

Известия РАН. Серия физическая, 2021, T. 85, № 9, стр. 1239-1244
О факторах стабильности/нестабильности биметаллических наноструктур ядро–оболочка
В. М. Самсонов 1, *, Н. Ю. Сдобняков 1, А. Ю. Колосов 1, И. В. Талызин 1, А. Ю. Картошкин 1, С. А. Васильев 1, В. С. Мясниченко 1, Д. Н. Соколов 1, К. Г. Савина 1, А. Д. Веселов 1, С. С. Богданов 1
1 Федеральное государственное бюджетное образовательное учреждение высшего образования
“Тверской государственный университет”
Тверь, Россия
* E-mail: samsonoff@inbox.ru
Поступила в редакцию 19.04.2021
После доработки 12.05.2021
Принята к публикации 28.05.2021
Аннотация
В соответствии с ранее выдвинутой гипотезой о взаимосвязи между спонтанной поверхностной сегрегацией одного из компонентов бинарных наночастиц А–В и стабильностью/нестабильностью наноструктур типа “ядро–оболочка” (A@B и B@A, где первый компонент отвечает ядру, а второй – оболочке частицы), стабильной должна быть та из двух наноструктур, оболочка которой отвечает спонтанно сегрегирующему к поверхности компоненту бинарных наночастиц. Проверка данной гипотезы и выявление возможных отклонений от этой закономерности осуществлялись с использованием атомистического моделирования наноструктур Co@Au, Au@Co, Ni@Cu и Cu@Ni.
ВВЕДЕНИЕ
Как отмечалось ранее в обзорах Р.А. Андриевского [1–3], а также в ряде других статей и монографий, наночастицы и наноматериалы в большинстве случаев нестабильны. Соответственно, усилия научных работников и технологов направлены на их стабилизацию или, по крайне мере, – прогнозирование стабильности/нестабильности. Несмотря на важность проблемы стабильности наноматериалов, к настоящему времени отсутствует даже классификация проявлений стабильности/нестабильности наночастиц и наносистем.
На протяжении последнего десятилетия большой интерес проявляется к биметаллическим наноструктурам, включая Янус-структуры и структуры ядро–оболочка, у которых центральная часть (ядро) представлено одним металлом, а оболочка – другим. В дальнейшем обозначение A@B будет отвечать биметаллическим наночастицам A–B с ядром из атомов сорта A и оболочке из атомов сорта B, а обозначение B@A ядру из атомов B и оболочке из атомов A.
Янус-структуры и структуры ядро-оболочка имеют перспективы применения в энергетике, медицине, катализе и других областях нанотехнологий. В частности, наночастицы Co(ядро)/Au(оболочка) имеют перспективы применения в магнитно-резонансной томографии, и оболочка из атомов Au должна защищать организм от токсического действия магнитного компонента, т.е. Co. В экспериментальной работе [4] было установлено, что отжиг наноструктур Au@Co, состоящих из 30 000 атомов, при 800 К в течение 1 ч приводит к разрушению структуры ядро-оболочка, тогда как наноструктуры Co@Au сохраняют свою стабильность.
В [5] нами была выдвинута и частично подтверждена гипотеза о взаимосвязи между степенью стабильности наноструктур A(ядро)@B(оболочка) и спонтанной поверхностной сегрегацией одного из компонентов в бинарных наночастицах A–B с первоначально равномерным распределением компонентов. В соответствии с этой гипотезой, стабильной или, по крайней мере, более стабильной будет та из двух возможных структур (A@B или B@A), оболочка которой соответствует компоненту, который спонтанно сегрегирует к поверхности бинарных наночастиц A–B. В частности, термодинамическое и атомистическое моделирование [5], показало, что в бинарных наночастицах Au–Co должна наблюдаться поверхностная сегрегация Au как низкоэнергетического по сравнению с Co компонента, характеризующегося более низкими значениями энергии связи и поверхностного натяжения. Соответственно в [5] был сделан вывод о том, что наноструктуры Co@Au должны быть более стабильными, чем Au@Co, и этот вывод, согласующийся с экспериментальной работой [4], был подтвержден молекулярно-динамическими результатами для наночастиц того же размера. Вместе с тем, наши результаты [6] атомистического моделирования наноструктур Co@Au и Au@Co меньшего размера (содержащих 3000 атомов) свидетельствуют о том, что возможны альтернативные сценарии проявлений нестабильности наноструктур ядро-оболочка. В [5, 6] эти сценарии не были должным образом проанализированы. Соответственно, целью данной работы была более детальная верификация сформулированной выше гипотезы, включая выявление возможных отклонений от нее и поиск альтернативных сценариев проявления нестабильности структур ядро–оболочка.
