

Известия РАН. Энергетика, 2021, № 2, стр. 97-118
Теоретическое обобщение и развитие математического аппарата неизотермической кинетики
Е. А. Бойко 1, *, А. В. Страшников 1, **
1 ФГАОУ ВО “Сибирский федеральный университет”
Красноярск, Россия
* E-mail: EBoiko@sfu-kras.ru
** E-mail: savtr@mail.ru
Поступила в редакцию 01.12.2020
После доработки 15.02.2021
Принята к публикации 24.02.2021
Аннотация
Теоретически обоснована и экспериментально подтверждена применимость математического аппарата неизотермической кинетики для оценки реакционной способности энергетических углей и разработаны практические рекомендации по его использованию для экстраполяции результатов комплексного термического анализа на условия реальных теплотехнологических процессов и установок. Получено решение обобщенного уравнения неизотермической кинетики в интегральной форме, позволяющее теоретически оценить скорость реакции различных стадий термохимического превращения угля произвольного порядка в зависимости от температуры процесса при скорости нагрева угольных частиц в широком диапазоне от 10–2 до 104 град/мин.
ВВЕДЕНИЕ
При определении кинетических параметров процесса горения твердого органического топлива с дифференцированной оценкой процессов испарения влаги, термического разложения, а также выгорания и газификации коксовой основы, целесообразнее всего использовать единый комплекс методов термического анализа, позволяющий получить исчерпывающую информацию при исследовании образца. Многочисленными работами [1–4] предлагается использовать низкотемпературную (менее 900°С) область в качестве рабочей зоны при определении кинетических параметров в условиях непрерывного подъема температуры с линейной скоростью нагрева. Тем самым исключается влияние внешней диффузии при подводе окислителя к поверхности горения.
Непрерывная оценка скорости протекания основных процессов термохимического превращения твердого топлива и тем более их отдельных составляющих при высоких скоростях нагрева, присущих условиям реальных энерготехнологических установок по переработке твердого органического топлива, на существующем уровне экспериментальной техники не осуществима. В связи с этим рассматривается подход расчета длительности протекания основных процессов термохимической обработки топлива (сушки, термической деструкции с раздельной оценкой смоловыделения и выхода индивидуальных парогазовых компонентов, выгорания и газификации коксовой (нелетучей) основы, термохимического превращения минеральной части) по результатам определения кинетических характеристик при медленном нагреве с использованием математического аппарата неизотермической кинетики. Обоснование правомочности реализации данного подхода базируется на закономерностях влияния скорости нагревания на процесс термохимического превращения твердых органических топлив и на доказательстве правомочности использования для описания данного процесса теоретических основ неизотермической кинетики [2].
ОСОБЕННОСТЬ ПРОТЕКАНИЯ ПРОЦЕССОВ ТЕРМОХИМИЧЕСКОГО ПРЕВРАЩЕНИЯ ТОПЛИВА В УСЛОВИЯХ РЕАЛЬНЫХ ТОПЛИВОИСПОЛЬЗУЮЩИХ УСТАНОВОК
Термохимическое превращение угольного вещества связано с независимым протеканием большого числа реакций и образованием радикалов ароматической природы [3]. Возможной рекомбинацией радикалов обусловлены процессы синтеза (поликонденсации) осколков, идущие одновременно с деструкцией и ответственные за формирование коксовой структуры [4].
Образование жидких и газообразных продуктов термической деструкции является следствием отщепления структурных групп и низкомолекулярных соединений, входящих в состав элементарных структурных единиц, и их дальнейших преобразований [5]. Можно предположить, что каждый из продуктов деструкции твердого органического топлива обязан своим отщеплением одной или нескольким реакциям, для которых характерны различные, но изменяющиеся в узких пределах, значения энергии активации. Зависимость между количеством реакционных групп данного типа и прочностью их связи подчиняется статистическому закону гауссового распределения. Замена узкого распределения реакций с близкими кинетическими параметрами одной индивидуальной реакцией с усредненным значением энергии активации, впервые рассмотренная в работе Б.В. Нелюбина [6], близка к концепции “реакционного комплекса” с узким распределением E и k0 Г. Юнтгена [7]. Гипотеза об узком распределении энергии связей позволяет обоснованно выделить индивидуальные последовательно-параллельные реакции образования паро- и газообразных компонентов по мере подъема температуры и уточнить характер выделения летучих соединений в зависимости от молекулярного строения угольного вещества [8].
С другой стороны, термическое разложение угольного вещества необходимо рассматривать как развитие конкурирующих процессов – деструкции и поликонденсации [9]. Паро- и газообразные продукты термического разложения являются результатом участия элементарных структурных единиц угольного вещества в указанных процессах (в противоположность точки зрения С. Драйдена о возможном точном соответствии летучих веществ алифатическим структурам [10]). Особенно отчетливо это проявляется при анализе выделения углеводородных газов. По Х. Ойлерту [11] лишь начальный период выделения метана в первом приближении коррелирует с отщеплением CH3-групп исходного угля. Основная же его доля выделяется не за счет отщепления реакционных CH-групп, а в результате участия водорода в промежуточных реакционных актах. Для большинства твердых горючих ископаемых характерно выделение углеводородных газов (этана, этилена, пропана, пропилена) с порядком реакции больше единицы (n > 1) и со смещением максимума скорости в сторону более высоких температур по сравнению с Tmax основного периода выделения летучих веществ [12]. Значения n > 1 указывают на многоступенчатый характер образования углеводородных газов, который в значительной степени объясняется тем, что выделение углеводородных газов обусловлено не непосредственной деструкцией боковых цепей структурных единиц, а является результатом разложения промежуточных продуктов в период смолообразования [13]. Это предположение подтверждается и увеличенными значениями температуры максимума скорости выхода углеводородных газов по сравнению с положением температурного максимума основного периода выделения летучих веществ [14].
Основной период термического разложения, характеризующийся максимальной скоростью процесса, обусловлен главным образом выделением смол [2]. Если образование и выделение газообразных продуктов CO, CO2, H2, CH4, а также пирогенетической влаги, является следствием множества последовательно-параллельных реакций, то смоловыделение в большей мере отражает статистический характер разрыва молекулярных связей с узким интервалом распределения энергий активации и описывается, в первом приближении, одной реакцией первого порядка. Следует отметить, что анализ работ К. Крюгера [15] и Ван-Кревелена [16], которыми охватывается круг вопросов, связанный с термическим разложением отдельных петрографических микрокомпонентов каменных углей различной степени метаморфизма, показал, что основное отличие между микрокомпонентами в период выделения летучих веществ заключается в динамике выделения и составе углеводородных соединений, главным образом смол. Этот вывод подтвержден неоднократно Г.П. Алаевым как на отобранных из пласта петрографических разновидностях, так и на фракциях, обогащенных отдельными микрокомпонентами [17]. Суммарное выделение углеводородных соединений снижается в ряду “липтинит–витринит” (Lt–Vt) [18].
Приведенные выше данные относятся к результатам термического разложения угольного вещества с низкими скоростями нагрева (менее 20 град/мин). Однако известно, что при горении пылевидного твердого топлива в топочных условиях процесс выделения летучих веществ необходимо рассматривать как комплекс первичных деструктивных изменений органической массы угольного вещества и вторичных взаимодействий продуктов термического разложения с твердой фазой (коксовой основой). С уменьшением скорости нагрева роль вторичных процессов возрастает. Даже в интервале 2–20 град/мин при исследовании Кузбасских углей (марки Г) это отчетливо наблюдается на примере выделения водорода H2 [18]. Следует отметить, что увеличение смоловыделения при высокоскоростном нагреве разных марок углей отмечалось в работах [19–21]. По данным В. Петерса [20], на углях средней стадии метаморфизма смоловыделение в 1.6–1.8 раз выше, чем при стандартном полукоксовании в реторте Фишера. Причем основная доля повышенного выделения смол приходится на пековую составляющую. По данным П. Агарвала [19], содержание смол при высокоскоростном разложении угля с Vdaf ≈ 33% возрастает до 27% (вместо 16% при медленном нагреве). Увеличение скорости нагрева до ~106 град/с радиационный нагрев угольных частиц размером 3–5 мкм [21] приводит к смещению максимума скорости выделения жидких продуктов в область температур более 1000°С.
