al., 2010).
Результаты экспериментов по окислению пирита обычно интерпретируются в рамках химического или электрохимического механизма, т.е. стадией, контролирующей скорость реакций (1) и (4), считается адсорбция молекул или перенос электрона на поверхности пирита (Chandra, Gerson, 2010). Оба механизма могут иметь одинаковое уравнение скорости, но электрохимическая реакция имеет дополнительную зависимость от электрического потенциала или плотности тока (Chandra, Gerson, 2010). В пользу электрохимического механизма свидетельствуют зависимости скорости реакции от концентраций оксидантов О2 и Fe3+ (Williamson, Rimstidt, 1994; Rimstidt, Vaughan, 2003), а также изотопные исследования (Reedy et al., 1991), которые показали, что кислород сульфата берется из воды, а не из О2 (оксиданта). При электрохимическом подходе реакции (1) и (4) рассматриваются как суммы полуреакций анодного окисления пирита и катодного восстановления Fe3+ или растворенного О2 (Яхонтова, Грудев, 1978). В работе (Rimstidt, Vaughan, 2003) каждая из полуреакций расписана по стадиям с образованием промежуточных поверхностных частиц.
Скорости окисления сульфидов в кислых растворах уменьшаются с уменьшением их электрохимического коррозионного потенциала (потенциала покоя) (рис. 1а). Однако в смеси эти минералы окисляются с другими скоростями, чем по отдельности, что объясняется образованием гальванических пар. Пирит имеет более высокий потенциал покоя, чем большинство сульфидов, поэтому в смеси с ними он действует как катод и растворяется медленнее, а другие сульфиды действуют как анод и растворяются быстрее (Heidel et al., 2013; Chopard et al., 2017). Скорости извлечения Cu из халькопирита, Zn, Cd, Mn из сфалерита в смеси с пиритом выше на 1–2 порядка по сравнению с окислением этих минералов по-отдельности (рис. 1д). Окисление пирита в смеси с этими минералами практически не происходит (Heidel et al., 2013). Некоторые несульфидные минералы также изменяют скорость окисления пирита. Например, в присутствии гематита она уменьшается, а в присутствии глинозема увеличивается (Tabelin et al., 2017a), что объясняется разным влиянием минералов на анодную и катодную полуреакции окисления пирита (Tabelin et al., 2017b). Оксиды Mn не только ускоряют окисление пирита, но и сами выступают в роли его окислителя (Qiu et al., 2016). В будущем эти гальванические эффекты нужно изучить более детально, чтобы учесть в предсказании генерирования кислых дренажных вод и перехода в них тяжелых металлов.
Чтобы приблизиться к условиям отвала, в некоторых опытах между зернами пирита находилась не только вода или раствор, но также воздух (Jerz, Rimstidt, 2004; León et al., 2004; Nicholson et al., 1990). В этих условиях начальная скорость окисления пирита была тоже пропорциональна $P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{0.5}}$ (Jerz, Rimstidt, 2004), но она была несколько выше, чем в воде (верхний ромб при рН 8 на рис. 1а), что может объясняться более высоким содержанием О2 в воздухе. Это предположение подтверждается увеличением скорости окисления пирита в ∼3 раза с увеличением доли воздуха в порах от 0.05 до 0.75 (рис. 1е).
Из рис. 1е и других работ (Jerz, Rimstidt, 2004; León et al., 2004; Nicholson et al., 1990; Williamson, Rimstidt, 1994) следует, что величина r со временем уменьшается. Это объясняется осаждением на поверхности зерен пирита слоя твердых продуктов реакции, которые препятствуют свободному доступу О2 к свежей поверхности пирита (Todd et al., 2003). Уменьшение скорости описывается моделью сокращающегося сферического ядра (Jerz, Rimstidt, 2004; Nicholson et al., 1990; Wang et al., 2019), в которой используется коэффициент диффузии кислорода (Ds) через корку продуктов реакции. По данным (Nicholson et al., 1990) значение Ds при рН 8 равно 3 × 10–16 м2/с, а по данным (Wang et al., 2019) оно составляет 1.2 × 10–15 м2/с при содержании воды в порах более 25% и уменьшается до 4.7 × 10–19 м2/с при уменьшении этого содержания до 0.1%.
Следует отметить, что появление корки вторичных минералов на поверхности пирита не обязательно должно вызывать замедление его растворения, если корка не сплошная или она достаточно пористая, чтобы обеспечить свободный доступ О2 к свежей поверхности пирита. В этом случае замедление растворения пирита со временем может быть связано с растворением активных ультратонких частиц и поверхностных дефектов, которые образовались при измельчении минерала и исчезли в результате растворения. После этого растворяются дефекты, равномерно расположенные в объеме зерен. На них образуются ямки травления, которые углубляются с постоянной скоростью, т.к. растворение идет на их дне, а боковые стенки ямок остаются инертными. Такой механизм типичен, например, для многих породообразующих минералов (Алексеев и др., 2005; Anbeek et al., 1994; Gautier et al., 2001), которые при длительном времени растворяются с постоянной скоростью, нормированной к геометрической площади поверхности исходных зерен. Для пирита также характерно преимущественное растворение поверхностных дефектов (McKibben, Barnes, 1986; Sun et al., 2015), а длительность опытов обычно не превышала 100 сут, что, возможно, было недостаточным, чтобы выйти на плато с постоянной скоростью растворения. Действительно, в более длительных опытах (до 410 сут) скорость растворения пирита уменьшалась в первые 100 сут, но потом она оставалась примерно на одном уровне, хотя ее колебания были довольно значительными (рис. 1ж). Для окончательного вывода о механизме окисления пирита нужны дополнительные более точные и очень длительные (годы) эксперименты при комнатной температуре в близнейтральных растворах без перемешивания.
ПРЕДСКАЗАНИЕ СОСТАВА ДРЕНАЖНЫХ ВОД
Величина рН растворов после дренирования отвалов определяется соотношением содержаний минералов, производящих и потребляющих кислоту, а также скоростями выветривания этих минералов. Низкие значения рН дренирующих растворов означает развитие процесса по наихудшему сценарию, т.к. в раствор переходят также тяжелые металлы в высоких концентрациях (Замана, Чечель, 2014; Рыбникова, Рыбников, 2019; Evangelou, Zhang, 1995). Наиболее благоприятны близнейтральные дренирующие растворы, т.к. тяжелые металлы, которые переходят в эти растворы при растворении пирита, сорбируются гидроксидом Fe(III) или соосаждаются вместе с ним. Однако возможны повышенные содержания сульфата и коллоидных форм гидроксида Fe(III) (Nordstrom et al., 2015). В щелочных растворах увеличивается мобильность элементов, которые стабильны в виде оксианионов, например, ${\text{SO}}_{4}^{{2 - }},$ ${\text{AsO}}_{4}^{{3 - }},$ ${\text{MoO}}_{4}^{{2 - }},$ ${\text{CrO}}_{4}^{{2 - }}.$ Для предсказания состава дренажных вод используют разные методы, среди которых наиболее распространены статические и кинетические тесты (Алексеев и др., 2011; Méndez-Ortiz et al., 2007). Основным статическим тестом является определение кислотно-щелочного баланса (Acid Basic Accounting), т.е. способности минеральных ассоциаций производить и нейтрализовать кислоту (Гаськова, Бортникова, 2007; Méndez-Ortiz et al., 2007; Sobek et al., 1978). Первая способность определяется содержанием пиритной серы, а вторая – содержанием минералов, нейтрализующих кислоту. Этот метод имеет ряд недостатков (Dold, 2017): 1) игнорируются другие сульфиды (кроме пирита), которые при окислении производят меньше кислоты или не производят ее вовсе (галенит, сфалерит); 2) не учитывается кислотный потенциал гидроксидов, сульфатов и сидерита; 3) коэффициент, используемый для расчета кислотного потенциала (31.25) занижен в 2 раза. В работе (Dold, 2017) предлагаются пути преодоления этих недостатков с помощью количественного минералогического анализа. Однако сохраняется еще один существенный недостаток этого метода, который заключается в игнорировании скоростей растворения минералов, которые сильно различаются (например, очень быстрое растворение карбонатных пород и очень медленное растворение пород гранитного состава).