МЕТОДЫ АТОМИСТИЧЕСКОГО МОДЕЛИРОВАНИЯ
В [5] при молекулярно-динамическом моделировании наноструктур Au@Co и Co@Au, содержащих 30 000 атомов в соотношении 1 : 1, т.е. наноструктур Au15 000@Co15 000 и Co15 000@Au15 000, мы использовали программу LAMMPS и метод погруженного атома с параметризацией, предложенной в работе [7]. Та же программа и потенциалы, предложенные в работе [7], были использованы для МД моделирования стабильности/нестабильности наноструктур Ni15 000@Cu15 000 и Cu15 000@Ni15 000. Кроме того, в развитие работы [6], для моделирования наноструктур Au2500@Co2500 и Co2500@Au2500, т.е. наноструктур Au@Co и Co@Au меньшего размера (состоящих из 5000 атомов) мы воспользовались другой разработанной нами компьютерной программой и альтернативным потенциалом – потенциалом сильной связи [8]. Был также использован альтернативный метод атомистического моделирования – методом Монте-Карло, апробация которого проводилась нами для биметаллических наночастиц Co–Au и Cu–Ni в [9‒11]. К сожалению, в отличие от LAMMPS наша программа по молекулярно-динамическому моделированию не позволяет наблюдать эволюцию систем, содержащих 30 000 атомов
Независимо от выбора метода атомистического моделирования и моделируемой системы, во всех случаях первоначально создавалась исходная структура ядро–оболочка, где половина атомов типа A или B находились в ядре частицы, а другая половина (B или A, соответственно) – в оболочке частицы. Затем наноструктуры A@B и B@A эволюционировали в условиях постепенного повышения температуры или при постоянной заданной температуре релаксации (отжига). Такого рода релаксация использовалась при МД моделировании наночастиц, состоящих из 30 000 атомов.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
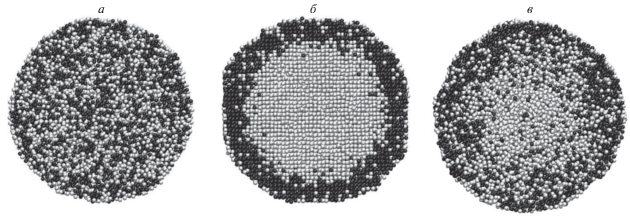
На рис. 1 сравниваются конфигурации наноструктур Co15 000@Au15 000 и Au15 000@Co15 000 после МД релаксации (отжига) в течение 1 нс. Этого малого времени релаксации оказалось достаточным, чтобы выявить различие в поведении указанных биметаллических структур, в которых первоначально ядро и оболочка были представлены атомами только одного из двух указанных металлов. Действительно, как видно из рис. 1а, атомы Co диффундируют в оболочку из атомов Au, но отсутствуют в наружном монослое, т.е. наноструктура Co15 000@Au15 000 сохраняет свою стабильность. В свою очередь, для системы Au15 000@Co15 000 характерны гораздо большие изменения: сечение демонстрирует признаки огранения, видны некие каналы, по которым атомы Au проникают в оболочку из атомов Co, в том числе в наружный монослой. Иными словами, рис. 1б демонстрирует признаки разрушения биметаллической структуры Au@Co. Выбранные значения числа атомов в рассматриваемых биметаллических частицах (30 000) и температуры (800 K) совпадают с условиями экспериментов [4], где, в отличие от наноструктур Co15 000@Au15 000, отжиг в течение часа наночастиц Au15 000@Co15 000 при T = 800 K привел к полному разрушению структуры ядро-оболочка. Разумеется, такое время отжига недоступно для МД экспериментов, и мы могли наблюдать лишь начальную стадию деградации наночастиц Au15 000@Co15 000.