При медленном нагреве вторичный крекинг смол обуславливает снижение их суммарного выделения; одновременно возрастает выход газа (и искажается его компонентный состав), а также интенсифицируется коксообразование. Очень быстрый отвод летучих продуктов в условиях высокоскоростного нагрева снижает до минимума их вторичные преобразования. Изложенное выше позволяет сделать важное предположение, что изменение количества и состава летучих веществ в зависимости от скорости нагрева связано с протеканием вторичных процессов, а не с изменением механизма деструкции угольного вещества. Механизм образования отдельных парогазовых компонентов летучих веществ, в конечном счете, определяется физико-химическим молекулярным строением исходного угольного вещества.
В условиях высокоскоростного нагрева индивидуальные реакции газовыделения с узким распределением кинетических параметров протекают практически единовременно, а выделение основной массы продуктов деструкции смещается в область повышенных температур. Выдвинутая З.Ф. Чухановым схема последовательно-параллельных реакций предполагает возможность разделения выхода отдельных компонентов летучих веществ по температуре процесса [22].
Схожесть механизма термического разложения угольного вещества при экстремально различных скоростях нагрева может быть строго подтвержден неизменностью кинетических характеристик смоловыделения и индивидуальных реакций деструкции. Выполненные Г.П. Алаевым экспериментальные исследования [18] показали, что кинетические характеристики E и k0 смоловыделения Кузбасского газового угля в интервале скоростей нагрева 2–24 град/мин не изменяют своих значений. Соотношение же между реакциями деструкции и синтеза, вследствие различия констант их скоростей, изменяется на этом интервале существенно.
Известно, что выделение низкомолекулярных газообразных продуктов термической деструкции при медленном нагреве связано с протеканием последовательных по температуре и частично налагающихся друг на друга индивидуальных реакций. Так, в работе [2] показано, что выделение метана CH4 является следствием 6-ти реакций с различными кинетическими параметрами. Выделение CO2, CO и H2O протекает по 4‑м “единичным” реакциям. И только дифференциальные кривые углеводородных газов CnHm, выделение которых связано с образованием смол, могут быть описаны одной реакцией. Экспериментами Г. Юнтгена и его сотрудников [7] были подтверждены практически неизменные значения E и k0 для углеводородных газов при скоростях нагрева угля от 10–2 до 104 град/мин.
К этому следует добавить, что прецизионный анализ выделения углеводородных газов (до C10) при термическом разложении широкой гаммы углей, выполненный Ж. Элдером [23], показал неизменность механизма их образования и для различных по степени метаморфизма топлив (в интервале Cdaf= 70–90% температура максимума скорости их выхода меняется незначительно).
В настоящей работе в качестве возможного варианта рассматривается неизменный механизм термической деструкции угольного вещества при переходе от медленного (эксперимент, ~5–20 град/мин) к высокоскоростному (например, топочные условия, ~104–106 град/с) нагреву. Учитывая невозможность осуществления непрерывной оценки скорости выделения летучих веществ при высокоскоростном нагреве при проведении прямого эксперимента, целесообразно использовать расчетное определение длительности периода выделения летучих веществ по результатам кинетической оценки экспериментальных данных с медленным нагревом. Необходимым условием такого расчета является максимальное приближение к непрерывному анализу и использование математического аппарата неизотермической кинетики.
Выделившиеся горючие газообразные летучие компоненты при достижении достаточной концентрации стехиометрической смеси реагируют в газовой фазе с кислородом воздуха до получения конечных продуктов CO2 и H2O [24]. Смолы при недостатке окислителя могут повторно деструктировать, образуя вторичные продукты, в частности, ацетилен, бензол, водород и сажу, которые, в свою очередь, также вступают в реакцию с кислородом воздуха, генерируя продукты полного (CO2 и H2O) и неполного горения (например, CO) с последующим их догоранием [25].
В процессе выделения летучих веществ и их последующего горения происходит нагрев угольной частицы, сопровождающийся инициализацией процесса горения ее нелетучей (коксовой) основы. Реакция протекает на поверхности угольной частицы, в результате которой из газового объема кислород воздуха адсорбируется на поверхности углерода. На ней атомы кислорода вступают в химическую реакцию, образуя сложные углерод-кислородные комплексы CxOy [24]. Последние затем распадаются с получением продуктов полного CO2 и неполного CO горения.
Водяные пары H2O и углекислый газ CO2, образующиеся в результате протекания процессов сушки, возгонки балластных летучих, горения горючих летучих и коксовой основы топлива, а также в результате догорания неполных продуктов сгорания могут реагировать с твердой фазой, образуя CO и H2. Эти реакции протекают по радикально-цепному механизму при непосредственном участии гидроксилов как промежуточных соединений с появлением комплекса HCOH, а затем и валентных донорно-акцепторных связей с атомарным углеродом [2]. Вследствие соударения атомов и молекул с вакантными местами и благодаря наличию валентных сил на активных местах поверхности образуются промежуточные комплексы. Последние стимулируют электронный перенос, обеспечивая хемосорбцию газообразных компонентов на поверхность коксовой основы.
Получающийся в результате вторичных реакций водород может вступать в химические реакции: с углеродом до образования неполного продукта горения CH4 и кислородом до H2O [14]. Метан в газовой фазе окисляется до CO2 и H2O. Продукты полного H2O и неполного CO горения могут взаимодействовать между собой, образуя CO2 и H2 [27].
НЕИЗОТЕРМИЧЕСКАЯ КИНЕТИКА ПРОЦЕССА ТЕРМИЧЕСКОЙ ДЕСТРУКЦИИ ТВЕРДЫХ ОРГАНИЧЕСКИХ ТОПЛИВ
Классическая кинетика при описании термохимических превращений веществ раздельно рассматривает влияние температуры и концентрации реагирующих веществ на скорость процесса. При этом не учитывается изменение концентрации в зависимости от температуры. Неизотермическая кинетика лишена этого недостатка [28].
Полагаем, что процесс термического разложения (деструкции) твердых горючих ископаемых протекает как гомогенная мономолекулярная реакция. Механизм мономолекулярного разложения обусловлен образованием активированной молекулы A+ с последующим ее разложением и дезактивацией:
Скорость процесса, представляющая собой первую производную концентрации разлагающегося вещества по времени, описывается уравнением:
где k – константа скорости реакции; $C_{{{{A}^{ + }}}}^{{}}$ – доля непрореагировавшего к данному моменту времени вещества (остаточная концентрация реагирующего вещества); τ – продолжительность (время) реакции; n – порядок реакции.Следует отметить, что возможность протекания мономолекулярной реакции по 1‑му порядку не очевидна [29]. При достаточно большой концентрации реагирующего вещества между активированными (за счет флуктуаций энергии колебания – комплекс M в уравнении (1)) и неактивированными молекулами существует статическое равновесие, т.е. процессы активации и дезактивации протекают с почти равными скоростями. Вследствие этого концентрация активных молекул, а, следовательно, и скорость реакции, пропорциональна общей концентрации реагирующих молекул. В таком случае будем иметь 1-й порядок реагирования.
С уменьшением концентрации реагирующего вещества, сопровождающееся увеличением интервала между актами активации, активированные молекулы успевают прореагировать, что приближает реакцию ко 2-му порядку.
Можно полагать, что видимый порядок реакции термической деструкции (а также отдельных ее стадий) находится в диапазоне 1 ≤ n ≤ 2. В частности, выделение паро- и газообразных продуктов CO, CO2, H2O при термической деструкции карбоновых кислот и их ангидридов протекает по 1-му порядку относительно концентрации реакционноспособных групп в твердой фазе [30]. Найденные при этом значения энергии активации E и предъэкспоненциального множителя k0 указывают на то, что разрыв химических связей (т.е. деструкция основных молекул) является определяющим фактором процесса.