Недостаток статических тестов частично преодолен в кинетических тестах, где используется гумидная ячейка или колонна с дробленой породой (Sapsford et al., 2009). Ячейка раз в неделю промывается водой на протяжении полугода и больше, а раствор после каждой промывки анализируется на рН и содержание элементов. В этих тестах учитывается кинетика реакций и динамика фильтрации воды, но в условиях, которые отличаются от условий в отвалах (температура, отношение масс жидкой и твердой фаз, размер кусков, доступность О2 и др.), т.е. результаты получаются искаженными (Erguler et al., 2014). Неисправимым недостатком кинетических тестов остается ограниченность во времени, т.к. их трудно выполнять в течение нескольких лет, а тем более десятилетий, которые необходимы для прогноза эволюции КДП.
Более перспективным может оказаться математическое моделирование процесса выветривания отвалов горных пород с использованием известных законов физики и химии с параметрами, измеренными экспериментально или непосредственно в отвалах для конкретных случаев. Преимущество этого метода заключается в возможности рассчитывать эволюцию процесса для больших интервалов времени (десятки лет) как в прошлое (проверка соответствия модели природному объекту), так и в будущее (предсказание поведения КДП). В качестве первого приближения иногда используют равновесно-кинетическую модель, в которой участвуют известные скорости растворения минералов и масса воды (с растворенным О2), прошедшая через отвал за определенное время (Sidkina et al., 2020). Однако при этом не учитывается влияние других процессов на производство КДП.
Отвалы горных пород имеют высокую пористость (0.2–0.4) и располагаются выше уровня грунтовых вод, что определяет ненасыщенность пор водой, т.е. в порах помимо воды присутствует воздух. Существуют разные уравнения для описания фильтрации воды в этих условиях. Наиболее известным является уравнение Ричардса, которое используется в разных формах (Bouchemella et al., 2015). Это нелинейное дифференциальное уравнение в частных производных, основная трудность решения которого заключается в отсутствии точных аналитических решений. Гидравлические свойства пористой среды в этом уравнении (кривая удержания воды и гидравлическая проводимость) описываются с помощью эмпирических моделей, коэффициенты в которых определяются экспериментально для конкретных пород (рис. 1з). Такое ограничение обусловлено тем, что эти функции зависят от многих параметров, которые трудно учесть расчетным путем. Это распределение пор по размеру, состав и структура поверхности пор, форма пор. Использование разных эмпирических моделей для описания гидравлических свойств пористой среды может приводить в некоторых случаях к существенно разным результатам моделирования фильтрации воды (Bouchemella et al., 2015).
Кислород, необходимый для окисления пирита, поставляется внутрь отвала в основном путем диффузии молекул О2 в воздушной или водной среде, заполняющей поры. В воздухе коэффициент диффузии кислорода и его содержание значительно больше, чем в воде. Это значит, что основной диффузионный перенос О2 происходит через воздух, заполняющий поры. Для пор с переменным содержанием воды и воздуха вычисляется эффективный коэффициент диффузии De. Для этого используют различные диффузионные модели с подгоночными параметрами, значения которых определяются экспериментально на конкретных породах (Aachib et al., 2004). С увеличением доли воды в порах от 0 до 1 величина De уменьшается на 5 порядков (рис. 1и), что соответствует пропорциональному уменьшению скорости окисления пирита (Elberling, Nicholson, 1996).
Перемещение воздуха внутрь отвала является значительно более эффективным механизмом поставки кислорода к пириту, чем диффузия. Оно происходит в более крупных порах под действием градиента давления, который возникает при ветре (Amos et al., 2009) или в результате расходования кислорода на окисление пирита (Binning et al., 2007). Другой движущей силой является градиент температуры, который возникает при выделении тепла во внутренней части отвала в результате окисления пирита. Например, внутри хорошо проницаемого отвала с содержанием пирита 7% температура достигала 65°С, что вызывало конвекцию воздуха в отвале (Lefebvre et al., 2001a). Двумерное моделирование этого процесса с учетом уравнений теплопереноса показало, что скорость окисления пирита в отвале сильно меняется при изменении температуры и скорости фильтрации воздуха (Lefebvre et al., 2001b).
Размер кусков в отвалах может колебаться от <0.1 мкм до >1 м (Anterrieu et al., 2010; Fala et al., 2005). Чтобы облегчить учет кинетики химических реакций в моделях, все куски в отвале рассматриваются в виде сфер одинакового размера (эквивалентный диаметр), суммарная площадь поверхности которых такая же, как для фактической смеси частиц разного размера (Aubertin et al., 1998). Дальнейшая эволюция этих сфер рассматривается в основном в рамках простой модели сокращающейся сферы (Bouffard et al., 2006) или более сложной модели пассивированной сокращающейся сферы (Chandra, Gerson, 2010; Molson et al., 2005). В последнем случае используют дополнительный коэффициент диффузии кислорода через корку вторичных минералов на поверхности зерен пирита. Однако величина коэффициента сильно различается (10–11–10–15 м2/с) в разных моделях (Gerke et al., 1998; Molson et al., 2005; Romano et al., 2003; Wunderly et al., 1996), что заставляет сомневаться в ее достоверности, особенно когда коэффициент диффузии используется в качестве калибровочного (подгоночного) параметра (Wilson et al., 2018a). Модель сокращающейся сферы использовалась при описании процессов гидрометаллургической обработки сульфидных руд и при выветривании хвостов, где зерна мелкие и каждое зерно состоит из одного минерала. Однако уже для кучного выщелачивания, где размер частиц достигает 12–25 мм, эта модель плохо подходит, т. к. мелкие зерна сульфидов располагаются в основном внутри более крупных кусков вмещающей породы (Ghorbani et al., 2011). Еще хуже эта модель соответствует действительности для отвалов, где размер кусков больше.
Чем меньше количество атмосферных осадков, тем ниже рН и выше концентрации растворенных металлов в дренажных водах (Liu et al., 2019). Это согласуется с периодическим увеличением поступления O2 в сухой сезон для одного и того же отвала (Lorca et al., 2016), т.к. диффузия О2 возрастает при уменьшении содержания воды в порах (рис. 1и). Возможно и более сложное влияние противоположно направленных факторов, причем с неочевидным результатом. Например, уменьшение размера зерен увеличивает площадь поверхности и, соответственно, скорость окисления пирита. Однако одновременно уменьшается и размер пор, что увеличивает содержание воды в порах, замедляет диффузию О2 и, соответственно, уменьшает скорость окисления пирита (Molson et al., 2005).
