

Химическая физика, 2019, T. 38, № 3, стр. 37-51
Смесевые композиции ультратонких волокон поли-3-гидроксибутирата с комплексом цинк–порфирин. Структура и свойства
С. Г. Карпова 1, *, А. А. Ольхов 1, 2, 3, П. М. Тюбаева 1, 2, Н. Г. Шилкина 3, А. А. Попов 2, А. Л. Иорданский 3
1 Институт биохимической физики им. Н.М. Эмануэля Российской академии наук
Москва, Россия
2 Российский экономический университет им. Г.В. Плеханова
Москва, Россия
3 Институт химической физики им. Н.Н. Семёнова Российской академии наук
Москва, Россия
* E-mail: karpova@sky.chph.ras.ru
Поступила в редакцию 23.04.2018
После доработки 19.06.2018
Принята к публикации 20.06.2018
Аннотация
Методом электроформования получены ультратонкие волокна биоразлагаемого полимера поли-3-гидроксибутирата (ПГБ) с инкапсулированным комплексом цинка с порфирином (0–5% от массы ПГБ). В работе использованы следующие методы: дифференциально-сканирующая калориметрия, зондовый метод ЭПР, сканирующая электронная микроскопия, ИК-спектроскопия. Показано, что при добавлении в волокна ПГБ порфирина наблюдается рост кристалличности и замедляется молекулярная подвижность в аморфных областях полимера. Температурное воздействие на волокна (отжиг при 140 °C) приводит к резкому росту кристалличности и росту молекулярной подвижности в аморфных областях волокон ПГБ и ПГБ с 1% порфирина. Добавление 3% порфирина в волокна ПГБ обуславливает резкое снижению кристалличности. Воздействие водной среды при 70 °С на структуру и динамику ультратонких волокон указывает на рост степени кристалличности. Озонирование волокон обуславливает замедление молекулярной подвижности при небольших временах воздействия (до 100 мин), при более глубоких степенях окисления наблюдается рост молекулярной подвижности.
ВВЕДЕНИЕ
Создание полимерных композиционных материалов на основе модификаций биологически активных соединений на сегодняшний день – активно развивающаяся область химической технологии и направлено на производство полимерных изделий со значимыми биомедицинскими свойствами. Комплексы металлов с порфиринами являются гомогенными катализаторами автоокисления ряда биогенных веществ. В этом процессе происходит промежуточное образование активных форм кислорода – супероксидного анион-радикала, пероксидного и гидроксильного радикалов, пероксида водорода, цитостатическая активность которых хорошо известна. Эти радикальные и ион-радикальные частицы вызывают окислительные деструктивные реакции в клетках, обуславливая бактерицидный эффект [1, 2]. В настоящее время материалы на основе наноразмерных и ультратонких биополимерных волокон, содержащих различные функциональные добавки, широко применяются в различных отраслях производства и жизнедеятельности человека: экологии, медицине, питании и др. [3]. Наиболее экономичным и удобным с технологической точки зрения методом формования таких волокнистых материалов является электроформование полимерных растворов и расплавов [4, 5]. C помощью электростатического формования (электроформования) можно создавать уникальные материалы на основе наноразмерных и микроволокон с высокими показателями удельной поверхности и пористости. Эти материалы находят широкое применение в производстве матриксов для выращивания колоний живых клеток, создания пролонгированных форм доставки лекарственных веществ в организм человека, конструирования различных биорезорбируемых имплантатов и др. Волокнистые нетканые материалы на основе биополимеров создают условия для свободной миграции и пролиферации клеток в трехмерном пространстве матрикса, а соответственно, и высокий уровень интеграционной способности материала в живых тканях организма [6].
Среди биополимеров медицинского применения лидируют полиокси-алканоаты (ПОА), наиболее распространенным из которых является поли-3-гидроксибутират (ПГБ). Этот биополимер, получаемый биотехнологическим путем, обладает перспективными свойствами, такими как высокий уровень биосовместимости и способность к биорезорбции без образования токсичных продуктов [7, 8]. В связи с этим ПГБ широко исследуется в настоящее время и используется для разработки целого ряда изделий медицинского назначения для хирургии, стоматологии, кардиохирургии, ортопедии и других областей [9].
Как известно, надмолекулярная структура полимерного волокна существенным образом влияет на комплекс физико-механических, диффузионных свойств, кинетику биорезорбции, а также механизм окислительной деградации [10]. На надмолекулярную структуру волокна оказывают влияние не только технологические параметры: молекулярная масса и концентрация полимера, динамическая вязкость, напряжение, расстояние от капилляра до формирующегося слоя, электропроводность, температура и др., но и наличие различных функциональных высокомолекулярных [11] и низкомолекулярных [12] добавок в формовочном растворе. Вводимые в формовочный полимерный раствор функциональные добавки (УФ-абсорберы, лекарственные вещества, факторы клеточного роста и др.) благодаря разной силе межмолекулярного взаимодействия могут тормозить или ускорять процессы кристаллизации, ориентации или релаксации полимерных макромолекул [13]. При этом может изменяться геометрия волокон. Поэтому получение на выходе волокон определенной морфологии – одна из основных проблем, актуальная и на сегодняшний день.