Принимая температурную зависимость константы скорости k, по уравнению Аррениуса [18], можно констатировать, что скорость реакции, описываемая по уравнению (2), определяется двумя величинами: температурой и концентрацией реагирующего вещества. Классическая кинетика, как уже отмечалось, предполагает первоначально оценивать влияние одного из них – концентрации – при T = const, а затем из опытов с различной температурой определяется влияние второго параметра – температуры процесса. В соответствии с этой схемой интегральный метод анализа уравнения (2) сводится к следующему:
а) замена параметра ${{C}_{{{{A}^{ + }}}}}$ на (C0 – C), где C0 – начальная концентрация реагирующего вещества, а C – его разложившаяся к моменту времени τ часть;
б) преобразование уравнения (2) до вида
с последующим его анализом.Разделив переменные и проинтегрировав, получим
(4)
$\frac{1}{{n - 1}}\left[ {\frac{1}{{{{{\left( {{{C}_{0}} - C} \right)}}^{{n - 1}}}}} - \frac{1}{{C_{0}^{{n - 1}}}}} \right] = k\tau $(5)
${{\left( {{{C}_{0}} - C} \right)}^{{n - 1}}} = \frac{{C_{0}^{{n - 1}}}}{{1 + \left( {n - 1} \right)C_{0}^{{n - 1}}k\tau }}.$Для реакции 1-го порядка имеем экспоненту
и, следовательно, после подстановки в уравнение (3) получимУравнением (7) описывается скорость реакции при n = 1 в зависимости от ее длительности τ при постоянной температуре T = const.
В уравнении (6) при решении его относительно текущей концентрации прореагировавшего вещества имеем
Значение C из уравнения (8) при оценке термического разложения приравнивается суммарному количеству летучих веществ, выделившихся к моменту времени τ. Учитывая, что в образовании летучих участвует не одна, а несколько групп реакций [31], предлагается для изотермических условий использовать следующее выражение:
(9)
$V = \sum\limits_{i = 1}^m {{{{\left( {{{C}_{0}}} \right)}}_{i}}\left( {1 - \exp \left( { - {{k}_{i}}\tau } \right)} \right)} ,$Уравнением (9) суммарная кинетика процесса термического разложения учитывается не однокомпонентной, многокомпонентной схемой расчета. Если температура является функцией времени, то уравнение (9) нередко решается в виде [32]
(10)
$С = {{С}_{0}}\left( {1 - \exp \left( { - \int\limits_0^{{\tau }} {{{k}_{0}}\exp \left( { - \frac{E}{{RT(\tau )}}} \right)d\tau } } \right)} \right).$Интеграл в показателе степени этого уравнения вычисляется М. Девидом [33] для случая линейного закона изменения температуры по времени.
Использование уравнения (10) для оценки температурной зависимости текущей концентрации $С$ некорректно. В условиях непрерывного нагрева изменяется не только константа скорости, но и, собственно, концентрация реагирующего вещества.
При обращении к неизотермической кинетике введем обозначение скорости нагрева (темп нагрева) $\beta = \frac{{dT}}{{d\tau }}.$
Преобразуем уравнение (3) до вида
(11)
$\frac{{dC}}{{dT}} = \frac{{{{k}_{0}}}}{\beta }\exp \left( { - \frac{E}{{RT}}} \right){{\left( {{{C}_{0}} - C} \right)}^{n}}.$Уравнение (11) является обобщенным уравнением неизотермической кинетики. Для большинства мономолекулярных реакций предэкспоненциальный множитель (частотный или вероятностный фактор) k0 имеет близкое к средней частоте колебания молекул (1013–1014, 1/с). Объяснимо и более высокое значение k0 – до 1017 1/с – в предположении, что определенные группы молекул в активизированных комплексах имеют вращательную степень свободы (преимущественно для реакций со свободными радикалами).
Из уравнения (11) следует, что скорость реакции, отнесенная к одному градусу, обратно пропорциональна скорости нагрева β. Следствием этого является сдвиг реакции в область повышенных температур с увеличением скорости нагрева. Одновременно расширяется температурная область протекания реакции и снижается максимальная скорость процесса [34].
Для определения основных кинетических параметров (n, E, k0) в процессе нагрева топлива с постоянной скоростью разделим переменные в уравнении (11):
(12)
$\frac{{dC}}{{{{{\left( {{{C}_{0}} - C} \right)}}^{n}}}} = \frac{{{{k}_{0}}}}{\beta }\exp \left( { - \frac{E}{{RT}}} \right)dT.$Проинтегрировав уравнение (12), получим [2]:
(13)
$\int\limits_0^C {\frac{{dC}}{{{{{\left( {{{C}_{0}} - C} \right)}}^{n}}}} = \frac{{{{k}_{0}}}}{\beta }\int\limits_0^T {\exp \left( { - \frac{E}{{RT}}} \right)dT} } .$Значение интеграла в левой части уравнения (13) зависит от порядка реакции
(14)
${\text{при}}\,\,\,\,n = {\text{1}}{\kern 1pt} :\,\,\int\limits_0^C {\frac{{dC}}{{{{{\left( {{{C}_{0}} - C} \right)}}^{n}}}} = \ln \frac{{{{C}_{0}}}}{{{{C}_{0}} - C}}} ,$(15)
${\text{при}}\,\,\,\,n = {\text{2}}{\kern 1pt} :{\text{ }}\int\limits_0^C {\frac{{dC}}{{{{{\left( {{{C}_{0}} - C} \right)}}^{n}}}}} = \frac{C}{{{{C}_{0}}\left( {{{C}_{0}} - C} \right)}}.$В общем случае,
(16)
${\text{при}}\,\,\,\,n \ne {\text{1}}{\kern 1pt} :\,\,\int\limits_0^C {\frac{{dC}}{{{{{\left( {{{C}_{0}} - C} \right)}}^{n}}}}} = \frac{1}{{n - 1}}\frac{{C_{0}^{{n - 1}} - {{{\left( {{{C}_{0}} - C} \right)}}^{{n - 1}}}}}{{{{{\left[ {{{C}_{0}}\left( {{{C}_{0}} - C} \right)} \right]}}^{{n - 1}}}}}.$Значение интеграла в правой части уравнения (16) не зависит от порядка реакции, но и не может быть получено в элементарных функциях. Приближенное значение $\int_0^T {\exp \left( { - \frac{E}{{RT}}} \right)dT} $ вычислялось Ван-Кревеленом [16] в предположении, что период основного процесса термического разложения ограничен узким температурным интервалом вблизи температуры, соответствующей максимальной скорости процесса Tmax, а именно – в интервале от 0.8 до 1.3 Tmax. При этом использовалось следующее последовательное приближение:
(17)
$\exp \left( { - \frac{E}{{RT}}} \right) \approx {{\left( {\exp \left( { - \frac{{{{T}_{{\max }}}}}{T}} \right)} \right)}^{{\frac{E}{{R{{T}_{{\max }}}}}}}} \approx {{\left( {x\frac{T}{{{{T}_{{\max }}}}}} \right)}^{{\frac{E}{{R{{T}_{{\max }}}}}}}}.$Константа x определяется по тангенсу угла наклона прямой в координатах $\exp \left( { - \frac{1}{T}} \right) = f(T)$ и численно равна 0.368, а интеграл $\int_0^T {\exp \left( { - \frac{E}{{RT}}} \right)dT} $ может быть решен в виде
(18)
$\int\limits_0^T {\exp \left( { - \frac{E}{{RT}}} \right)dT} = \int\limits_0^T {{{{\left( {x\frac{T}{{{{T}_{{\max }}}}}} \right)}}^{{\frac{E}{{R{{T}_{{\max }}}}}}}}dT} \, = \,\int\limits_0^T {{{{\left( {\frac{x}{{{{T}_{{\max }}}}}} \right)}}^{{\frac{E}{{R{{T}_{{\max }}}}}}}}{{T}^{{\frac{E}{{R{{T}_{{\max }}}}}}}}dT} = {{\left( {\frac{x}{{{{T}_{{\max }}}}}} \right)}^{{\frac{E}{{R{{T}_{{\max }}}}}}}}\frac{{{{T}^{{\frac{E}{{R{{T}_{{\max }}}}} + 1}}}}}{{\frac{E}{{R{{T}_{{\max }}}}} + 1}}.$Порядок реакции, описываемой уравнением (16), может быть найден из зависимости $\ln \int_0^C {\frac{{dC}}{{{{{\left( {{{C}_{0}} - C} \right)}}^{n}}}}} = f\left( {\ln T} \right).$
Прямая линия в этих координатах отвечает истинному порядку реакции. Однако для оценки n можно использовать и приближенное аналитическое решение [16].