В моделировании КДП вместо уравнения скорости окисления пирита иногда ошибочно (см. раздел “Кинетика…”) используется уравнение скорости реакции (2) окисления Fe2+ в растворе (Gerke et al., 1998; Kohfahl et al., 2007). Иногда экспериментальное уравнение скорости окисления пирита вообще не рассматривается, учитывается лишь стехиометрия реакции. В этом случае контролирующей стадией всего процесса считается медленная диффузия О2 в воздушно-водной среде пор и в корке вторичных минералов на поверхности зерен пирита (Molson et al., 2005). Однако медленная поставка О2 к пириту и быстрое начальное потребление О2 пиритом означает уменьшение концентрации О2 у пирита, что, согласно уравнения (6), должно приводить к замедлению окисления пирита (пунктир на рис. 1а). Более реальным здесь представляется устойчивое состояние с равными малыми скоростями поставки и потребления О2, т.е. в моделировании нужно учитывать и то, и другое. К сожалению, в моделировании КДП почти не используется кинетика растворения породообразующих минералов (одно из редких исключений – работа (Sidkina et al., 2020)), хотя таких данных накоплено довольно много (например, Palandri, Kharaka, 2004). Их использование позволило бы оценить эффективность нейтрализации КДП для конкретных случаев, т.е. приблизить модели к природным объектам.
Тем не менее, моделирование показало свою эффективность в выявлении дополнительных факторов, ускоряющих или замедляющих образование КДП. В частности, моделирование подтвердило эффективность использования мелко-зернистого покрытия в качестве капиллярного барьера, замедляющего доступ О2 (Molson et al., 2008). Если же горизонтальные мелко-зернистые слои находятся внутри отвала, они задерживают вышележащую воду лишь до определенного предела, а затем вода прорывается в локальных местах в нижележащий крупно-зернистый слой, образуя каналы, а окисление пирита происходит в основном между каналами в крупных порах, заполненных воздухом (Molson et al., 2005). В случае наклонных мелко-зернистых слоев прорыв не происходит, т.к. вода не накапливается, а перемещается по слою и сливается по склону отвала. Характерной особенностью отвалов является их неоднородность по размеру кусков и минеральному составу, причем крупные и мелкие куски породы могут иметь существенно разное (на порядок) содержание сульфидов (Ghorbani et al., 2011). Неоднородность отвалов вызывает неопределенность прогнозов КДП. Возможное решение этой проблемы заключается в стохастическом подходе с использованием коэффициентов корреляции между кислотообразующими и кислотопотребляющими минералами (Pedretti et al., 2017, 2020). Аналогичное моделирование показало, что пространственная корреляция гидравлических свойств влияет на распределение влаги внутри отвала и в некоторых случаях тоже создает преимущественные пути потока (Fala et al., 2013).
В моделировании КДП существует большое число уравнений и параметров со своими погрешностями, которые суммируются и могут привести к неверным решениям (Brookfield et al., 2006). Поэтому важной задачей моделирования остается уточнение используемых уравнений и параметров. Основным критерием правильности математической модели обычно считается ее соответствие природному объекту. Но такого соответствия нетрудно добиться и искусственным путем, меняя значения параметров, упрощая, корректируя или игнорируя зависимости, которые кажутся несущественными или не укладываются в модель. Более правильный подход заключается в использовании параметров, измеренных независимым способом (температура, количество осадков, гидравлическая проводимость, коэффициент диффузии, пористость, площадь поверхности минералов и др.). Тогда может появиться возможность одной и той же программой, но с разными значениями параметров, создавать адекватные модели разного масштаба: от небольших лабораторных ячеек до крупных контейнеров и еще более крупных отвалов (Wilson et al., 2018a, 2018b).
ПРЕДОТВРАЩЕНИЕ И РЕКУЛЬТИВАЦИЯ КИСЛЫХ ДРЕНАЖНЫХ ВОД
Этот раздел стоит несколько в стороне от главной темы статьи и присутствует здесь в кратком виде лишь для полноты картины с использованием данных, приведенных в обзорах (Naidu et al., 2019; Park et al., 2019; Pozo-Antonio et al., 2014; Skousen et al., 2019). Одним из самых распространенных способов предотвращения КДП является ограничение доступа О2 в шахтные отвалы путем создания плохо проницаемого для О2 барьера. В качестве барьера используется водное или сухое покрытие. Вода замедляет поступление О2 потому, что коэффициент диффузии О2 в ней на много порядков ниже, чем в воздухе (рис. 1и). Например, шахтные хвосты одного из Zn-Cu рудников в Канаде исследовались после 60-и летнего выветривания (Moncur et al., 2015). Непокрытые водой участки имели толстую (40–60 см) зону окисления, поровая вода была кислой (рН 1.9–4.2) с высокими концентрациями Fe (3.5–20 г/л) и S (7.9–59 г/л). Участки, покрытые слоем воды в 1 м, имели тонкую (6 см) зону окисления, поровая вода была близнейтральной с низкими концентрациями Fe (0.16–1.35 мг/л) и S (1.6–1.7 г/л). Однако этот способ неприменим в аридном климате, где испарение воды преобладает над ее осаждением.
Сухой барьер для О2 создается путем покрытия отвала цементом, битумом, глиной (Pozo-Antonio et al., 2014). В случае с глиной и другими тонко-пористыми материалами (хвосты без сульфидов, ил, зола, шлам, шлак) используется свойство воды задерживаться в мелких порах под действием капиллярных сил, препятствуя тем самым проникновению О2 в отвал. Например, в миниатюрном отвале, покрытом илистой почвой, через 6 мес концентрация О2 уменьшилась с 21 до 1.5%, что вызвало уменьшение скорости окисления пирита на 93% (Igarashi et al., 2006). Как показали длительные лабораторные и полевые тесты (500 сут и 4 г.), смесь шлама (размер частиц 25 мкм) с почвой тоже эффективно подавляет образование КДП (Demers et al., 2017). В качестве покровного материала для отвала может быть использован и органический материал (осадок сточных вод, компост, опилки), который при разложении потребляет О2 (Park et al., 2019). В этом случае, однако, возможно восстановительное растворение оксигидроксида Fe(III) и переход в раствор адсорбированных токсичных элементов (Ribet et al., 1995).
Если нейтрализующий потенциал породы невысок, в отвал добавляют щелочные материалы: известь, известняк, доломит, гипс, золу, шлак, цементную пыль, фосфаты, сахарную пену, боксит (Hakkou et al., 2009; Mylona et al., 2000; Park et al., 2019; Pérez-López et al., 2009; Zhou et al., 2017). Преимущество многих из этих материалов не только в повышении рН (т.е. в уменьшении содержания металлов и активности бактерий), но и в образовании цементирующего слоя, который уменьшает диффузию О2 и инфильтрацию воды. При большом избытке щелочных материалов, однако, рН раствора может повыситься до 10 и больше, что увеличит растворимость Al, Cu, Ni, Pb, Zn (Tabelin et al., 2018). Более эффективно предварительное смешивание и совместное размещение в отвалах сульфид-содержащей породы и материалов, потребляющих кислоту (Park et al., 2019). Однако при негомогенной смеси в отвалах могут формироваться пути преимущественного течения кислой воды.
Для подавления активности железоокисляющих бактерий используют бактерициды: анионные ПАВ, чистящие средства, органические кислоты, пищевые консерванты (Sobek et al., 1990). Бактерии существуют в кислой среде (рН < 3), но внутри клетки рН ∼ 7. ПАВ позволяют протонам свободно проникать внутрь клетки, что вызывает ее гибель (Evangelou, Zhang, 1995). Например, распыление додецилбензолсульфоната натрия в одном из мест утилизации мусора (4 раза в течение года) понизило концентрации Fe и Mn в сточных водах на 82 и 90% (Parisis et al., 1994). Недостаток метода заключается в необходимости повторных обработок бактерицидами.