В предыдущих работах мы уже представляли исследования волокнистых материалов на основе ПГБ, содержащих дипиридамол [14, 15] и комплексы железа(III) с хлорпорфирином [16]. В этих работах было показано влияние низкомолекулярных веществ на структуру кристаллической и аморфной фаз ПГБ-волокон. Разница в структуре полимерных волокон, содержащих одинаковые концентрации вышеназванных веществ, может объясняться различием в силе межмолекуляроного взаимодействия между низкомолекулярным веществом и полярными группами полимера. При этом структура материала будет иметь различную устойчивость к внешним факторам: окислению, гидролизу, термодеструкции и др. Поэтому исследование поведения внутренней структуры полимерных ультратонких волокон, модифицированных различными функциональными добавками, при воздействии внешних факторов является актуальной и практически значимой задачей.
Цель настоящей работы – изучение влияния термического отжига, водного воздействия и озонолиза на надмолекулярную структуру ультратонких волокон ПГБ, модифицированных комплексами цинк–тетрафенилпорфирин (Zn-ТФП).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Объекты и методы исследования
Комплекс цинка с тетрафенилпорфирином был получен по известной методике [17] и предоставлен А.В. Лобановым (ИХФ РАН). Цинковый комплекс тетрафенилпорфирина представляет собой бордово-коричневый кристаллический порошок. Он хорошо растворяется в хлорорганических и ароматических растворителях, диметилформамиде (ДМФА), диметилсульфоксиде (ДМСО), но нерастворим в воде.
Для получения композитных волокон готовили растворы Zn-ТФП в хлороформе (1, 3 или 5 мас.%). Далее эти растворы добавляли к растворам ПГБ в хлороформе. Выборка была представлена серией из пяти образцов одного состава.
Для получения волокон ПГБ использовали природный биоразлагаемый полимер полигидроксибутират серии 16F, полученный методом микробиологического синтеза компанией “Biomer ®” (Германия). Средневязкостная молекулярная масса ПГБ составляла 2.06 ⋅ 105, плотность ρ = = 1.248 г/см3, Тпл = 177 °С, степень кристалличности ~63%.
В качестве модифицирующего вещества для создания волокнистых матриксов с антисептическими свойствами применяли комплекс цинка с порфирином (Zn-ТФП). Готовили формовочные растворы ПГБ и ПГБ/Zn-ТФП в хлороформе при температуре 60 °C, используя автоматическую магнитную мешалку. Концентрация ПГБ в растворе составляла 7 мас.%, содержание Zn-ТФП в формовочном растворе – 1, 3 и 5 мас.% относительно массы ПГБ. Волокна получали методом электроформования с помощью однокапиллярной лабораторной установки со следующими параметрами: диаметр капилляра – 0.1 мм, напряжение электрического тока – 12 кВ, расстояние между электродами – 18 см, электропроводность раствора – 10 мкСм/см [18].
ЭПР-спектры (Х-диапазона) регистрировали на автоматизированном спектрометре ЭПР-В (Институт химической физики РАН, Москва). Значение микроволновой мощности в резонаторе, во избежание эффектов насыщения не превышало 7 мВт. При записи спектров амплитуда модуляции всегда была существенно меньше ширины резонансной линии и не превышала 0.5 Гс. В качестве зонда использовали стабильный нитроксильный радикал ТЕМПО. Радикал вводили в волокна из газовой фазы при температуре 50 °C.
Концентрацию радикала в полимере определяли с использованием программного обеспечения фирмы “Bruker–winer and simfonia” (эталоном служил ССl4 с известной концентрацией радикала, которая не превышала 10–3 моль/л). Экспериментальные спектры спинового зонда в области медленных движений (τ > 10–10 с) анализировали в рамках модели изотропного броуновского вращения с помощью программы, описанной в работе [19]. При моделировании спектров применяли следующие главные значения g-тензора и тензора сверхтонкого взаимодействия радикала: gxx = 2.0096, gyy = 2.0066, gzz = 2.0025, Ахх = 7.0 Гc, Ayy = 5.0 Гc, Azz = 35.0 Гc. Величину Azz определяли экспериментально из ЭПР-спектров нитроксильного радикала, находящегося в полимере, при –216 °C; она не сильно отличалась от значения, приведенного в работе [20].
Значения времени корреляции вращения зонда, τ, в области быстрых вращений (5 ⋅ 10–11 < τ < 10–9 с) находили из спектров ЭПР по формуле [21]
(1)
$\tau = {\text{ }}\Delta {{H}_{ + }}\left[ {{{{\left( {{{{{I}_{ + }}} \mathord{\left/ {\vphantom {{{{I}_{ + }}} {{{I}_{--}}}}} \right. \kern-0em} {{{I}_{--}}}}} \right)}}^{{0.5}}}--{\text{ }}1} \right] \cdot 6.65 \cdot {{10}^{{--10}}},$Исследование образцов методом дифференциально-сканирующей калориметрии (ДСК) осуществляли на приборе DSC 204 F1 фирмы “Netzsch” в среде аргона cо скоростью нагревания 10 град/мин. Среднестатистическая ошибка измерения тепловых эффектов составляла ±3%. Энтальпию плавления рассчитывали по программе “NETZSCH Proteus”. Термический анализ проводили по стандартной методике [22]. Разделение пиков осуществляли с помощью программного обеспечения “NETZSCH Peak Separation 2006.01”. Вычисления проводили комбинированным методом Гаусса–Ньютона, в котором метод Marquardt скомбинирован с оптимизацией длины шага итерации [23].
Образцы подвергали отжигу при температуре (140 ± 1) °C в термошкафу и воздействию дистиллированной воды при (70 ± 1) °C различных значениях времени выдержки. Перед введением радикала образцы, подвергнутые воздействию воды, сушили до постоянной массы. Окисление образцов проводили в атмосфере озоно-кислородной смеси с парциальной концентрацией озона 5 ⋅ 10–5 моль/л. Степень окисления определяли методом ИК-спектроскопии на спектрометре “Bruker IFS 48” (США) по изменению полосы поглощения карбонильной группы в области 1650–1700 см–1.