В общем случае, при n ≠ 1:
(19)
$\ln \int\limits_0^C {\frac{{dC}}{{{{{\left( {{{C}_{0}} - C} \right)}}^{n}}}}} \approx \ln \left( {\frac{{1 - {{{\left( {{{C}_{0}} - C} \right)}}^{{1 - n}}}}}{{1 - n}}} \right)$(20)
$\ln \left( {\frac{{1 - {{{\left( {{{C}_{0}} - C} \right)}}^{{1 - n}}}}}{{1 - n}}} \right) = \left( {\frac{E}{{R{{T}_{{\max }}}}} + 1} \right)\ln \frac{T}{{{{T}_{{\max }}}}}.$По данным Ван-Кревелена, экспериментальная оценка термического разложения каменного угля с Vdaf = 25% в температурном интервале 300–500°С при скорости нагрева 2 град/мин – использовался термогравиметрический анализ с графическим дифференцированием суммарной кривой потери веса – показала, что основной период термического разложения (смоловыделение) подчиняется реакции 1-го порядка: в координатах $\ln \int_0^C {\frac{{dC}}{{{{{\left( {{{C}_{0}} - C} \right)}}^{n}}}}} = f\left( {\ln T} \right)$ этот процесс представлен прямой линией.
Значение интеграла $\int_0^T {\exp \left( { - \frac{E}{{RT}}} \right)dT} $ в правой части уравнения (13) может быть найдено также путем использования вспомогательной переменной $j = \frac{E}{{RT}}$ и интегрирования его по частям [35]:
(21)
$\int\limits_0^T {\exp \left( { - \frac{E}{{RT}}} \right)dT} = T\exp \left( { - \frac{E}{{RT}}} \right) + \frac{E}{R}{{\xi }_{i}}\left( { - \frac{E}{{RT}}} \right).$(22)
${{\xi }_{i}}\left( { - j} \right) = - \frac{{\exp \left( { - j} \right)}}{j}\left( {1 - \frac{{1!}}{j} + \frac{{2!}}{{{{j}^{2}}}} - \frac{{3!}}{{{{j}^{3}}}} + ...} \right).$В работе [36] на примере термодеструкции полимеров показано, что точность значений ξi – функций, приведенных в различных пособиях по численным методам [37], достаточна для кинетических расчетов. Это же было подтверждено и авторами работы [38] при исследовании кинетики термического разложения углей.
Опыт практических вычислений показал, что в уравнении (22) можно пренебречь всеми членами в скобках, кроме двух первых
(23)
${{\xi }_{i}}\left( { - j} \right) = - \frac{{\exp \left( { - j} \right)}}{j}\left( {1 - \frac{{1!}}{j} + \frac{{2!}}{{{{j}^{2}}}}} \right).$С учетом уравнения (3.23) получим:
(24)
$\int\limits_0^T {\exp \left( { - \frac{E}{{RT}}} \right)dT} \approx \frac{{R{{T}^{2}}}}{E}\left( {1 - \frac{{2RT}}{E}} \right)\exp \left( { - \frac{E}{{RT}}} \right).$При больших значениях j можно оставить только первый член в скобках, тогда выражение (24) упростится до вида
(25)
$\int\limits_0^T {\exp \left( { - \frac{E}{{RT}}} \right)dT} \approx \frac{{R{{T}^{2}}}}{E}\exp \left( { - \frac{E}{{RT}}} \right).$В отличие от уравнения (18) значение интеграла $\int_0^T {\exp \left( { - \frac{E}{{RT}}} \right)dT} ,$ найденное по уравнениям (24) или (25), не связано с температурой максимума дифференциальной кривой и носит более обобщенный характер [39].
Решение обобщенного уравнения неизотермической кинетики в интегральной форме (уравнение 13) позволяет теоретически оценить скорость реакции термической деструкции любого порядка в зависимости от температуры процесса. Найденное по уравнению (13) значение C – текущей концентрации разложившегося вещества при температуре T – после подстановки в исходное уравнение (11) позволяет представить обобщенное уравнение неизотермической кинетики в окончательном виде. С учетом уравнений (14–16) и (24) получим:
(26)
${\text{при}}\,\,\,\,n = 0{\kern 1pt} :\,\,\,\frac{{dC}}{{dT}} = \frac{{{{k}_{0}}}}{\beta }\exp \left( { - \frac{E}{{RT}}} \right),$(27)
$\begin{gathered} {\text{при}}\,\,\,\,n = {\text{1}}{\kern 1pt} :\,\,\,\frac{{dC}}{{dT}} = \frac{{{{k}_{0}}}}{\beta }\exp \left( { - \frac{E}{{RT}}} \right){{C}_{0}}\exp \left( { - \frac{{{{k}_{0}}}}{\beta }\int\limits_0^T {\exp \left( { - \frac{E}{{RT}}} \right)dT} } \right) = \\ = \frac{{{{k}_{0}}{{C}_{0}}}}{\beta }\exp \left\{ { - \left( {\frac{E}{{RT}} + \frac{{{{k}_{0}}}}{\beta }\frac{{R{{T}^{2}}}}{E}\left( {1 - \frac{{2RT}}{E}} \right)\exp \left( { - \frac{E}{{RT}}} \right)} \right)} \right\}, \\ \end{gathered} $(28)
$\begin{gathered} {\text{при}}\,\,\,n = {\text{2}}{\kern 1pt} :\,\,\,\frac{{dC}}{{dT}} = \frac{{{{k}_{0}}}}{\beta }\exp \left( { - \frac{E}{{RT}}} \right)\frac{1}{{{{{\left( {\frac{1}{{{{С}_{0}}}} + \frac{{{{k}_{0}}}}{\beta }\int\limits_0^T {\exp \left( { - \frac{E}{{RT}}} \right)dT} } \right)}}^{2}}}} = \\ = \frac{{{{k}_{0}}C_{0}^{2}}}{\beta }\frac{{\exp \left( { - \frac{E}{{RT}}} \right)}}{{{{{\left[ {1 + \frac{{{{k}_{0}}{{C}_{0}}}}{\beta }\frac{{R{{T}^{2}}}}{E}\left( {1 - \frac{{2RT}}{E}} \right)\exp \left( { - \frac{E}{{RT}}} \right)} \right]}}^{2}}}}. \\ \end{gathered} $В общем случае,
(29)
${\text{при}}\,\,n \ne {\text{1}}{\kern 1pt} :\,\,\,\frac{{dC}}{{dT}} = \frac{{{{k}_{0}}}}{\beta }\exp \left( { - \frac{E}{{RT}}} \right)\frac{{C_{0}^{n}}}{{{{{\left[ {1 + \left( {n - 1} \right)\frac{{{{k}_{0}}C_{0}^{{n - 1}}}}{\beta }\frac{{R{{T}^{2}}}}{E}\left( {1 - \frac{{2RT}}{E}} \right)\exp \left( { - \frac{E}{{RT}}} \right)} \right]}}^{{\frac{n}{{n - 1}}}}}}}.$При любом порядке реакции ее скорость определяется температурой. Из уравнений (26–29) следует, что во всех случаях имеется сомножитель $\frac{{{{k}_{0}}}}{\beta }\exp \left( { - \frac{E}{{RT}}} \right),$ учитывающий температурную зависимость скорости реакции по уравнению Аррениуса. С увеличением температуры его величина возрастает от нуля до предельного значения $\frac{{{{k}_{0}}}}{\beta }$ при условии, что T → ∞. Остальные члены этих уравнений (кроме, разумеется, уравнения нулевого порядка) представляют собой 2-ой сомножитель, отражающий зависимость скорости процесса от концентрации, которая к тому же сама является функцией температуры. Именно это обстоятельство не учитывает формальное уравнение (10), полученное на основе описания изотермического процесса. Влияние этого сомножителя в значительной мере определяется порядком реакции. Оно тем меньше, чем выше значение n, но всегда отлично от нуля, поскольку при T → ∞ и $\int_0^T {\exp \left( { - \frac{E}{{RT}}} \right)dT} \to \infty .$
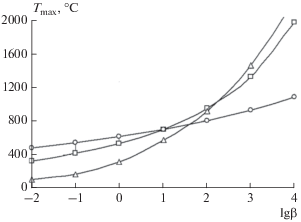
Во всех случаях, кроме n = 0, скорость процесса в зависимости от температуры имеет максимум; при n = 0 она изменяется по экспоненте. Этот случай нетипичен для термической деструкции, вследствие чего реакции нулевого порядка можно в дальнейшем исключить из рассмотрения. Детальный анализ уравнений (26–29) позволяет выявить влияние параметров n, E, k0, β на характер изменения скорости реакции в неизотермических условиях. В работах [2, 40] показано, что с увеличением скорости нагрева, β, возрастает и скорость реакции при неизменных k0, E и n. Никакого изменения концентрации разлагающегося вещества при изменении скорости нагрева не наблюдается, происходит лишь смещение реакции в область повышенных температур. При этом температура, соответствующая максимальной скорости выделения летучих веществ, возрастает. На рисунке 1 приведена зависимость Tmax= f(lg(β)) применительно к выделению различных газовых компонентов с индивидуальными значениями кинетических характеристик с различными скоростями нагрева.