Многие органические вещества (олеат натрия, гуминовая кислота, фосфолипиды, диэтилентриамин (DETA), триэтилентетрамин (TETA) и др.) образуют на поверхности пирита пассивирующий слой, который препятствует его растворению. Например, олеат натрия соединяется с Fe2+ и Fe3+ на поверхности пирита и образует электрохимически пассивный слой Fe-олеата, который сдерживает окисление пирита в течение по крайней мере 30 сут (Jiang et al., 2000). Гуминовая кислота тоже сдерживает окисление пирита, т. к. она адсорбируется на его поверхности почти необратимо из-за сильного сродства к продуктам окисления (Ačai et al., 2009). Молекулы фосфолипидов адсорбируются на поверхности пирита своими гидрофильными головами так, что их гидрофобные углеводородные хвосты направлены наружу (Elsetinow et al., 2003; Zhang et al., 2003). Эксперименты при рН < 2 показали уменьшение скорости окисления пирита в присутствии фосфолипидов на 60–80% и увеличение их эффективности с увеличением количества и длины хвостов. Однако в присутствии силикатов пассивирующая способность липидов уменьшается (Kargbo et al., 2004). Исследования с органическими веществами пока ограничены лишь кратковременными экспериментами, поэтому их длительная стабильность в природных условиях остается под вопросом, т.к. микроорганизмы могут разлагать органику.
Микроинкапсулирование, т.е. покрытие зерен сульфидов протектирующей оболочкой, было предложено сначала как суммарное действие трех компонентов (Huang, Evangelou, 1992): Н2О2 быстро окисляет Fe(II) до Fe(III) на поверхности пирита, фосфат калия реагирует с Fe(III) и образует поверхностный слой фосфата Fe(III), ацетат натрия поддерживает рН ∼ 5 (область стабильности фосфата Fe(III)). Определены оптимальные соотношения этих компонентов, при которых протектирующий эффект максимален (Kollias et al., 2015). Однако фосфор в природных условиях опасен тем, что может вызвать сильный рост биологической продуктивности водоемов. Замена его силикатом натрия дала еще более выраженный протектирующий эффект (Fan et al., 2017): скорость окисления пирита при рН 7.4 снизилась на 97%. Компонент Н2О2 является довольно дорогим и нестабильным, а без него протектирующий слой не образуется. Для преодоления этого недостатка было предложено использовать органосиланы, которые состоят из неорганического кремния и органических функциональных групп СН3О- и СН3СН2О-. Например, n-пропилтриметоксисилан (NPS) снижает скорость химического и биологического окисления пирита на 95% (Diao et al., 2013) в результате образования плотной сети связей Fe–O–Si на поверхности пирита.
Содержание сульфидов в отвалах и хвостах обычно не превышает 10%, поэтому микроинкапсулирование сульфидов требует излишнего расхода дорогих компонентов. Чтобы уменьшить этот расход, разрабатываются методики, нацеленные только на сульфиды. Например, для покрытия арсенопирита в экспериментах использован комплекс Ti-катехол (Park et al., 2018a). Он устойчив в растворе, но на поверхности сульфидов адсорбируется, под действием электрохимических реакций окисляется, разлагается и осаждается в виде TiO2. В работе (Jha et al., 2012) использован более дешевый Si-катехол, который окислительно разлагается на поверхности пирита с образованием стабильного SiO2. Препарат эффективен в кислых и близнейтральных растворах даже при низких концентрациях. Комплекс Al-катехол, возможно, является наиболее эффективным, т. к. уменьшает скорость окисления пирита на 98% (Park et al., 2018b). В работе (Seng et al., 2019) гальваническое взаимодействие с пиритом обеспечивали металлические частицы Fe и Al (20–40 мкм), которые имели меньший потенциал покоя, чем пирит. В контакте с пиритом они действовали как анод и преимущественно растворялись, а пирит (катод) был гальванически защищен от растворения. Однако пассивирующий эффект был ограничен временем нахождения частиц на поверхности пирита. Добавление фосфата усиливало и продлевало этот эффект в результате образования железо-фосфатного покрытия. Использование микроинкапсулирования в экспериментах показало хорошие результаты в предохранении сульфидов от окисления. Однако для окончательной оценки эффективности этих методов нужны дополнительные исследования в условиях, близких к природным (присутствие других минералов, длительное время, циклы высыхания-смачивания).
Если превентивные меры не сработали или не были применены вообще, а КДП образуется, приходится использовать более дорогие методы рекультивации, которые имеют дело с кислыми дренирующими растворами (Naidu et al., 2019; Skousen et al., 2019). В основе активных методов, которые требуют постоянного контроля и регулирования, лежит добавление нейтрализаторов кислоты, аэрация и осаждение гидроксидов металлов. В основе пассивных методов (биореакторов) лежит сульфат-редукция органическим веществом и осаждение сульфидов металлов. В последнее время наметилась тенденция применения методов, которые позволяют извлекать полезные компоненты из КДП (H2SO4, Fe, Cu, Zn, Ni, РЗЭ) и очищать воду для ее повторного использования (Naidu et al., 2019). Это прямой и обратный осмос, нанофильтрация, мембранная дистилляция, диализ, ионный обмен и др.
Перспективным направлением рекультивации может стать переработка сульфид-содержащих отходов (в первую очередь шахтных хвостов с малым размером частиц) в конструкционный или геополимерный материал (Park et al., 2019). Отходы предлагают использовать при строительстве дорог, в качестве добавки в асфальт, цемент, для производства геополимеров. В последнем случае хвосты подвергаются автоклавной обработке при 60–120°С и 1–350 бар в присутствии воды и щелочи. Образуется монолитный прочный материал (кирпичи, блоки) с аморфной полимерной структурой – Si–O–Al–O–Si-, устойчивой к кислотам (Ren et al., 2015). В результате такой обработки выщелачиваемость металлов из хвостов уменьшается на 98% (Kiventerӓ et al., 2018). Стойкость геополимера к выщелачиванию повышается при Na/Al ∼1 и Si/Al от 1 до 5 (Duxon et al., 2007). Нужны дополнительные экспериментальные исследования выщелачиваемости вредных элементов из геополимеров.
ЗАКЛЮЧЕНИЕ
Образование КДП при выветривании отвалов сульфид-содержащих пород является актуальной экологической проблемой, которая изучается с помощью экспериментальных, расчетных и природных исследований. Основное внимание в этой проблеме уделяют пириту, т. к. он является самым распространенным сульфидным минералом. Анализ опубликованных работ позволил суммировать факторы, влияющие на скорость окисления пирита r (табл. 1). Величина r контролируется реакциями на поверхностных дефектах и зависит от температуры, рН, содержания окислителей (O2, Fe3+). Эти зависимости неоднократно изучались экспериментально, в результате чего получены уравнения скорости. Одной из стадий окисления пирита является реакция окисления Fe2+ в растворе, которая при низких рН протекает очень медленно и поэтому могла бы контролировать скорость всего процесса. Однако это не происходит, т. к. реакция ускоряется железоокисляющими бактериями, которые всегда присутствуют в природных условиях. Окисление пирита может сильно замедляться в присутствии некоторых лигандов, а также других сульфидов, окисление которых при этом ускоряется вследствие гальванического эффекта. Со временем скорость окисления пирита в экспериментах уменьшается, что может быть связано с растворением поверхностных дефектов или с образованием протектирующего поверхностного слоя продуктов реакции. Для однозначного выявления причины требуются более точные и длительные опыты.
Таблица 1.