Геометрические параметры волокнистых материалов исследовали с помощью сканирующего электронного микроскопа “Hitachi TM-3000” (Япония) при ускоряющем напряжении 20 кВ. На поверхность образца нетканого волокнистого материала напыляли слой золота толщиной 100–200 Å.
В работе использовали программно-аппаратный комплекс на основе ИК-фурье-спектрометра Vertex 70 с модулем комбинационного рассеяния RAM II. Спектральный диапазон, см–1: 8000–350; спектральное разрешение, см–1: не более 0.4; воспроизводимость волнового числа, см–1: не более 0.01; пределы допускаемой абсолютной погрешности измерений по шкале волновых чисел, см–1: не более ±0.1; максимальная скорость сканирования, мм/с: 100. А также использовали ИК-фурье-спектрометр, способный регистрировать спектр в интервале длин волн от 600 до 4000 см–1 с разрешением не менее 4 см–1, с приставкой нарушенного полного внутреннего отражения (НПВО, ATR) с кристаллом из селенида цинка.
Проведение испытаний: исследуемый образец помещают на кристалл приставки НПВО и механически прижимают к поверхности кристалла. ИК-спектр регистрируют в соответствии с инструкцией по эксплуатации прибора.
Влияние концентрации Zn-ТФП на геометрические параметры волокна
На рис. 1 представлены микрофотографии нетканых волокнистых мате-риалов на основе ПГБ и комплексов Zn-ТФП, сделанные на оптическом микроскопе Micromed Polar 3 ToupCam 5.1 MP в отраженном свете при увеличении в 200 раз. Исходные волокна ПГБ характеризуются наличием веретеноподобных утолщений, причиной появления которых является неоптимальная электропроводность формовочного раствора (менее 1 мкСм/см). Размер утолщений в поперечном направлении составляет от 75 до 150 мкм, в продольном направлении – 10–25 мкм. Средний диаметр основного волокна колеблется от 2.5 до 5 мкм. Как видно из рис. 1б–г, при добавлении комплекса Zn-ТФП утолщения на волокнах ПГБ исчезают. Это связано, по нашему мнению, с ростом электропроводности формовочных растворов (от 1 до 10 мкСм/см). Увеличение электропроводности приводит к равномерному вытягиванию капли полимерного формовочного раствора в поле действия электростатических сил [18]. Подобные эффекты нами были замечены ранее на волокнах ПГБ, содержащих хитозан [13], дипиридамол [11] и комплексы Fe(III)Cl – порфирин [16]. Заметное увеличение электропроводности связано с добавлением в раствор ПГБ полярных низкомолекулярных компонентов. Средний диаметр волокна ПГБ + 1% Zn-ТФП колеблется в интервале 5–12 мкм. При этом по длине волокна наблюдаются небольшие локальные утолщения. Волокна имеют толстые (10–12 мкм) и тонкие участки (5–7 мкм), что свидетельствует о расщеплении первичной струи полимерного раствора. Волокна с 3% комплексов Zn-ТФП характеризуются более стабильным диаметром, около 7–8 мкм, и небольшим количеством волокон с диаметром около 5 мкм, свидетельствующим о незначительном эффекте расщепления первичной струи. Волокна с 5% комплексов Zn-ТФП имеют практически постоянный диаметр, около 7 мкм, по всей длине волокна, что говорит о полном отсутствии расщеплений первичной струи в поле действия электростатических сил.
Рис. 1.
Микрофотографии волокнистых материалов ПГБ (а), ПГБ + 1% Zn-ТФП (б), ПГБ + 3% Zn-ТФП (в), ПГБ + + 5% Zn-ТФП (г).

Как видно на рис. 1, морфология нетканых материалов в зависимости от состава волокна различна. С добавлением полярных комплексов Zn-ТФП изменяется плотность упаковки и ориентация волокон в нетканом материале. Видно, что самая рыхлая упаковка наблюдается в случае исходных ПГБ-волокон, что связано с наличием веретеноподобных уплотнений. По мере роста концентрации Zn-ТФП уплотнения исчезают, колебания диаметра волокна уменьшаются, и волокна укладываются более плотно с преимущественной пространственной ориентацией относительно соседних волокон, как хорошо видно на рис. 1г для волокон ПГБ + 5% Zn-ТФП.
Таким образом, введение полярных комплексов Zn-ТФП приводит к изменению геометрических параметров волокон, что является следствием уплотнения и анизотропии нетканого волокнистого материла в целом. При этом мы ожидали изменений в структуре полимерного волокна.
Термофизические характеристики кристаллической фазы композиций ПГБ/Zn-ТФП
Вначале рассмотрим влияние Zn-ТФП на кристаллическую фазу волокон ПГБ, используя метод ДСК. Следует отметить, что концентрация порфирина в аморфных областях полимера будет значительно выше, чем исходно заданные значения в 1, 3 и 5%, так как этот расчет проводили для всей массы образца, а частицы Zn-ТФП находятся только в аморфной прослойке, и доля таких областей уменьшается (как будет показано ниже) с ростом концентрации комплекса в полимере. Серия экспериментов с образцами различного состава показала, что как при первом, так и при втором температурном сканировании все они имеют схожие по форме термограммы с одним максимумом, который соответствует температуре плавления ПГБ. Термограммы кристаллизации этих волокон переходят в максимумы бимодальной формы.