Рис. 1.
Экваториальные сечения наноструктур Co15 000@Au1 5000 (а) и Au15000@Co15000 (б) после релаксации при температуре T = 800 K в течение 1 нс. Здесь и на последующих рисунках атомы Au изображены темными шарами.

Рис. 2 демонстрирует результаты МД моделирования наноструктур Co@Au и Au@Co, содержащих на порядок меньшее число атомов (5000). Как видно из рис. 2а, даже при достаточно высокой температуре 1000 K оболочка из атомов Au свою стабильность, хотя отдельные атомы Co появляются в наружном монослое. При повышении температуры оболочка из атомов Au постепенно истончается, и лишь при температурах, превышающих макроскопическую температуру плавления Au (1360 K), структура ядро–оболочка почти полностью переходит в бинарные наночастицы Au–Co с однородным распределением компонентов. Постепенное уменьшение толщины оболочки из атомов Au коррелирует с заметным уменьшением поверхностной сегрегации Au, которое предсказывает как атомистическое, так и термодинамическое моделирование [5, 12, 13]. Более сложным оказалось поведение системы Au2500(ядро)@Co2500 (рис. 2б): при температурах, превышающих температуру плавления Au, т.е. металла оболочки, атомы Au начинают смещаться по поверхности ядра в одном направлении, что приводит к формированию некоторой Янус-структуры с неполным разделением атомов Au и Co между противоположными половинками такой биметаллической частицы. На первый взгляд, этот эффект может показаться артефактом. Однако в экспериментальной работе [4] отмечается, что отжиг наноструктур Au15 000@Co15 000 приводил к разделению атомов Au и Co между противоположными сторонами частиц и, соответственно, к формированию плоской границы раздела между ними. Таким образом, хотя в [4] термин “Янус-структура” не используется, фактически речь идет о переходе наноструктуры ядро–оболочка в Янус-структуру. Таким образом, результаты, представленные на рис. 2а, согласуются с данными [4], хотя в последней указанный переход наблюдался при гораздо более низкой температуре (800 вместо 1500 K). При дальнейшем повышении температуры в рассматриваемых МД экспериментах (выше 1500 K) наночастицы Au–Co теряли целостность, т.е. распадались на кластеры меньшего размера. В МК экспериментах оба типа наночастиц Co2500@Au2500 (рис. 3а) и Au2500(ядро)@Co2500 (рис. 3б) сохраняли структуру ядро-оболочка вплоть до T = 1800 K. Более адекватными нам представляются МД результаты, демонстрирующие более высокую стабильность наноструктур Co@Au по сравнению со структурами Au@Co.
Рис. 2.
Конфигурации наночастиц Co2500@Au2500 (а) и Au2500(ядро)@Co2500 (б) после МД эволюции в течение времени порядка 1 нс до достижения температур 1000 и 1500 K, соответственно.

Рис. 3.
Конфигурации наночастиц Co2500@Au2500 (а) и Au2500(ядро)@Co2500 (б), полученные при моделировании по методу МК (T = 1800 K).