Факторы, влияющие на скорость окисления пирита (r) в отвалах пород
| Факторы | Описание |
|---|---|
| Т, pH, O2, Fe3+ | Увеличение температуры, рН и концентрации окислителей увеличивает r (установлены количественные зависимости r от этих параметров) |
| Изоморфные примеси в пирите | Вызывают изменение r в пределах порядка |
| Стадия окисления Fe2+ в растворе | Уменьшает r, но только в отсутствие железоокисляющих бактерий |
| Железоокисляющие бактерии | Увеличивают r в кислых растворах на ∼порядок |
| Лиганды PO43-, C2O42- | Уменьшают r |
| Другие сульфиды | Уменьшают r, но ускоряют свое окисление (гальванический эффект) |
| Время | Уменьшение r вследствие растворения поверхностных дефектов или образования поверхностного протектирующего слоя |
| Минералы, нейтрализующие кислоту | Величина r чуть возрастает, но растворы становятся близнейтральными с низким содержанием тяжелых металлов |
| Размер пор в отвалах | По мелким порам, заполненным водой, диффузия О2 к зернам пирита происходит на 5 порядков медленнее (r пропорционально уменьшается), чем по крупным порам, заполненным воздухом |
| ΔР, ΔТ | Градиенты давления и температуры стимулируют быструю конвективную доставку О2 через крупные поры (r значительно увеличивается) |
| Размер зерен пирита и кусков породы | Уменьшение размера зерен пирита увеличивает r, но размещение зерен внутри кусков породы препятствует окислению пирита |
| Неоднородность отвалов по размеру кусков и пор | Слои мелкозернистого материала задерживают воду, что препятствует проникновению О2 в отвал (образуется капиллярный барьер, который уменьшает r) |
| Покрытие отвала или зерен пирита изолирующим материалом | Препятствует проникновению О2 и воды в отвал или к зернам пирита (уменьшение r) |
В зависимости от содержания в отвале минералов, способных производить и нейтрализовать кислоту, а также их скоростей окисления и растворения, процесс выветривания отвала может протекать при разных рН раствора. Наиболее опасны растворы с низкими рН, т. к. они еще содержат и тяжелые металлы в высоких концентрациях. Для прогнозирования поведения отвала при выветривании обычно используют статические методы, которые, однако, имеют ряд недостатков. Основным из них является игнорирование скоростей растворения минералов. В кинетических тестах этот недостаток преодолевается, но результаты получаются искаженными из-за различий лабораторных и природных условий.
Математическое моделирование выветривания отвала, основанное на детальных геохимических и минералогических исследованиях, способно быстро рассчитать эволюцию процесса для больших интервалов времени. Однако неизбежные упрощения модели, большое число уравнений и параметров со своими погрешностями не позволяют пока уверенно создавать модели, адекватные природным объектам. Тем не менее, моделирование позволяет выявить дополнительные факторы, влияющие на скорость окисления сульфидов в отвалах, и оценить их эффективность. Отвалы обладают высокой пористостью, причем поры сильно различаются по размеру и имеют разное содержание воды и воздуха. Скорость диффузии О2 в воде на много порядков меньше, чем в воздухе, поэтому доставка О2 к зернам сульфидов по мелким порам, заполненным водой, происходит значительно медленнее, чем по крупным порам, заполненным воздухом. Смена медленного диффузионного механизма доставки О2 внутрь отвала на быстрый конвективный механизм возможна в крупных порах при возникновении достаточного градиента температуры или давления.
Модель сокращающейся сферы, которая используется в кинетических расчетах, плохо соответствует действительности, т. к. сульфиды в отвалах представлены не отдельными зернами, а рассеяны в более крупных кусках породы. В моделировании выветривания отвалов обычно используются не те уравнения, которые в действительности контролируют окисление пирита, а часто обходятся вообще без них, предполагая (без достаточных оснований), что окисление пирита целиком контролируется скоростью диффузии О2 в порах и в корке вторичных минералов на поверхности зерен пирита. К недостаткам моделирования следует отнести также ограниченное использование кинетических данных по растворению породообразующих минералов, что препятствует адекватной оценке эффективности этих минералов в нейтрализации КДП.
Для предотвращения КДП отвалы покрывают трудно проницаемым для О2 и воды материалом, добавляют в них щелочные материалы и бактерициды, изолируют отдельные сульфидные зерна от доступа О2 протектирующим слоем. Последний метод назван микроинкапсуляцией и получил интенсивное развитие в экспериментальном опробовании различных протектирующих материалов: органических веществ, фосфатов, кремнезема, катехолатов Ti, Si, Al. Если КДП уже образовался, растворы нейтрализуют щелочными материалами, аэрируют и осаждают гидроксиды металлов. Другой подход заключается в создании восстановительных болотных условий (биореакторы), где происходит сульфат-редукция с помощью органики и осаждаются сульфиды металлов. В последнее время наметилась тенденция к извлечению всех полезных компонентов кислых дренирующих растворов и к переработке сульфидсодержащих отходов в конструкционный или геополимерный материал. Влияние большинства факторов на окисление пирита (табл. 1) изучено в основном на качественном и полуколичественном уровне. Это ограничивает возможность использования зависимостей для построения предсказательных математических моделей, но не препятствует их эффективному использованию для предотвращения и рекультивации КДП.
Автор благодарит М.В. Мироненко за стимулирование данной работы и анонимного рецензента за конструктивные критические замечания.
Список литературы
Алексеев В.А., Кочнова Л.Н., Бычкова Я.В., Кригман Л.В. (2011) Экспериментальное исследование извлечения нормируемых элементов водой из загрязненных пород. Геохимия. (12), 1317-1342.
Alekseyev V.A., Kochnova L.N., Bychkova Ya.V., Krigman L.V. (2011) Extraction of hazardous elements by water from contaminated rocks: An experimental study. Geochem. Int. 49 (12), 1239-1262.
Алексеев В.А., Рыженко Б.Н., Шварцев С.Л., Зверев В.П., Букаты М.Б., Мироненко М.В., Чарыкова М.В., Чудаев О.В. (2005) Геологическая эволюция и самоорганизация системы вода–порода. Т. I: Система вода–порода в земной коре: взаимодействие, кинетика, равновесие, моделирование. Новосибирск, СО РАН, 244с.
Гаськова О.Л., Бортникова С.Б. (2007) К вопросу о количественном определении нейтрализующего потенциала вмещающих пород. Геохимия. (4), 461-464.
Gas’kova O.L., Bortnikova S.B. (2007) On the quantitative evaluation of the neutralizing potential of host rocks. Geochem. Int. 45(4), 409-412.
Замана Л.В., Чечель Л.П. (2014) Геохимия дренажных вод горнорудных объектов вольфрамового месторождения Бом-Горхон (Забайкалье). Химия в интересах устойчивого развития. 22, 267-273.
Рыбникова Л.С., Рыбников П.А. (2019) Закономерности формирования качества подземных вод на отработанных медноколчеданных рудниках Левихинского рудного поля (Средний Урал, Россия). Геохимия. 64(3), 282-299.
Rybnikova L.S., Rybnikov P.A. (2019) Regularities in the evolution of groundwater quality at abandoned copper sulfide mines at the Levikha ore field, Central Urals, Russia. Geochem. Int. 57 (3), 298-314.
Халезов Б.Д. (2009) Исследования и разработка технологии кучного выщелачивания медных и медно-цинковых руд. Автореферат дис. … докт. технич. наук. Екатеринбург: Ин-т металлургии и материаловедения им. А.А. Байкова РАН, 53 с.
Яхонтова Л.К., Грудев А.П. (1978) Зона гипергенеза рудных месторождений. М.: Изд. МГУ. 229 с.
Aachib M., Mbonimpa M., Aubertin M. (2004) Measurement and prediction of the oxygen diffusion coefficient in unsaturated media, with applications to soil covers. Water, Air, Soil Pollut. 156(1–4), 163-193.
Abraitis P.K., Pattrick R.A.D., Vaughan D.J. (2004) Variations in the compositional, textural and electrical properties of natural pyrite: a review. Int. J. Miner. Process. 74(1–4), 41-59.
Ačai P., Sorrenti E., Gorner T., Polakovič M., Kongolo M., de Donato P. (2009) Pyrite passivation by humic acid investigated by inverse liquid chromatography. Colloids Surf. A. 337, 39-46.