На рис. 2 представлены данные по изменению энтальпии плавления (∆Н) в образцах с ростом концентрации добавки. Видно, что энтальпия плавления смесевой композиции ПГБ с порфирином достаточно резко возрастает при добавлении 1% Zn-ТФП, добавление 3 и 5% порфирина обуславливает более плавное изменение этого параметра. На рис. 3 представлены термограммы плавления ПГБ с 1% Zn-ТФП. Аналогичные термограммы были получены для всех изучаемых образцов. Термограммы как 1-го так и 2-го сканирований имеют одиночные максимумы плавления, симметричную форму, практически одну температуру плавления для каждой серии, что свидетельствует о достаточной однородной кристаллической фазе. Данные по энтальпии плавления представлены в табл. 1. Следует отметить, что значения ∆Н, полученные методом ДСК, дают информацию как о доле кристаллической фазы, так и о паракристаллических системах в волокне. Из полученных данных мы делаем вывод, что порфирин оказывает пластифицирующий эффект на процесс кристаллизации волокна в процессе электроформования. Межмолекулярное расстояние растет при добавлении порфирина, подвижность цепей увеличивается, и процесс ориентации протекает еще эффективнее. В результате доля кристаллитов и паракристаллических структур возрастает. Важно отметить, что температуры плавления кристаллитов на кривых ДСК лежат в диапазоне от 140 до 184 °C.
Рис. 2.
Зависимость энтальпии плавления ΔН от состава волокна: 1 – исходный образец, 2 – после отжига при 140 °C в течение 0.5 ч, 3 – после отжига в течение 3 ч.
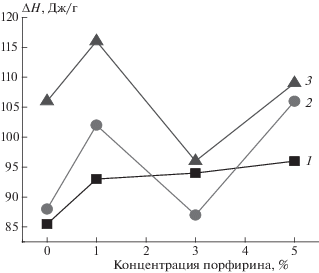
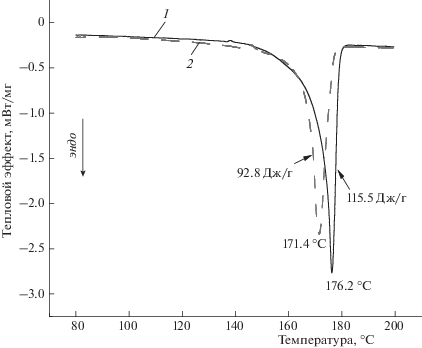
Таблица 1.
Энтальпия (∆Н) и температура плавления Тпл ультратонких волокон смесей ПГБ/Zn-ТФП, исследованных методом ДСК
| Смесь | ∆Н, Дж/г | Тпл, °С | ∆Нкр, Дж/г | Ткр, °С | ||
|---|---|---|---|---|---|---|
| первое сканирование | второе сканирование | первое сканирование | второе сканирование | |||
| ПГБ | 85.5 (88) (106)' |
84.4 (77) (86)' |
175.5 (176) (176)' |
172.3 (172) (172)' |
70 (74.7) (69)' |
96 (98.3) (78.4)' |
| ПГБ + 1% Zn-ТФП | 93 (102) (116)' |
90 (92) (93)' |
175.4 (174.2) (176)' |
171.8 (171) (171)' |
76.2 (97) (120)' |
92, 70 (95.5) (87, 76, 59)' |
| ПГБ + 3% Zn-ТФП | 94 (87) (96)' |
85 (76) (80)' |
175.3 (175) (176)' |
173 (171) (170)' |
75 (83) (39)' |
94.4 (88) (85, 70, 39) |
| ПГБ + 5% Zn-ТФП | 96 (106) (109)' |
94 (90) (92)' |
177 176 (176)' |
172.6 (172) (170)' |
74.8 (65) (71)' |
87.7 (84) (91, 81)' |
Кристаллиты и аморфные области в общем случае не отвечают минимуму свободной энергии; волокна ПГБ имеют неравновесную структуру, что приводит к незавершенности процесса кристаллизации. Стремление аморфно-кристаллической системы к минимуму свободной энергии облегчается при температуре отжига полимера ниже температуры плавления, когда макромолекулы получают достаточную подвижность. Из термодинамики кристаллизации следует, что абсолютному минимуму свободной энергии соответствует бесконечно большой продольный размер кристаллита. По этой причине кристаллиты стремятся увеличить продольный размер и тем самым уменьшить свою свободную энергию. Увеличение продольного размера кристаллита проявляется в процессе изотермического утолщения. Для приведения системы в термодинамическое равновесие был использован метод термического отжига при температурах ниже температуры плавления. Поэтому мы подвергали образцы отжигу при 140 °C в течение 0.5 и 3 ч, затем резко охлаждали их до комнатной температуры.