Рис. 1.
Температурное положение максимума скорости выделения газообразных компонентов с индивидуальными кинетическими параметрами при термическом разложении угля в зависимости от скорости нагрева: ⚪ – E = 180 кДж/моль, k0 = 109 1/с; ◻ – E = 75 кДж/моль, k0 = 103 1/с; Δ – E = 25 кДж/моль, k0 = 1.5 1/с.
Изменение E, k0 и β однотипно отражается на дифференциальных кривых термической деструкции – максимум кривой ${{\left( {\frac{{dC}}{{dT}}} \right)}_{{\max }}}$ смещается в сторону более высоких температур с увеличением E и β и с уменьшением k0, при одновременном снижении абсолютных значений максимума и увеличении ширины профиля скорости процесса (независимо от значений порядка реакции, n). Изменения температуры максимума, Tmax и абсолютной величины максимума дифференциальной кривой при постоянных E, k0 и n = 1 связаны прямолинейной зависимостью в координатах $\lg {{\left( {\frac{{dC}}{{dT}}} \right)}_{{\max }}} = f\left( {\lg {{T}_{{\max }}}} \right).$
Наклон же прямой определяется значениями E и k0. В связи с этим сопоставление реакций 1-го порядка с различными кинетическими параметрами в указанной зависимости несколько затруднено. Иначе обстоит дело, если преобразовать ее до вида $\lg {{\left( {\frac{{dC}}{{d\tau }}} \right)}_{{\max }}} = f\left( {\lg {{\tau }_{{\max }}}} \right),$ тем самым учитывая непосредственно скорость нагрева, β. Действительно, $\frac{{dC}}{{d\tau }} = \frac{{dC}}{{dT}}\beta $ и ${{\tau }_{{\max }}} = \frac{{{{T}_{{\max }}}}}{\beta },$ где τmax – время, длительность нагрева от 0°С до Tmax. В координатах $\lg {{\left( {\frac{{dC}}{{d\tau }}} \right)}_{{\max }}} = f\left( {\lg {{\tau }_{{\max }}}} \right)$ изменение кинетических характеристик E и k0 приводит лишь к параллельному смещению прямой, не меняя ее наклона, который при одинаковой масштабности абсциссы и ординаты графика, всегда остается равным 45° (рис. 2). На данном рисунке приводятся результаты обработки гипотетических реакций выделения летучих веществ с различными кинетическими характеристиками при n = 1 и β = var.
Рис. 2.
Зависимость максимальной скорости реакции 1-го порядка от времени нагрева до значений Tmax: ⚪ – E = 160 кДж/моль, k0 = 109 1/с; Δ – E = 200 кДж/моль, k0 = 108 1/с.
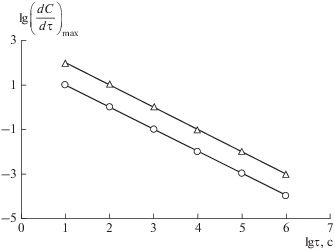
Зависимость $\lg {{\left( {\frac{{dC}}{{d\tau }}} \right)}_{{\max }}} = f\left( {\lg {{\tau }_{{\max }}}} \right)$ является весьма важной. Так с ее помощью можно сделать вывод о том, что в случае реакции 1-го порядка максимальная скорость ${{\left( {\frac{{dC}}{{d\tau }}} \right)}_{{\max }}}$ меняется в зависимости от времени нагрева до Tmax по гиперболе. Появляется возможность определения абсолютных значений ${{\left( {\frac{{dC}}{{d\tau }}} \right)}_{{\max }}}$ при любых скоростях нагрева, следующих линейному закону, по одной единственной экспериментальной точке при условии реагирования по 1-му порядку и постоянства кинетических параметров [41].
Этот вывод действителен именно для максимальной скорости реакции, так как в связи с увеличением ширины профиля дифференциальной кривой и увеличением ее асимметричности с ростом скорости нагрева β, промежуточные значения на ее крыльях отклоняются от гиперболического закона.
Способов оценки порядка реакции в практике используется достаточно много, однако, на наш взгляд, предпочтение следует отдать методу определения n, исходя из предположения о связи порядка реакции с остаточной концентрацией непрореагировавшего вещества в точке максимума дифференциальной кривой [42].
Исходное уравнение неизотермической кинетики – уравнение (11) после интегрирования можно представить в виде
(30)
$\frac{{{{{\left( {\frac{{{{C}_{0}} - C}}{{{{C}_{0}}}}} \right)}}^{{1 - n}}} - 1}}{{n - 1}} = \frac{{{{k}_{0}}}}{\beta }C_{0}^{{n - 1}}\int\limits_0^T {\exp \left( { - \frac{E}{{RT}}} \right)dT} .$С другой стороны, для максимума дифференциальной кривой, описываемой в общем случае уравнением 29, после дифференцирования этого уравнения по температуре и приравнивания второй производной нулю, можно установить связь между k0, E, Tmax и β:
(31)
$\frac{\beta }{{RT_{{\max }}^{2}}} = \frac{{{{k}_{0}}C_{0}^{{n - 1}}}}{E}\exp \left( { - \frac{E}{{R{{T}_{{\max }}}}}} \right).$Уравнение для реакции 1-го порядка полученное аналогичным способом имеет вид
(32)
$\frac{{{{k}_{0}}}}{\beta } = \frac{E}{{RT_{{\max }}^{2}}}\exp \left( {\frac{E}{{R{{T}_{{\max }}}}}} \right),$(33)
${{\left[ {\frac{{{{C}_{0}} - C}}{{{{C}_{0}}}}} \right]}_{{{{T}_{{\max }}}}}} = {{n}^{{\frac{1}{{1 - n}}}}}.$Графическая зависимость $\left[ {\frac{{{{C}_{0}} - C}}{{{{C}_{0}}}}} \right]$ при Tmax как функция от порядка реакции n, полученная расчетным путем, является “градуировочным” графиком (рис. 3), позволяющим определять порядок реакции по остаточной концентрации непрореагировавшего вещества в точке максимума экспериментальной дифференциальной кривой.