Acero P., Cama J., Ayora C. (2007a) Sphalerite dissolution kinetics in acidic environment. Appl. Geochem. 22(9), 1872-1883.
Acero P., Cama J., Ayora C. (2007b) Rate law for galena dissolution in acidic environment. Chem. Geol. 245(3–4), 219-229.
Amos R.T., Blowes D.W., Smith L., Sego D.C. (2009), Measurement of wind-induced pressure gradients in a waste rock pile. Vadose Zone J. 8, 953-962.
Anbeek C., Van Breemen N., Meijer E.L., Van Der Plas L. (1994) The dissolution of naturally weathered feldspar and quartz. Geochim. Cosmochim. Acta. 58(21), 4601-4613.
Anterrieu O., Chouteau M., Aubertin M. (2010) Geophysical characterization of the largescale internal structure of a waste rock pile from a hard rock mine. Bull. Geol. Eng. Environ. 69, 533-548.
Aubertin M., Ricard J.-F., Chapuis R.P. (1998) A predictive model for the water retention curve: application to tailings from hard-rock minesю Can. Geotech. J. 35, 55-69 (with Erratum 36, 401).
Binning P.J., Postma D., Russell T.F., Wesselingh J.A., Boulin P.F. (2007) Advective and diffusive contributions to reactive gas transport during pyrite oxidation in the unsaturated zone. Water Resour. Res. 43(2), art. no. W02414.
Blackmore S., Vriens B., Sorensen M., Power I.M., Smith L., Hallam S.J., Mayer K.U., Beckie R.D. (2018) Microbial and geochemical controls on waste rock weathering and drainage quality. Sci. Total Env. 640–641, 1004-1014.
Bouchemella S., Seridi A., Alimi-Ichola I. (2015) Numerical simulation of water flow in unsaturated soils: Comparative study of different forms of Richards’s equation. Eur. J. Env. Civil Eng. 19(1), 1-26.
Bouffard S.C., Rivera-Vasquez B.F., Dixon D.G. (2006) Leaching kinetics and stoichiometry of pyrite oxidation from a pyrite-marcasite concentrate in acid ferric sulfate media. Hydrometallurgy. 84(3-4), 225-238.
Bowell R.J., Williams K.P., Connelly R.J., Sadler P.J.K., Dodds J.E. (1999) Chemical containment of mine waste. Geol. Soc. Special Publ. 157, 213-240.
Brookfield A.E., Blowes D.W., Mayer K.U. (2006) Integration of field measurements and reactive transport modelling to evaluate contaminant transport at a sulfide mine tailings impoundment. J. Contam. Hydrol. 88(1–2), 1-22.
Caldeira C.L., Ciminelli V.S.T., Osseo-Asare K. (2010) The role of carbonate ions in pyrite oxidation in aqueous systems. Geochim. Cosmochim. Acta. 74(6), 1777-1789.
Chandra A.P., Gerson A.R. (2010) The mechanisms of pyrite oxidation and leaching: A fundamental perspective. Surf. Sci. Rep. 65(9), 293-315.
Chopard A., Plante B., Benzaazoua M., Bouzahzah H., Marion P. (2017) Geochemical investigation of the galvanic effects during oxidation of pyrite and base-metals sulfides. Chemosphere. 166, 281-291.
Demers I., Mbonimpa M., Benzaazoua M., Bouda M., Awoh S., Lortie S., Gagnon M. (2017) Use of acid mine drainage treatment sludge by combination with a natural soil as an oxygen barrier cover for mine waste reclamation: Laboratory column tests and intermediate scale field tests. Minerals Eng. 107, 43-52.
Diao Z., Shi T., Wang S., Huang X., Zhang T., Tang Y., Zhang X., Qiu R. (2013) Silane-based coatings on the pyrite for remediation of acid mine drainage. Water Res. 47(13), 4391-4402.
Dockrey J.W., Lindsay M.B.J., Mayer K.U., Beckie R.D., Norlund K.L.I., Warren L.A., Southam G. (2014) Acidic microenvironments in waste rock characterized by neutral drainage: Bacteria–mineral interactions at sulfide surfaces. Minerals. 4(1), 170-190.
Dold B. (2017) Acid rock drainage prediction: A critical review. J. Geochem. Explor. 172, 120-132.
Duxson P., Provis J.L., Lukey G.C., van Deventer J.S.J. (2007) The role of inorganic polymer technology in the development of ‘green concrete’. Cement Concr. Res. 37(12), 1590-1597.
Elbefling B., Nicholson R.V. (1996) Field determination of sulphide oxidation rates in mine tailings. Water Resour. Res. 32(6), 1773-1784.
Elsetinow A.R., Borda M.J., Schoonen M.A.A., Strongin D.R. (2003) Suppression of pyrite oxidation in acidic aqueous environments using lipids having two hydrophobic tails. Adv. Env. Res. 7(4), 969-974.
Erguler G.K., Erguler Z.A., Akcakoca H., Ucar A. (2014) The effect of column dimensions and particle size on the results of kinetic column test used for acid mine drainage (AMD) prediction. Miner. Eng. 55, 18-29.
Evangelou V.P., Zhang Y.L. (1995) A review: Pyrite oxidation mechanisms and acid mine drainage prevention. Crit. Rev. Env. Sci. Technol. 25(2), 141-199.
Fala O., Molson J., Aubertin M., Bussière B. (2005) Numerical modelling of flow and capillary barrier effects in unsaturated waste rock piles. Mine Water Environ. 24(4), 172-185.
Fala O., Molson J., Aubertin M., Dawood I., Bussière B., Chapuis R.P. (2013) A numerical modelling approach to assess long-term unsaturated flow and geochemical transport in a waste rock pile. Int. J. Mining, Reclamation and Environment. 27(1), 38-55.
Fan R., Short M.D., Zeng S.-J., Qian G., Li J., Schumann R.C., Kawashima N., Smart R.S.C., Gerson A.R. (2017) The formation of silicate-stabilized passivating layers on pyrite for reduced acid rock drainage. Environ. Sci. Technol. 51(19), 11317-11325.
Gautier J.-M., Oelkers E.H., Schott J. (2001) Are quartz dissolution rates proportional to B.E.T. surface areas? Geochim. Cosmochim. Acta. 65(7), 1059–1070.
Gerke H.H., Molson J.W., Frind, E.O. (1998) Modelling the effect of chemical heterogeneity on acidification and solute leaching in overburden mine spoils. J. Hydrol. 209, 166-185.
Ghorbani Y., Becker M., Mainza A., Franzidis J.-P., Petersen J. (2011) Large particle effects in chemical/biochemical heap leach processes – A review. Miner. Eng. 24, 1172-1184.
Hakkou R., Benzaazoua M., Bussière B. (2009) Laboratory Evaluation of the Use of Alkaline Phosphate Wastes for the Control of Acidic Mine Drainage. Mine Water and the Environment. 28(3), 206.
Heidel C., Tichomirowa M., Junghans M. (2013) Oxygen and sulfur isotope investigations of the oxidation of sulfide mixtures containing pyrite, galena, and sphalerite. Chem. Geol. 342, 29-43.
Huang X., Evangelou, V.P. (1992) Abatement of Acid Mine Drainage by Encapsulation of Acid Producing Geologic Materials. U.S. Department of the Interior, Bureau of Mines, Contact No. J0309013.
Igarashi T., Saito R., Sarashina M., Asakura K. (2006) Impoundment of excavated pyrite-bearing rock using silty covering soil. Clay Sci. 12, 143-148.