Наноструктуры Cu@Ni и Ni@Cu так же, как и структуры Co@Au и Au@Co, отвечают случаю, когда компоненты бинарных металлических наночастиц заметно различаются по энергиям связи [14] и, соответственно, по поверхностным натяжениям как в жидком, так и в твердом состояниях [15]. Термодинамическая теория [5, 12, 13] предсказывает для таких наночастиц выраженную поверхностную сегрегацию компонента, характеризующегося меньшим значением поверхностного натяжения, т.е. Au в бинарных наночастицах Au–Co и Cu в наночастицах Cu–Ni. Для наноструктурированных поликристаллов Cu–Ni термодинамическое моделирование [16] также предсказывает сегрегацию Cu к границам зерен, что согласуется с экспериментом [17]. Таким образом, в соответствии со сформулированной выше гипотезой, наноструктуры Ni@Cu должны быть более стабильными, чем наноструктуры Cu@Ni. Однако результаты МД моделирования, представленные на рис. 4, свидетельствуют о том, что применительно к наносистемам Ni15 000@Cu15 000 и Cu15 000@Ni15 000 сформулированная выше гипотеза не выполняется. Как видно из этого рисунка, вплоть до определенной температуры оба типа наноструктур Cu@Ni и Ni@Cu сохраняют свою стабильность. Однако при достижении температуры T = 1280 K структура Ni15 000@Cu15 000 (рис. 4а) переходит в бинарную металлическую наночастицу Cu@Ni с однородным распределением компонентов, т.е. исходная структура ядро–оболочка теряет свою стабильность. Вместе с тем, при той же температуре структура Cu15 000@Ni15 000 сохраняет свою стабильность (рис. 4б), и только при температуре T = 1290 K оболочка из атомов Ni почти полностью разрушается (рис. 4в). Примечательно также, что рис. 4в демонстрирует наружный монослой атомов, представленный в основном атомами Cu, т.е. в рассматриваемой системе имеет место поверхностная сегрегация Cu, которая проявляется лишь в наружном монослое частицы. Очевидно, такая сегрегация и связанная с ней диффузия атомов Cu к поверхности наночастиц выступает в роли фактора нестабильности оболочки, но этот фактор проявляется при более высокой температуре, чем температура нестабильности наноструктур Ni15 000@Cu15 000. Очевидно, причина более высокой стабильности структур Cu@Ni по сравнению с Ni@Cu заключается в том, что Ni характеризуется гораздо более высокой температурой плавления $T_{m}^{\infty }$ = 1728 K (для объемной фазы) по сравнению с температурой плавления Cu (1358 K). Иными словами, оболочку из атомов Ni можно уподобить твердой корке, обеспечивающей более высокую стабильность структур Cu@Ni.
Рис. 4.
Результаты МД эволюции биметаллических наноструктур Ni@Cu и Cu@Ni, состоящих из N = 30 000 атомов (экваториальные сечения): Ni@Cu при T = 1280 K и времени релаксации – 10 нс (а), Cu@Ni при той же температуре и времени релаксации – 200 нс (б), Cu@Ni, при T = 1290 K и времени релаксации – 7.9 нс (в). Атомы Cu изображены белыми, а атомы Ni – черными шарами.
ЗАКЛЮЧЕНИЕ
Таким образом, результаты атомистического моделирования наноструктур Co@Au и Au@Co подтверждают гипотезу о взаимосвязи стабильности/нестабильности наноструктур A@B и B@A со спонтанной поверхностной сегрегацией одного из компонентов бинарных наночастиц A–B с первоначально равномерным, в некотором приближении, распределением компонентов. Действительно, ранее с использованием термодинамического и атомистического моделирования нами была предсказана поверхностная сегрегация Au в бинарных наночастицах Au–Co, и наноструктуры Co@Au оказались более стабильными, чем наноструктуры Au@Co. Вместе с тем, для наноструктур Cu@Ni и Ni@Cu сформулированная выше гипотеза не выполняется: более высокую стабильность продемонстрировали наноструктуры Cu@Ni, а не Ni@Cu, хотя экспериментальные и теоретические результаты предсказывают поверхностную сегрегацию Cu. Мы объяснили более высокую стабильность Cu@Ni тем, что никель имеет более высокую температуру плавления ($T_{m}^{{\left( \infty \right)}}$ = 1728 К) чем медь (1358 К). Соответственно, при температурах $T \ll T_{m}^{{\left( \infty \right)}}$ оболочка из атомов Ni ведет себя как стабильная твердая корка. Однако остается не ясным, почему же Co@Au являются более стабильными, чем Au@Co, хотя Co также имеет более высокую температуру плавления (1768 К), чем Au (1337 К).