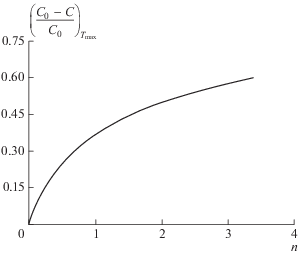
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ НЕИЗОТЕРМИЧЕСКОЙ КИНЕТИКИ ПРОЦЕССА ГОРЕНИЯ КОКСОВОЙ ОСНОВЫ ТВЕРДЫХ ОРГАНИЧЕСКИХ ТОПЛИВ
Преимущества неизотермической кинетики (одновременный учет изменения температуры процесса и концентрации реагирующих компонентов) делают перспективным ее применение и для реакций, протекающих на поверхности топливной частицы. В области низких температур взаимодействие коксового остатка с окислителем протекает одновременно и на внешней поверхности частицы и на внутренней поверхности доступных окислителю пор. Диффузионное торможение подводу окислителя сведено к минимуму и скорость процесса определяется скоростью химического реагирования. Принимая в качестве окислителя кислород воздуха, горение коксовой основы при температурах ниже 800°С можно рассматривать как химическую активированную, протекающую в объеме частицы, реакцию [43].
Во многих научных публикациях [2, 44] экспериментально показано, что в первом приближении можно принимать 1-й порядок реагирования по кислороду, но при этом вводятся соответствующие коррективы на величину участвующей в процессе внутренней поверхности реагирования.
Однако следует отметить, что в большинстве экспериментов, как правило, моделируются изотермические условия выгорания с последующей оценкой температурной зависимости из опытов с различными температурами изотермических выдержек. При этом оценивается и изменение внутренней реагирующей поверхности в зависимости от степени выгорания.
Одна из авторитетных работ, посвященных вопросу оценки реакционной способности коксового остатка натуральных углей, выполненная по “классической” схеме кинетических исследований С. Датте и К. Вэном [45], трактует процесс выгорания кокса в низкотемпературной области как результат реакции в объеме частицы, а выражение суммарной скорости выгорания при T = const принимается в виде следующей зависимости:
Предполагается 1-й порядок реагирования по кислороду (прямая пропорциональность парциальному давлению кислорода O2). В уравнении (34) C – отношение выгоревшего к данному моменту времени углерода кокса (убыль массы образца) к общей, потенциально возможной, потери веса (на горючую массу) – C0. Коэффициент α отражает относительную, доступную реагированию, поверхность пор, являющуюся функцией степени выгорания или, в конечном счете, – времени.
Сложность оценки константы скорости процесса, K, в этом случае очевидна. В первую очередь она сопряжена с экспериментальными трудностями при оценке величины α и недостаточно корректна при построении температурной зависимости lgK = f(1/T), поскольку не учитывает изменение количества (начальной концентрации) реагирующих с O2 активных центров на поверхности при изменении температуры (переход на другую температурную ступень).
Учитывая специфику реагирования коксовой основы в низкотемпературной области (реакция в объеме частицы) и отсутствие внешнего диффузионного торможения подводу окислителя, при соответствующем выборе условий эксперимента представляется возможным использование неизотермического режима для определения константы скорости процесса взаимодействия окислителя с коксовой основой натуральных топлив. В условиях одновременного химического реагирования и диффузии в пористой частице неизотермические исследования предполагают обязательными низкие скорости нагрева и малые размеры самой частицы.
Преимущества применения неизотермической кинетики для оценки кинетических процессов, протекающих на внутренней поверхности реагирования в объеме частицы, определяются не только такими факторами как корректность температурной зависимости константы скорости и сокращение объема экспериментальных работ – кинетические параметры могут быть найдены по результатам одного опыта, но, что самое главное, можно исключить необходимость экспериментальной оценки определения изменения поверхности реагирования в процессе выгорания. При непрерывном возрастании температуры влияние развития поверхности реагирования раскладывается на всю температурную область эксперимента и учитывается текущими значениями константы скорости K реакции.
Неизотермическая кинетика позволяет уточнить механизм взаимодействия окислителя частицами натурального угля. В отличие от выгорания коксового остатка в этом случае, в условиях линейного нагрева, представляется возможным выделить температурные области, соответствующие периодам образования и разложения угольно-кислородных комплексов, выделения летучих веществ в окислительной среде, инициирования воспламенения коксовой основы и ее непосредственного выгорания. Информация в указанном объеме может быть получена при организации эксперимента с непрерывной оценкой скорости всех вышеперечисленных последовательных стадий процесса характеризующих термоокислительную деструкцию угольного вещества. При этом появляется возможность оценить роль летучих веществ в формировании реакционных свойств (химической активности) коксового остатка. Каждый из перечисленных этапов описывается реальными значениями кинетических параметров [46].
С точки зрения реальных процессов топливоиспользующих устройств представляет несомненный интерес оценка температуры начала интенсивности выгорания коксовой основы, формировавшейся в условиях непрерывного взаимодействия с окислителем. Эта температура намного выше температуры, соответствующей температуре максимальной скорости процесса выделения летучих веществ. Как показали работы Г. Юнтгена и В. Петерса [7], с увеличением скорости нагрева и температура начала интенсивного выгорания нелетучего остатка (в первом приближении, температура воспламенения коксовой основы) и температура максимума скорости реакции выделения летучих веществ возрастает; однако разница между ними остается, хотя и несколько сокращается. При скорости нагрева β ≈ 104 град/с эта разница составляет 40–50°С (рис. 4).
Рис. 4.
Совместная оценка температур воспламенения коксовой основы и максимальной скорости выделения летучих в зависимости от темпа нагрева (по данным [17], угольV daf = 35%).
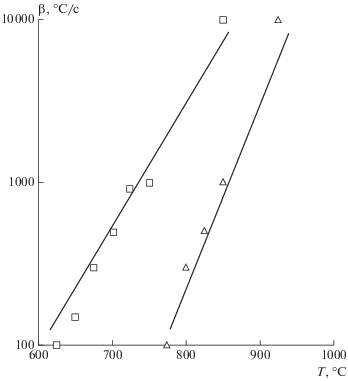
Смещение Tвоспл и Tmax в сторону повышенных температур при этом определяется кинетическим параметрами основного периода выделения летучих и выгорания коксового остатка.
Определение кинетических характеристик на всех стадиях взаимодействия окислителя с угольным веществом в неизотермических условиях основано на обработке зависимостей по типу lgK = f(1/T), получаемых при непрерывной регистрации изменения скорости процесса от начального момента нагрева до полного выгорания образца в условиях проведения комплексного термического анализа.
Эффективность данных о реакционной способности угля, получаемых с помощью комплексного термического анализа, будет значительно выше, если будет иметься возможность их переноса на условия реальных топливоиспользующих установок с последующей оценкой и прогнозом. Разработка таких методик является в настоящее время самостоятельной научной задачей [47].
Ниже приводится методика экстраполяции данных термического анализа, получаемых в условиях низких темпов нагрева (β = 5–20 град/мин), на любые скорости нагрева при условии, что порядок реакции n = 1.