Jaynes D.B., Rogowski A.S., Pionke H. B. (1984) Acid mine drainage from reclaimed coal strip mines. I. Model description. Water Resour. Res. 20(2), 233-242.
Jerz J.K., Rimstidt J.D. (2004) Pyrite oxidation in moist air. Geochim. Cosmochim. Acta. 68(4), 701-714.
Jha R.K.T., Satur J., Hiroyoshi N., Ito M., Tsunekawa M. (2012) Suppression of pyrite oxidation by carrier microencapsulation using silicon and catechol. Miner. Process. Extr. Metal. Rev. 33(2), 89-98.
Jiang C.L., Wang X.H., Parekh B.K. (2000) Effect of sodium oleate on inhibiting pyrite oxidation. Int. J. Miner. Process. 58, 305-318.
Kargbo D.M., Atallah G., Chatterjee S. (2004) Inhibition of pyrite oxidation by a phospholipid in the presence of silicate. Environ. Sci. Technol. 38, 3432-3441.
Kimball B.E., Rimstidt J.D., Brantley S.L. (2010) Chalcopyrite dissolution rate laws. Appl. Geochem. 25(7), 972-983.
Kiventerӓ J., Lancellotti I., Catauro M., Poggetto F.D., Leonelli C., Illikainen M. (2018) Alkali activation as new option for gold mine tailings inertization. J. Clean. Prod. 187, 76-84.
Kohfahl C., Greskowiak J., Pekdeger A. (2007) Effective diffusion and microbiologic activity as constraints describing pyrite oxidation in abandoned lignite mines. Appl. Geochem. 22(1), 1-16.
Kollias K., Mylona E., Papassiopi N., Xenidis A. (2015) Conditions favoring the formation of iron phosphate coatings on the pyrite surface. Desalin. Water Treat. 56(5), 1274-1281.
Lefebvre R., Hockley D., Smolensky J., Gélinas P. (2001a) Multiphase transfer processes in waste rock piles producing acid mine drainage: 1. Conceptual model and system characterization. J. Contam. Hydrol. 52(3–4), 137-164.
Lefebvre R., Hockley D., Smolensky J., Lamontagne A. (2001b) Multiphase transfer processes in waste rock piles producing acid mine drainage: 2. Applications of numerical simulation. J. Contam. Hydrol. 52(3–4), 165-186.
León E.A., Rate A.W., Hinz C., Campbell G.D. (2004) Weathering of sulphide minerals at circum-neutral-pH in semi-arid/arid environments: Influence of water content. SuperSoil 2004: 3rd Australian New Zealand Soils Conference, University of Australia, 7p.
Liu Z.-S., Huang C., Ma L., Dy E., Xie Z., Tufa K., Fisher E.A., Zhou J., Morin K., Aziz M., Meints C., O’Kane M., Tallon L. (2019) The characteristic properties of waste rock piles in terms of metal leaching. J. Contam. Hydrol. 226, 103540.
Lorca M.E., Mayer K.U., Pedretti D., Smith L., Beckie R.D. (2016) Spatial and temporal fluctuations of pore-gas composition in sulfidic mine waste rock. Vadose Zone J. 15(10), 1-13.
McKibben M.A., Barnes H.L. (1986) Oxidation of pyrite in low temperature acidic solutions: Rate laws and surface textures. Geochim. Cosmochim. Acta. 50(7), 1509-1520.
Méndez-Ortiz B.A., Carrillo-Chávez A., Monroy-Fernández M.G. (2007) Acid rock drainage and metal leaching from mine waste material (tailings) of a Pb-Zn-Ag skarn deposit: environmental assessment through static and kinetic laboratory tests. Revista Mexicana Ciencias Geológicas. 24(2), 161-169.
Mielke R.E., Pace D.L., Porter T., Southam G. (2003) A critical stage in the formation of acid mine drainage: colonization of pyrite by Acidithiobacillus ferrooxidans under pH-neutral conditions. Geobiology. 1, 81-90.
Millero F.J., Sotolongo S., Izaguirre M. (1987) The oxidation kinetics of Fe(II) in seawater. Geochim. Cosmochim. Acta. 51(4), 793-801.
Molson J.W., Fala O., Aubertin M., Bussiere B. (2005) Numerical simulations of pyrite oxidation and acid mine drainage in unsaturated waste rock piles. J. Contam. Hydrol. 78(4), 343-371.
Molson J., Aubertin M., Bussière B., Benzaazoua M. (2008) Geochemical transport modelling of drainage from experimental mine tailings cells covered by capillary barriers. Appl. Geochem. 23(1), 1-24.
Moncur M.C., Ptacek C.J., Lindsay M.B.J., Blowes D.W., Jambor J.L. (2015) Long-term mineralogical and geochemical evolution of sulfide mine tailings under a shallow water cover. Appl. Geochem. 57, 178-193.
Mylona E., Xenidis A., Paspaliaris I. (2000) Inhibition of acid generation from sulphidic wastes by the addition of small amounts of limestone. Miner. Eng. 13(10–11), 1161-1175.
Naidu G., Ryu S., Thiruvenkatachari R., Choi Y., Jeong S., Vigneswaran S. (2019) A critical review on remediation, reuse, and resource recovery from acid mine drainage. Environ. Pollut. 247, 1110-1124.
Nicholson R.V., Gillham R.W., Reardon E.J. (1990) Pyrite oxidation in carbonate-buffered solution: 2. Rate control by oxide coatings. Geochim. Cosmochim. Acta. 54(2), 395-402.
Nordstrom D.K. (1985) The rate of ferrous iron oxidation in a stream receiving acid mine effluent. In: Selected Papers in the Hydrologic Sciences (ed. S. Subitzky). U.S. Geological Survey Water Supply Paper 2270, Washington, DC, 113-119.
Nordstrom D.K., Blowes D.W., Ptacek C.J. (2015) Hydrogeochemistry and microbiology of mine drainage: An update. Appl. Geochem. 57, 3-16.
Palandri J.L., Kharaka Y.K. (2004) A Compilation of Rate Parameters of Water-Mineral Interaction Kinetics for Application to Geochemical Modeling. Open file report 2004-1068. Menlo Park, U.S. Geological Survey.
Parisi D., Horneman J., Rastogi V. (1994) Use of bactericides to control acid mine drainage from surface operations. In: Proceedings of the International Land Reclamation and Mine Drainage Conference. U.S. Bureau of Mines SP 06B-94, Pittsburgh, PA, 319-325.
Park I., Tabelin C.B., Magaribuchi K., Seno K., Ito M., Hiroyoshi N. (2018a) Suppression of the release of arsenic from arsenopyrite by Carrier-microencapsulation using Ti-catechol complex. J. Hazard Mater. 344, 322-332.
Park I., Tabelin C.B., Seno K., Jeon S., Ito M., Hiroyoshi N. (2018b) Simultaneous suppression of acid mine drainage formation and arsenic release by Carrier-microencapsulation using aluminum-catecholate complexes. Chemosphere 205, 414-425.
Park I., Tabelin C.B., Jeon S., Li X., Seno K., Ito M., Hiroyoshi N. (2019) A review of recent strategies for acid mine drainage prevention and mine tailings recycling. Chemosphere. 219, 588-606.
Pedretti, D., Mayer, K.U., Beckie, R.D. (2017) Stochastic multicomponent reactive transport analysis of low quality drainage release from waste rock piles: Controls of the spatial distribution of acid generating and neutralizing minerals. J. Contam. Hydrol. 201, 30-38.
Pedretti, D., Mayer, K.U., Beckie, R.D. (2020) Controls of uncertainty in acid rock drainage predictions from waste rock piles examined through Monte-Carlo multicomponent reactive transport. Stochastic Environ. Res. Risk Assessment. 34(1), 219-233.