В табл. 1 и на рис. 2 представлены полученные данные. Из кинетической теории кристаллизации известно, что размер кристаллитов L при изотермическом отжиге зависит от времени отжига по закону L ∼ lg t, где t – время отжига. Поэтому в волокнах ПГБ и ПГБ с 1% порфирина после отжига в течение 0.5 и 3 ч наблюдается рост энтальпии плавления, причем с ростом времени отжига этот эффект достаточно сильно возрастает на величину ∼26%. Интересно, что для волокон ПГБ с 3% порфирина наблюдается резкое снижение ∆Н после отжига. Термограммы всех волокон имеют одиночные максимумы плавления и симметричную форму, за исключением образцов ПГБ + 3% Zn-ТФП, имеющих бимодальный пик кристаллизации, что обусловлено, на наш взгляд, образованием кристаллитов с дефектами. Наблюдаемый эффект, по нашему мнению, можно объяснить внедрением частиц порфирина в паракристаллические структуры и в дефектные кристаллические области, так как после 140 °С начинаются процессы плавления таких структур. Порфирин проникает в эти структуры и фиксируется в них вследствие резкого охлаждения волокна. Молекулы порфирина остаются в этих паракристаллических областях полимера, что препятствует процессу кристаллизации, и, как результат, энтальпия плавления снижается. Чем выше концентрация добавки, тем в большей степени должны протекать процессы аморфизации кристаллических областей при отжиге. При добавлении 1% Zn-ТФП степень кристалличности после отжига при 140 °С имеет наиболее высокие значения, так как концентрация порфирина невелика и, как результат, низка доля деформированных порфирином кристаллических областей. Добавление 3% Zn-ТФП обуславливает резкое снижение степени кристалличности после отжига как в течение 0.5, так и 3 ч, что обусловлено высокой долей деформированных порфирином кристаллических областей. Однако при добавлении 5% Zn-TФП имеет место повышение ∆Н (относительно образца ПГБ с 3% Zn-TФП). По нашему мнению, такие изменения обусловлены сегрегацией частиц порфирина в частицы большего объема, когда проницаемость в расплавленные кристаллические области затруднена, поэтому деформации такими частицами подвергается более низкая доля кристаллитов и паракристаллических структур. В результате отжиг таких волокон приводит к росту энтальпии плавления (рис. 2).
Совокупность результатов исследований особенностей геометрической структуры волокон и их термофизического поведения позволяет полагать, что при отсутствии порфирина в ПГБ образуются веретеноподобные неравновесные структуры. По мере возрастания концентрации порфирина как низкомолекулярной добавки эти структуры исчезают; цилиндрические тонкие волокна теперь характеризуются более совершенной и равновесной структурой с более высокой степенью кристалличности. Исследования кристаллических областей ультратонких волокон ПГБ с порфирином показало, что с ростом концентрации порфирина увеличивается доля кристаллических и паракристаллических структур; наиболее сильные изменения имеют место при добавлении 1% Zn-TФП. Термограммы при втором сканировании также имеют одиночные максимумы плавления и симметричную форму, что указывает на достаточно высокую скорость релаксации системы (рис. 3).
Динамические характеристики аморфной фазы ультратонких волокон
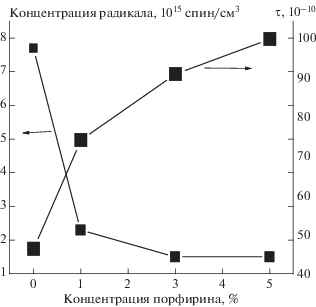
Структура аморфных областей в значительной степени определяется долей кристаллической фазы в волокне. Вследствие этого при добавлении малой доли Zn-ТФП, когда растет степень кристалличности ПГБ, имеет место изменение структурно-динамического состояния аморфных областей. Молекулярная динамика этих областей была изучена методом ЭПР с использованием стабильных нитроксильных радикалов. Спектры ЭПР радикала ТЕМПО в матрице ПГБ имеют сложный вид и представляют суперпозицию двух спектров, соответствующих двум популяциям радикалов с различающимися временами корреляции τ1 и τ2, где τ1 характеризует молекулярную подвижность в более плотных, а τ2 – в менее плотных аморфных областях (рис. 4). Используя специальную программу NLSL, аналогично процедуре, описанной ранее в работе [19], была проведена оценка соотношения концентраций различных по плотности областей и рассчитаны времена корреляции в плотных и рыхлых областях путем математической обработки спектров. Результаты таких расчетов представлены в табл. 2. На рис. 4 представлены спектры быстрой и медленной составляющих волокна ПГБ. Из данных табл. 2 видно, что с ростом концентрации порфирина в волокне доля плотных областей возрастает в ∼6 раз. Время корреляции в плотных областях также увеличивается. Причем наиболее резкое изменение τ с ростом концентрации Zn-ТФП имеет место при добавлении 1% порфирина в волокно; дальнейший рост концентрации Zn-ТФП обуславливает более плавный рост времени корреляции (табл. 2). Важно отметить, что аналогичный ход кривой имеет место и при изменении ∆Н с ростом концентрации добавки (рис. 2). В работе был сделан расчет времени корреляции τ2 из спектров ЭПР (5 ⋅ 10–11 < τ2 < 10–9 с). Зависимость τ2 от концентрации Zn-ТФП также имеет нелинейный характер (рис. 5). Сделано предположение, что рост времени корреляции в смесевых композициях обусловлен замедлением молекулярной подвижности вследствие уплотнения аморфной фазы. Рост кристалличности всегда сопровождается увеличением доли выпрямленных цепей в аморфной прослойке и, как следствие, замедлением молекулярной подвижности. Такие изменения в аморфной фазе сопровождаются уменьшением концентрации радикала. Был сделан расчет равновесной концентрации радикала, адсорбированного в одинаковых по массе образцах волокон ПГБ/Zn-ТФП, с использованием программного обеспечения фирмы “Bruker–winer and simfonia”. На рис. 5 представлены такие зависимости. Видно, что замедление молекулярной подвижности происходит симбатно с изменением концентрации радикала в волокне.
Рис. 4.
а – Экспериментальные (1) и теоретические (2) спектры ЭПР нитроксильного радикала ТЕМПО образца ПГБ; б – медленная (1) и быстрая (2) составляющие спектра.