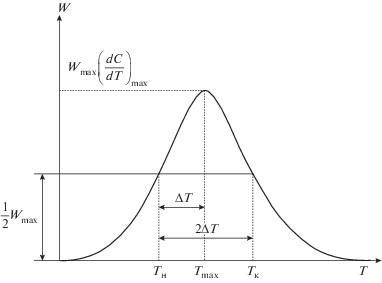
Если дополнить отражаемую уравнением (32) связь основных кинетических характеристик еще одним параметром – шириною максимума дифференциальной кривой 2ΔT на половине его высоты (т.е. на $\frac{1}{2}{{W}_{{\max }}},$ см. рис. 5) – то появится возможность оценить количественно длительность выделения летучих веществ при любой скорости нагрева [40]:
Для максимума дифференциальной кривой первого порядка (уравнение 27), после дифференцирования этого уравнения и приравнивания второй производной к нулю, наряду с уравнением (31), можно установить зависимость начальной концентрации C0 и температурой, соответствующей максимальной скорости реакции Tmax:
(36)
${{С}_{0}} = {{W}_{{\max }}}\frac{{RT_{{\max }}^{2}}}{E}\exp \left( {1 - \frac{{2R{{T}_{{\max }}}}}{E}} \right).$Перепишем обобщенное уравнение неизотермической кинетики (27) для температуры, соответствующей $\frac{1}{2}{{W}_{{\max }}}$ – Tн (либо Tк):
(37)
$\frac{1}{2}{{W}_{{\max }}} = \frac{{{{k}_{0}}{{C}_{0}}}}{\beta }\exp \left\{ { - \left( {\frac{E}{{R{{T}_{{\text{н}}}}}} + \frac{{{{k}_{0}}}}{\beta }\frac{{RT_{{\text{н}}}^{2}}}{E}\left( {1 - \frac{{2R{{T}_{{\text{н}}}}}}{E}} \right)\exp \left( { - \frac{E}{{R{{T}_{{\text{н}}}}}}} \right)} \right)} \right\}.$Подставив в уравнение (37) значение $\frac{{{{k}_{0}}}}{\beta }$ из уравнения (32) и С0 из уравнения (36), получим:
(38)
$\exp \left\{ {\frac{E}{R}\left( {\frac{{{{T}_{{\text{н}}}} - {{T}_{{\max }}}}}{{{{T}_{{\text{н}}}}{{T}_{{\max }}}}}} \right) + 1 - \frac{{2R{{T}_{{\max }}}}}{E} - \frac{{T_{{\text{н}}}^{2}}}{{T_{{\max }}^{2}}}\left( {1 - \frac{{2R{{T}_{{\text{н}}}}}}{E}} \right)\exp \left( {\frac{E}{R}\left( {\frac{{{{T}_{{\text{н}}}} - {{T}_{{\max }}}}}{{{{T}_{{\text{н}}}}{{T}_{{\max }}}}}} \right)} \right)} \right\} - \frac{1}{2} = 0.$Учитывая, что |Tн – Tmax| = ΔT (асимметричностью дифференциальной кривой пренебрегаем) получаем окончательное выражение:
(39)
$\exp \left\{ {1 + \frac{E}{R}\frac{{\Delta T}}{{{{T}_{{\text{н}}}}{{T}_{{\max }}}}} - \frac{{2R{{T}_{{\max }}}}}{E} - \frac{{T_{{\text{н}}}^{2}}}{{T_{{\max }}^{2}}}\left( {1 - \frac{{2R{{T}_{{\text{н}}}}}}{E}} \right)\exp \left( {\frac{E}{R}\frac{{\Delta T}}{{{{T}_{{\text{н}}}}{{T}_{{\max }}}}}} \right)} \right\} - \frac{1}{2} = 0.$Методика использования выведенных зависимостей для экстраполяции данных термического анализа заключается в следующем: по заданным (определенным по результатам КТА) значениям E, k0 и β первоначально по уравнению (32) определяем величину Tmax, а затем по уравнению (39) величину – ΔT, которая в последствии используется для оценки длительности выделения летучих по уравнению (35). Следует отметить, что выражения (32) и (39) являются трансцендентными уравнениями, которые приходится решать соответствующими численными методами (например, методом Ньютона [48]).
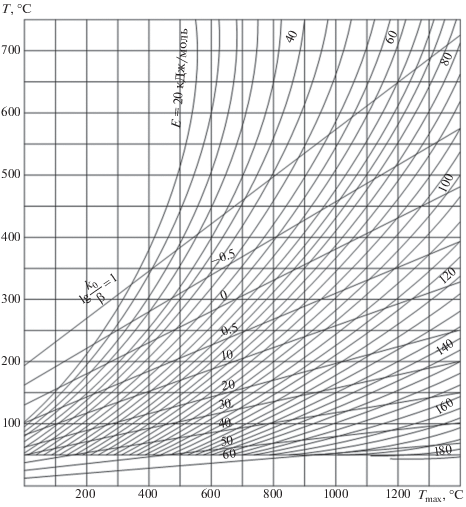
В практике выполнения инженерных расчетов для определения величин Tmax, ΔT и τл целесообразней использовать номограмму, которая связывает между собой эти величины с кинетическими параметрами E, k0 и β. Подобная номограмма, основанная на экспериментальных данных для узкого интервала изменения Tmax и ΔT, приведена в работах Г. Юнтгена и Н. Карра [7, 12].
На рисунке 6 представлена номограмма, построенная по результатом автоматизированных расчетов с помощью зависимостей (37) и (39). Номограмма составлена применительно к следующим условиям: а) пределы изменяемых величин E = 20–180 кДж/моль, $\lg \frac{{{{k}_{0}}}}{\beta }$ = –1–10, Tmax = 50–1400°С, ΔT = 50–750°С; точность расчетов составляет 0.001°С; реализован комбинированный метод, основанный на использовании сочетания методов бисекции, линейной и квадратичной интерполяции [48].
Рис. 6.
Номограмма для определения температуры максимума Tmax и модуля разности температур ΔT = = |Tk – Tmax| дифференциальной кривой выделения летучих веществ при различных значениях энергии активации E, вероятностного фактора k0 и скорости нагревания β.
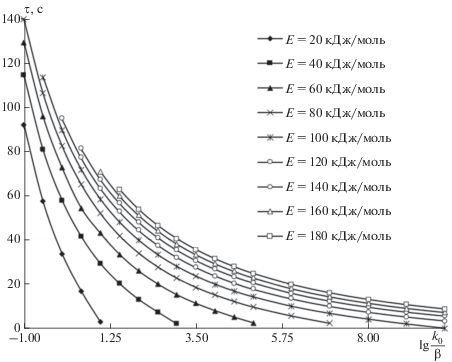
На рисунке 8 приведена номограмма, позволяющая напрямую выполнять оценку длительности протекания процесса выхода летучих веществ на различные скорости нагрева при нагревании твердого энергетического топлива. Данная номограмма составлена на идентичные условия, что и номограмма на рис. 6.
На рисунках 7 и 8 представлены результаты автоматизированного расчета величины ΔT по выражению (39) и длительности выделения летучих веществ на различные скорости нагрева.
Рис. 7.
Зависимость значений полуширины дифференциальной кривой процесса выделения различных газообразных компонентов летучих веществ от скорости нагрева: ο – E = 180 кДж/моль, k0 = 109 1/с; ◻ – E = 75 кДж/моль, k0 = 103 1/с; Δ – E = 25 кДж/моль, k0 = 1.5 1/с.
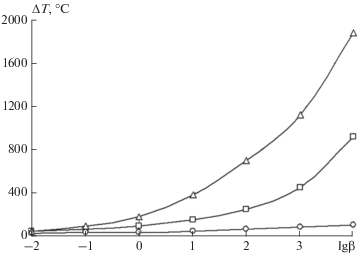
Рис. 8.
Зависимость длительности выделения различных газообразных компонентов летучих веществ от скорости нагрева.
Разработанная методика определения длительности выделения летучих веществ, в частности, индивидуальных газообразных компонентов при термическом разложении твердого топлива создает предпосылки для расчета скорости и оптимизации условий его выгорания в условиях реальных топливоиспользующих установок.
ЗАКЛЮЧЕНИЕ
Теоретически обоснована и экспериментально подтверждена применимость математического аппарата неизотермической кинетики для оценки реакционной способности энергетических углей, разработаны практические рекомендации по его использованию для экстраполяции результатов комплексного термического анализа на условия реальных теплотехнологических процессов и установок. Получено решение обобщенного уравнения неизотермической кинетики в интегральной форме, позволяющее теоретически оценить скорость реакции термохимического превращения угля произвольного порядка в зависимости от температуры процесса.
Выведена универсальная система нелинейных кинетических уравнений для расчетной экстраполяции данных комплексного термического анализа на условия высокоскоростного нагрева пылеугольных частиц. Выполнено решение указанной системы трансцендентных уравнений комбинированным численным методом, и по результату аналитического эксперимента построены номограммы для определения длительности выделения летучих веществ при любых скоростях нагрева угольных частиц, в том числе присущих реальным топочным условиям. Прикладное значение номограммы заключается в возможности оценки температурных интервалов и длительности термического разложения твердого топлива в топочной камере котла или в реакционном пространстве любой огнетехнической установки.
Установлено, что с ростом скорости нагрева угольных частиц на 5–6 порядков происходит сдвиг термохимической реакции в область повышенных температур с одновременным расширением температурной области протекания реакции и уменьшением значений максимальной скорости процесса. Получено, что при высоких скоростях нагрева выделение летучих веществ может лимитировать длительность выгорания углей (выделяется 60–80% от общего количества) и создавать диффузионные осложнения горению коксового остатка.