Представленные в работе результаты демонстрируют условность как понятий стабильности и нестабильности одной из наноструктур A@B или B@A, так и представлений о более и менее стабильных структурах. Действительно, до определенной температуры стабильными могут быть обе структуры, A@B и B@A. В данной работе мы предложили считать менее стабильной или просто нестабильной ту из двух альтернативных структур, которая теряет стабильность при более низкой температуре. При этом возможны различные сценарии потери стабильности. В наших МД экспериментах наблюдались три такие сценария:
1) переход структуры ядро–оболочка в бинарную наночастицу с равномерным, в некотором приближении, распределении компонентов;
2) переход наноструктуры A@B в Янус-структуру;
3) распад наночастицы A@B на нанокластеры меньшего размера
Как правило, нестабильность наноструктур – крайне нежелательное явление, и основной задачей исследователей в этой области является выяснение температурных интервалов и других условий стабильности. Однако нестабильность наноструктур может быть также целенаправленно использована как метод получения других наноструктур на основе того же наносплава, например, получение структур ядро–оболочка с использованием спонтанной сегрегации и получения Янус-структур путем деградации структуры ядро–оболочка.
Исследования выполнены при поддержке Министерства науки и высшего образования Российской Федерации в рамках выполнения государственного задания в сфере научной деятельности (проект № 0817-2020-0007) и Российского фонда фундаментальных исследований (проекты № 18-03-00132, № 20-37-70007, № 20-33-90192).
Список литературы
Андриевский Р.А. // Усп. хим. 2002. Т. 71. №. 10. С. 967; Andrievskiy R.A. // Russ. Chem. Rev. 2002. V. 71. No. 10. P. 853.
Андриевский Р.А. // Усп. хим. 2014. Т. 83. № 4. С. 365; Andrievskiy R.A. // Russ. Chem. Rev. 2014. V. 83. No. 4. P. 365.
Andrievskiy R.A. // J. Mat. Sci. 2014. V. 49. № 4. P. 1449.
Sato K., Matsushima Y., Konno T.J. // AIP Advances. 2017. V. 7. No. 6. Art. No. 065309.
Samsonov V.M., Talyzin I.V., Kartoshkin A.Yu., Vasilyev S.A. // Appl. Nanosci. 2019. V. 9. No. 1. P. 119.
Сдобняков Н.Ю., Самсонов В.М., Колосов А.Ю. и др. // Физ.-хим. асп. изуч. класт. наностр. и наноматер. 2019. Т. 11. С. 520.
Zhou X.W., Johnson R.A., Wadley H.N.G. // Phys. Rev. B. 2004. V. 69. No. 14. Art. No. 144113.
Cleri F., Rosato V. // Phys. Rev. B. 1993. V. 48. № 1. P. 22.
Sdobnyakov N., Khort A., Myasnichenko V. et al. // Comput. Mat. Sci. 2020. V. 184. Art. No. 109936.
Myasnichenko V., Sdobnyakov N., Kirilov L. et al. In: Recent advances in computational optimization. Studies in computational intelligence. V. 838. Cham: Springer, 2020. P. 133.
Myasnichenko V., Fidanova S., Mikhov R. et al. In: Advances in high performance computing. HPC 2019. Studies in computational intelligence. V. 902. Cham: Springer, 2021. P. 61.
Samsonov V.M., Bembel A.G., Kartoshkin A.Y. et al. // J. Therm. Analyt. Calorim. 2018. V. 133. No. 2. P. 1207.
Самсонов В.М., Талызин И.В., Картошкин А.Ю., Самсонов М.В. // Физ. мет. и металловед. 2019. Т. 120. № 6. С. 630; Samsonov V.M., Talyzin I.V., Kartoshkin A.Yu., Samsonov M.V. // Phys. Met. Metallogr. 2019. V. 120. No. 6. P. 578.
Киттель Ч. Введение в физику твердого тела. М.: Наука, 1978. 791 с.
Alchagirov A.B., Alchairov B.B., Taova T.M., Khoko-nov Kh.B. // Transact. JWRI. 2001. V. 30. P. 287.
Kaptay G. // J. Mater. Sci. 2016. V. 51. № 4. P. 1738.
Pellicer E., Varea A., Sivaraman K.M. et al. // ACS Appl. Mater. Inter. 2011. V. 3. No. 7. P. 2265.
Дополнительные материалы отсутствуют.
Инструменты
Известия РАН. Серия физическая