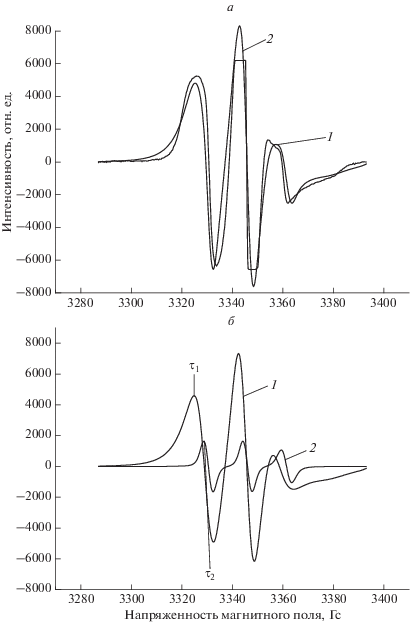
Таблица 2.
Время корреляции (τ) и доля аморфных областей (α) в волокнах ПГБ/порфирин
| Содержание Zn-ТФП, % |
τ ⋅ 10–10 с | τ ⋅ 10–10 с, отжиг при 140 °С | α(1)/α(2), % | |||
|---|---|---|---|---|---|---|
| плотная область | рыхлая область |
плотная область |
рыхлая область |
до отжига | после отжига | |
| 0 | 80.7 | 5.7 | 73 | 6 | 3.1 | 2.6 |
| 1 | 107 | 6 | 56.6 | 7.5 | 6.2 | 1.6 |
| 3 | 117 | 6.1 | 59.3 | 7.9 | 11 | 1.5 |
| 5 | 125 | 6.9 | 58 | 8.1 | 19 | 1.5 |
Представляет интерес изменение молекулярной подвижности после отжига при 140 °С, когда система переходит в равновесное состояние. Радикал вводили в волокна после отжига. На рис. 6 представлено изменение параметра τ/τ0 (τ0 – исходное, τ − текущее значения) с ростом времени отжига при 140 °С для изучаемых смесевых композиций. Видно, что уже на первых минутах отжига происходит резкое снижение времени корреляции, и только после отжига в течение 30 мин этот процесс резко замедляется. Кинетика изменения времени корреляции в ПГБ была получена с помощью программы NLSL и времени, рассчитанного из спектров ЭПР (рис. 7). Видно, что более интенсивные изменения времени корреляции при отжиге имеют место в плотных слоях аморфной фазы.
Рис. 6.
Зависимость параметра τ/τ0 от времени отжига при 140 °С: ◼ – 0% порфирина, ⚫ – 1%, ▲ – 3%, ▼ – 5%.
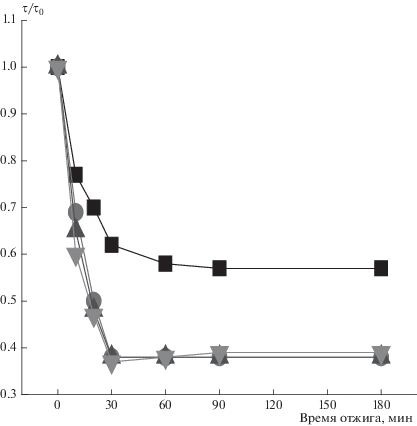
Рис. 7.
Зависимость τ от времени отжига при 140 °С в ПГБ, рассчитанное из спектров ЭПР (1) и по программе NLSL (2).
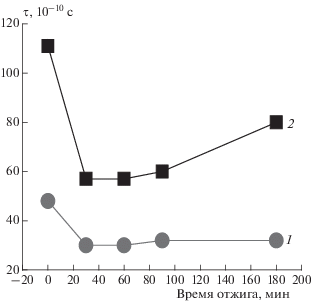
Рост степени кристалличности при отжиге, как отмечалось выше, обуславливает достаточно сильный рост плотности цепей в аморфной фазе, и радикал теряет способность проникать в наиболее плотные аморфные структуры. Именно поэтому молекулярная подвижность возрастает с ростом времени отжига.
Регистрируемые концентрации радикала в волокнах также свидетельствуют о резком уменьшении этой величины после отжига в течении 2 ч. Так, например, в волокнах ПГБ концентрация радикала составляла 8 ⋅ 1015 спин/г, после отжига в течение 2 ч – 1.4 ⋅ 1015 спин/г. Аналогичная картина наблюдается для всех изучаемых волокон. Были рассчитаны также времена корреляции с помощью программы NLSL после отжига в течении 2 ч. Эти данные представлены в табл. 2. Видно, что подвижность радикала в плотных областях, а следовательно, и молекулярная подвижность в аморфной фазе волокна разного состава (за исключением ПГБ) изменяются чрезвычайно слабо. Доля плотных аморфных областей при этом также резко уменьшается по сравнению с исходными образцами.
Таким образом, можно сделать вывод, что введение Zn-ТФП в ПГБ приводит к уплотнению аморфной фазы полимера в процессе формования волокна и, соответственно, к замедлению молекулярной подвижности (уплотнение аморфной фазы не столь велико, радикал сорбируется в таких плотных структурах). Отжиг при 140 °С обуславливает рост степени кристалличности и резкое уплотнение аморфных областей. Часть аморфной составляющей приобретает плотность, непроницаемую для радикала, и, как следствие, уменьшается концентрация радикала в волокне. Зонд теряет способность сорбироваться в таких плотных прослойках, и, как результат, подвижность зонда, а следовательно, и молекулярная подвижность цепей резко возрастают.