Преимущества неизотермической кинетики (одновременный учет изменения температуры процесса и концентрации реагирующих компонентов) обуславливают перспективность ее применение и для оценки скоростей гетерогенных реакций. Впервые предложен формальный прием для обработки ДТГ-кривой гетерогенного процесса горения коксовых частиц с оценкой ее кинетических параметров на основе обобщенного математического аппарата неизотермической кинетики в размерностях, принятых для мономолекулярных реакций, т.е. без учета изменения размера коксовых частиц. Подставляя полученные значения E, кДж/моль и k0, 1/с в уравнение (7), осуществляется построение дифференциальных кривых убыли массы образца при экстремальных скоростях нагрева, выполняется решение обратной кинетической задачи с учетом диаметра коксовых частиц, и определяются кинетические параметры процесса горения нелетучей основы (E и k0) с общепринятыми для гетерогенных реакций размерностями.
Список литературы
Хитрин Л.Н. Физика горения и взрыва. М.: Изд. МГУ, 1957. С. 442.
Бойко Е.А. Реакционная способность энергетических углей. Красноярск: ИПЦ СФУ, 2011. С. 606.
Глущенко И.М. Термический анализ твердых топлив. М.: Металлургия, 1968. С 192.
Уэндланд У. Термические методы анализа. М.: Мир, 1978. С. 526.
Turns S.R. An introduction to combustion. N.Y.: McGraw-Hill, 1996. P. 537.
Нелюбин Б.В. К вопросу оценки кинетики реакций газообразования при пиролизе угля // Химия твердого топлива. 1969. № 6. С. 18–23.
Juntgen H. Coal characterization in relation to coal combustion // Erdol und Kohle-Erdgas-Petrochem. 1987. 40. № 4. P. 153–165.
Липович В.Г. Химия и переработка углей. М.: Химия, 1988. С. 336.
Померанцев В.В. Основы практической теории горения. Л.: Энергоатомиздат, 1986. С. 312.
Draiden S. The chemical Reactivity of Carbons // Carbon, V. 6. 2008. P. 213–221.
Oilert H. Use of inert gas to prevent dust explosions // Combust. Inst. Eur. Symp. London–N.Y. 2003. P. 189–194.
Carr N.J. Decomposition reaction of solids (an experiment in reviewing) // Thermochimica. Acta. 1990. V. 79. № 3. P. 323–370.
Lopez F.C.R., Tannous K. Coconut fiber pyrolysis decomposition kinetics applying single- and multi-step reactions models // Thermochimica Acta. 2020. № 691. P. 1–12.
Сорокопуд Л.М. К вопросу о механизме воспламенения летучих в пылеугольной аэросмеси // Теплоэнергетика. 1991. № 2. С. 8–16.
Choi S. An experimental investigation of early stages of pulverized coal combustion – enhanced devolatilization in an oxidizing environment // Sydney, Int. Conf. Coal. Sci. 2015. P. 355–358.
Ван-Кревелен Д.В. Наука об угле. М.: ГНТП литературы по горному делу, 1960. С. 394.
Алаев Г.П. Определение степени выгорания пылеугольного факела в вихревой камере горения котла Е-500-140ВЖ // Известия СО АН. Серия технические науки. 1990. Вып. № 6. С. 103–105.
Алаев Г.П. Методика расчета длительности выделения летучих при сжигании твердого топлива // Проблемы энергосбережения, 1990. Вып. № 5. С. 56–59.
Agarwal P. A single particle model for the evolution and combustion of coal volatiles // Fuel. 1996. № 6. P. 803–810.
Peters A.A. Product distribution and kinetic predictions of Greek lignite pyrolysis // Fuel. 2010. № 10. P. 1304–1308.
Solomon P.R. The relationship between char reactivity and physical and chemical structural features // Abst. Pap., 194th ACS Nat. Meet. (Amer. Chem. Soc.) New Orleans. Washington. 1987. P. 500.
Чуханов З.Ф. Разделение процессов прогрева и полукоксования топливных частиц. М.: ДАН СССР, 1950. Т. 72. № 4. С. 17–26.
Elder J.P. Coal pyrolysis kinetics by non-isotermal thermogravimetry // React. Solids. 1997. № 4. P. 347–358.
Федосеев А.С. Кинетическая модель реакции углерода с кислородом // Химия твердого топлива. 1990. № 2. С. 111–114.
Smoot L.D. Fundamentals of coal combustion. Amsterdam. Oxford. N.Y. Elsevier. 1993. P. 621.
Головина Е.С. Высокотемпературное горение и газификация углерода. М.: Энергоатомиздат, 1983. С. 176.
Babiskiy P., Labojko G., Kotyczka-Moranska M., Shiazko M. Kinetics of pressurized oxy-combustion of coal chars // Thermochimica Acta. 2019. № 682. P. 569–588.
Elder J.P. Coal pyrolysis kinetics by non-isotermal thermogravimetry // React. Solids. 1987. № 4. P. 347–358.
Boiko E.A. Research on kinetics of the thermal processing of brown coals of various oxidative ageing degree using the non-isothermal methods // Thermochimica Acta. 2000. № 348. P. 97–104.
Улановский М.Л. Проблемы дифференциально-термического анализа углей // Кокс и химия. 1990. № 10. С. 52–53.
MoGraw Michael. Coal-water slurries are ready for utility boilers // FRI News, 1989, № 29. P. 1–5.
Бойко Е.А. Экспериментально-расчетная методика оценки кинетических процессов термохимического превращения твердых органических топлив // Физика горения и взрыва. 2005. № 1. С. 55–65.
David M. Formulas for calculating the calorific value of coal: development, tests and uses // Fuel Processing Technology. 1983. № 7. P. 11–22.
Rosdam-Abadi M. Combustion studies of coal derived solid fuels by thermogravimetric analysis. III. Correlation between burnout temperature and carbon combustion efficiency // Thermochimica Acta. 2017. № 4. P. 358–367.
Eloler J.P. Proximate analysis by automated thermogravimetry // Fuel. 1993. V. 62. P. 17–23.
Бойко Е.А. Комплексный термический анализ процессов термолиза и горения нелетучих продуктов канско-ачинских углей разной степени окисленности // Журн. прикладной химии. 2003. Т. 76. № 4. С. 605–610.
Волков Э.П. Моделирование горения твердого топлива. М.: Наука, 1994. С. 320.
Wong G.S. High pressure centrifugal coal slurry pump development // 6th Int. Symp. Coal Slurry Combust. And Technol. Orlando, 1999. P. 1027–1039.
Blackburn P.R. Direct ignition of pulverized coal with electric arc heated air // Proc. Amer. Power conf. Chicago, 1999. V. 41. P. 1050–1063.
Бойко Е.А. Диффузионно-кинетическая модель горения и тепломассообмена пылеугольных частиц в газовом потоке // Химия твердого топлива. 2008. № 6. С. 3–13.
Tang T.B. Analysis of dynamics kinetic data from a single stage in the decomposition of solids // Thermochimica Acta. 2002. V. 57. № 1. P. 93–98.
Jerez A. A modification to the Freeman and Carroll method for analysis of the kinetics of non-isothermal process // J. Thermal Analysis. 1993. V. 26. № 2. P. 315–318.
Iamaluddin A.S. Estimation of kinetic parameters for char oxidation // Fuel. 1999. № 3. P. 317–320.
Smoot L.D. Fundamentals of coal combustion // Amsterdam. Oxford. N.Y. Elsevier, 1993. P. 621.
Datte S. Analysis and development of effective invariant kinetic parameters finding method based on the non-isothermal data // Thermocimica. Acta. 2005. V. 92. № 1. P. 157–160.
Blumental G. Approximation heterogenkinetischer Modellfunktionen durh die Avrami-Erofeev-Funktion // Z. Chem. 2012. Bd. 22. № 2. S. 49–51.
Warnatz J. Detailed studies of combustion chemistry // EC. Bruxelles, Processing of the contractors meeting on EC combustion research. 1988. P. 172.
Амосов А.А. Вычислительные методы для инженеров. М.: Высшая школа, 1994. С. 554.
Дополнительные материалы отсутствуют.
Инструменты
Известия РАН. Энергетика