Pérez-López R., Cama J., Nieto J.M., Ayora C., Saaltink M.W. (2009) Attenuation of pyrite oxidation with a fly ash pre-barrier: Reactive transport modelling of column experiments. Appl. Geochem. 24(9), 1712-1723.
Pesic B., Oliver D.J., Wichlacz P. (1989) An electrochemical method of measuring the oxidation rate of ferrous to ferric iron with oxygen in the presence of Thiobacillus ferrooxidans. Biotechnol. Bioeng. 33, 428-439.
Pozo-Antonio S., Puente-Luna I., Lagüela-López S., Veiga-Ríos M. (2014) Techniques to correct and prevent acid mine drainage: A review. DINA. 81(184), 73-80.
Qiu G., Luo Y., Chen C., Lv Q., Tan W., Liu F., Liu C. (2016) Influence factors for the oxidation of pyrite by oxygen and birnessite in aqueous systems. J. Environ. Sci. 45, 164-176.
Reedy B.J., Beattie J.K., Lowson R.T. (1991) A vibrational spectroscopic 18O tracer study of pyrite oxidation. Geochim. Cosmochim. Acta. 55, 1609-1614.
Ren X., Zhang L., Ramey D., Waterman B., Ormsby S. (2015) Utilization of aluminum sludge (AS) to enhance mine tailings-based geopolymer. J. Mater. Sci. 50, 1370-1381.
Ribet I., Ptacek C.J., Blowes D.W., Jambor J.L. (1995) The potential for metal release by reductive dissolution of weathered mine tailings. J. Contam. Hydrol. 17, 239-273.
Rimstidt J.D., Vaughan D.J. (2003) Pyrite oxidation: a state-of-the-art assessment of the reaction mechanism. Geochim. Cosmochim. Acta. 67(5), 873-880.
Romano C.G., Mayer K.U., Jones D.R., Ellerbroek D.A., Blowes D.W. (2003) Effectiveness of various cover scenarios on the rate of sulfide oxidation of mine tailings. J. Hydrol. 271(1–4), 171-187.
Sapsford D.J., Bowell R.J., Dey M., Williams K.P. (2009) Humidity cell tests for the prediction of acid rock drainage. Miner. Eng. 22(1), 25-36.
Sasaki K., Tsunekawa M., Hasebe K., Konno H. (1995) Effect of anionic ligands on the reactivity of pyrite with Fe(III) ions in acid solutions. Colloids Surf. A. 101(1), 39-49.
Scharer J.M., Garga V., Smith R., Halbert B.E. (1991) Use of steady state models for assessing acid generation in pyritic mine tailings. In: The Second National Conference on the Abatement of Acidic Drainage. V. 2. Montreal, Canada, September 16 to 18, 1991, 211.
Seng S., Tabelin C.B., Kojima M., Hiroyoshi N., Ito M. (2019) Galvanic microencapsulation (GME) using zero-valent aluminum and zero-valent iron to suppress pyrite oxidation. Mater. Trans. 60(2), 277-286.
Sidkina E.S., Mironenko M.V., Cherkasova E.V. (2020) Application of equilibrium-kinetic modeling for predicting the chemical composition of subdump waters of the Udokan deposit (Russia). Geochem. Int. 58(13), 1419-1429.
Singer P.C., Stumm W. (1970) Acid mine drainage – the rate limiting step. Science. 167(3921), 1121-1123.
Skousen J.G., Ziemkiewicz P.F., McDonald L.M. (2019) Acid mine drainage formation, control and treatment: Approaches and strategies. Extr. Indust. Society. 6(1), 241-249.
Sobek A.A., Rastogi V., Benedetti D.A. (1990) Prevention of water pollution problems in mining: the bactericide technology. Int. J. Mine Water. 9(1–4), 133-148.
Sobek A.A., Schuller W.A., Freeman J.R., Smith R.M. (1978) Field and laboratory methods applicable to overburdens and minesoils. US EPA publication: EPA-600/2–78–054. Washington. 203 p.
Stumm W., Lee G.F. (1961) Oxygenation of ferrous iron. Ind. Eng. Chem. 53, 143-146.
Sun H., Chen M., Zou L., Shu R., Ruan R. (2015) Study of the kinetics of pyrite oxidation under controlled redox potential. Hydrometallurgy. 155, 13-19.
Tabelin C.B., Veerawattananun S., Ito M., Hiroyoshi N., Igarashi T. (2017a) Pyrite oxidation in the presence of hematite and alumina: I. Batch leaching experiments and kinetic modeling calculations. Sci. Total Environ. 580, 687-698.
Tabelin C.B., Veerawattananun S., Ito M., Hiroyoshi N., Igarashi T. (2017b) Pyrite oxidation in the presence of hematite and alumina: II. Effects on the cathodic and anodic half-cell reactions, Sci. Total Environ. 581–582, 126-135.
Tabelin C.B., Igarashi T., Villacorte-Tabelin M., Park I., Opiso E.M., Ito M., Hiroyoshi N. (2018) Arsenic, selenium, boron, lead, cadmium, copper, and zinc in naturally contaminated rocks: a review of their sources, modes of enrichment, mechanisms of release, and mitigation strategies. Sci. Total Environ. 645, 1522-1553.
Todd E.C., Sherman D.M., Purton J.A. (2003) Surface oxidation of pyrite under ambient atmospheric and aqueous (pH = 2 to 10) conditions: electronic structure and mineralogy from X-ray absorption spectroscopy. Geochim. Cosmochim. Acta. 67(5), 881-893.
Wang H., Dowd P.A., Xu C. (2019) A reaction rate model for pyrite oxidation considering the influence of water content and temperature. Miner. Eng. 134, 345-355.
Williamson M. A., Rimstidt J. D. (1994) The kinetics and electrochemical rate-determining step of aqueous pyrite oxidation. Geochim. Cosmochim. Acta. 58(24), 5443-5454.
Williamson M.A., Kirby C.S., Rimstidt J.D., (2006) Iron dynamics in acid mine drainage. In: 7th International Conference on Acid Rock Drainage 2006, ICARD. V. 3. American Society of Mining and Reclamation, St. Louis, 2411-2423.
Wilson D., Amos R.T., Blowes D.W., Langman J.B., Ptacek C.J., Smith L., Sego D.C. (2018a) Diavik Waste Rock Project: A conceptual model for temperature and sulfide content dependent geochemical evolution of waste rock – Laboratory scale. Appl. Geochem. 89, 160-172.
Wilson D., Amos R.T., Blowes D.W., Langman J.B., Smith L., Sego D.C. (2018b) Diavik Waste Rock Project: Scale-up of a reactive transport model for temperature and sulfide-content dependent geochemical evolution of waste rock. Appl. Geochem. 96, 177-190.
Wunderly M.D., Blowes D.W., Frind E.O., Ptacek C.J. (1996) Sulfide mineral oxidation and subsequent reactive transport of oxidation products in mine tailings impoundments: A numerical model. Water Resour. Res. 32(10), 3173-3187.
Yu J.-Y., Park M., Kim J. (2002) Solubilities of synthetic schwertmannite and ferrihydrite. Geochem. J. 36, 119-132.
Zhang X., Borda M.J., Schoonen M.A.A., Strongin D.R. (2003) Adsorption of phospholipids on pyrite and their effect on surface oxidation. Langmuir. 19, 8787-8792.
Zhou Y., Short M.D., Li J., Schumann R.C., Smart R.S.C., Gerson A.R., Qian G. (2017) Control of acid generation from pyrite oxidation in a highly reactive natural waste: A laboratory case study. Minerals 7, Art. 89.
Дополнительные материалы отсутствуют.