Наблюдаемые изменения молекулярной динамики радикала, а следовательно, и сегментарной подвижности при отжиге образцов мы объясняем процессами доориентации цепей. Отжиг при 140 °C приводит к росту доли предельно выпрямленных цепей. Природа такого роста заключается в том, что часть сегментов с высокой степенью выпрямленности β, большей критического значения h/L = $\sqrt {{2 \mathord{\left/ {\vphantom {2 k}} \right. \kern-0em} k},} $ где h – расстояние между концами цепей, а L – длина цепи, k – длина сегмента, при отжиге стремится принять предельно выпрямленную конфигурацию, поэтому при повышенных температурах мы наблюдаем увеличение степени кристалличности и доли паракристаллических структур в волокне, а также рост доли аморфной фазы с высокой степенью выпрямленности цепей [24]. Как результат концентрация радикала уменьшается и повышается подвижность радикала, а следовательно, растет и молекулярная подвижность, фиксируемая зондом, так как проницаемость радикала в плотные аморфные прослойки затруднена.
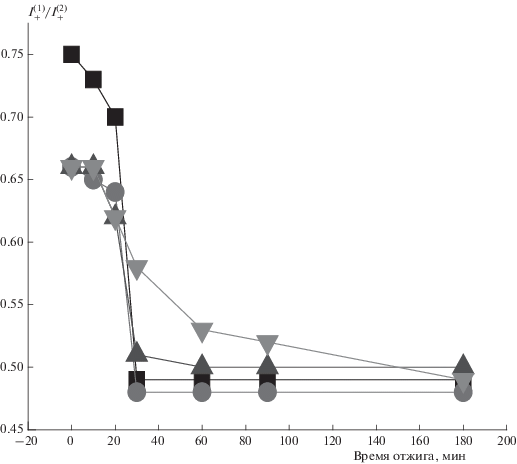
В работе также были получены данные об изменении параметра ${{I_{ + }^{{\left( 1 \right)}}} \mathord{\left/ {\vphantom {{I_{ + }^{{\left( 1 \right)}}} {I_{ + }^{{\left( 2 \right)}}}}} \right. \kern-0em} {I_{ + }^{{\left( 2 \right)}}}}$ (отношение амплитуд медленной и быстрой составляющих молекулярной динамики в спектре ЭПР) с ростом времени отжига (рис. 8). Видно, что при этом доля медленной составляющей уменьшается, тем самым подтверждая вывод о сорбции радикала в более рыхлых областях волокна.
Рис. 8.
Зависимость параметра ${{I_{ + }^{{\left( 1 \right)}}} \mathord{\left/ {\vphantom {{I_{ + }^{{\left( 1 \right)}}} {I_{ + }^{{\left( 2 \right)}}}}} \right. \kern-0em} {I_{ + }^{{\left( 2 \right)}}}}$ от времени отжига при 140 °С. Обозначения те же, что и на рис. 6.
Температурное воздействие на молекулярную динамику в волокнах ПГБ/Zn-ТФП
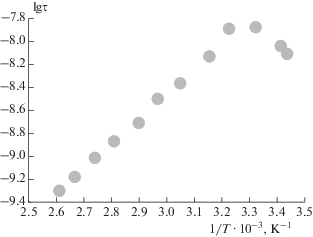
Дополнительная информация о динамическом поведении образцов ПГБ/Zn-ТФП различного состава была получена при исследовании температурной зависимости скорости вращения радикала и определении соответствующих значений энергии активации Eа. Характерной особенностью зависимости Eа от процентного содержания порфирина в ПГБ является рост этого параметра с ростом содержания Zn-ТФП в образце, эта величина в ПГБ составляет 35.7 кДж/моль, в ПГБ + 1% Zn-ТФП – 40.5 кДж/моль, в ПГБ + 3% Zn-ТФП – 42 кДж/моль и в ПГБ + 5% Zn-ТФП – 43 кДж/моль.
Характерно, что и зависимость энергии активации вращения зонда от концентрации радикала также имеет нелинейный характер. При введении 1% порфирина наблюдается достаточно резкий рост Еа, и дальнейший рост концентрации Zn-ТФП обуславливает более плавное изменение этой величины. Наблюдаемые изменения Еа связаны с ростом степени упорядоченности межкристаллитных областей.
Как уже отмечалось ранее, с ростом концентрации порфирина в волокне растет и доля более плотных образований. Характерной особенностью полученных зависимостей является их нелинейность (рис. 9). Для всех волокон на начальном этапе повышения температуры (до 40 °C) происходит рост времени корреляции, и только при более высоких температурах зависимость приобретает линейный характер, что свидетельствует о “размораживании” все более плотных участков аморфной фазы и проникновении радикала в эти области.
Воздействие водной среды на структуру и динамику ультратонких волокон ПГБ/Zn-ТФП
Отжиг при 70 °С – стандартизированная процедура, позволяющая провести сравнительный анализ различных систем. Выбор температуры обработки волокнистых материалов в воде в этом случае связан с необходимостью увеличения гидрофильности ПГБ, поскольку после удаления большей части воды сушкой меньшая ее часть остается в полимере в связанном виде (водородные связи) и способна к десорбции при значительно более высоких температурах. Даже небольшое увеличение гидрофильности полимерных матриц влияет на диффузионные процессы транспорта лекарственных веществ, которые, как правило, полярны. При комнатной температуре диффузия воды в полимер протекает долго, до нескольких суток. Мы не проводили эксперименты при более высокой температуре из-за возможности гидролитической деструкции полимерной матрицы. При выдержке в воде происходит изменение молекулярной и надмолекулярной структуры полимера. Необходимо было установить степень этих структурных изменений.
Так как представленные полимеры используются в медицинских целях, они часто подвергаются воздействию инертной низкомолекулярной жидкости при повышенной температуре. Были получены спектры ЭПР образцов ПГБ/Zn-ТФП, экспонированных в дистиллированной воде при 70 °C. Радикал вводили в волокна после отжига в водной среде (сушили образец в течение 2 сут). Результаты расчета значений параметра τ/τ0 (τ0 − исходное и τ − текущее значения), представленные на рис. 10, указывают на изменение подвижности радикала, а следовательно, молекулярной подвижности макромолекул в зависимости от продолжительности экспозиции при различном соотношении компонентов. Так, выдержка в течение 15 мин волокон ПГБ в водной среде при 70 °С обуславливает рост времени корреляции, отжиг образца ПГБ + 1% ZnТФП отжиг не приводит к изменению подвижности радикала, и в волокнах ПГБ с 3 и 5% порфирина наблюдается достаточно резкое снижение подвижности радикала, а следовательно, и подвижности молекул полимера.
Рис. 10.
Зависимость параметра τ/τ0 от времени отжига при 70 °С в водной среде для образцов ПГБ/Zn-ТФП: 1 – 0%, 2 – 1%, 3 – 3%, 4 – 5% порфирина.
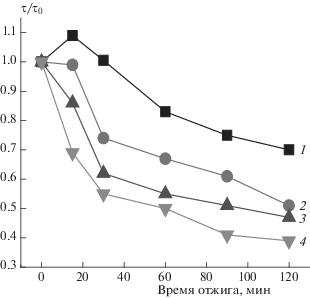
При более длительном воздействии на волокна наблюдается снижение значения параметра τ/τ0 для всех изучаемых образцов, что свидетельствует, об увеличении подвижности радикала, а следовательно, и молекулярной подвижности в аморфной фазе изучаемых волокон. Ранее в работе [15] было показано, что для пленочных образцов смешение хитозана с ПГБ приводит к гидрофилизации системы. Ее сорбционная емкость благодаря высокому сродству воды к молекулам данного полисахарида резко возрастает, что одновременно влечет усиление пластифицирующего действия воды на ПГБ. В этом случае экспоненциально возрастают коэффициенты диффузии воды и низкомолекулярных соединений. По-видимому, аналогичные процессы возникают в волокнах на основе смеси ПГБ/Zn-ТФП. Пластифицирующее действие воды, наиболее интенсивно реализующееся в аморфных областях ПГБ, в комбинации с повышенной температурой создает благоприятные условия для завершения его дополнительной кристаллизации и приводит к более упорядоченному состоянию межкристаллитных областей. После удаления воды в волокне остаются гидратированные комплексы, которые разрыхляют аморфную структуру в большей степени, чем частицы негидратированные, поэтому наблюдается снижение времени корреляции (рис. 10).
С ростом времени выдержки волокна в водной среде доля гидратированных комплексов изменяется и плотность аморфных областей все в большей степени уменьшается, что и способствует снижению τ. После отжига в течение 2 ч степень кристалличности волокон (по данным ДСК) увеличилась на ∼7%, в то время как после отжига при 140 °С эта величина возросла на ∼26%. Так же как и при отжиге при 140 °С, в данном эксперименте имеет место распрямление цепей ПГБ. Такие процессы обуславливают сильное уплотнение аморфной фазы волокна. В результате радикал не проникает в такие области и концентрация его должна уменьшаться, что подтверждается экспериментально на примере с ПГБ (рис. 11). Причем при сопоставлении данных по изменению концентрации радикала после отжига при 140 и 70 °С в водной среде видно, что скорость изменения концентрации в первом случае (расчет концентрации радикала проводился с использованием программного обеспечения фирмы “Bruker–winer and simfonia”) имеет более высокие значения. Если сопоставить характер изменения молекулярной подвижности при отжиге при 140 °С и комбинированном воздействии воды и температуры при 70 °С (рис. 6 и 10), видно, что в первом случае происходит более резкое изменение τ, поэтому можно заключить, что именно при 140 °С более интенсивно протекают процессы уплотнения аморфных областей.
Рис. 11.
Изменение концентрации радикала в ПГБ от времени отжига при 140 °С (1) и при 70 °С в водной среде (2).
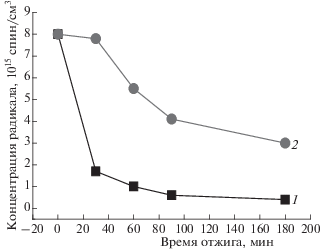
Таким образом, воздействие температуры и продолжительности выдержки в водной среде на ультратонкие волокна, согласно динамическому поведению радикала, достаточно сильно влияет на структуру межкристаллитных областей изучаемых волокон. Изменение значений времени корреляции при воздействии воды указывает на рост жесткости макромолекул и их уплотнение в волокнах. Наиболее сильное воздействие оказывает отжиг в водной среде на волокна ПГБ + 3 и 5% Zn-ТФП. Это может быть обусловлено достаточно высокой проницаемостью молекул воды в аморфные области композиции благодаря наличию в межмолекулярном пространстве полимера гидратированных комплексов, которые могут значительно увеличивать межмолекулярный объем в аморфных областях полимера. Радикалы, находящиеся в этих областях, получают возможность вращаться быстрее.
Пластифицирующий эффект наступает при более высоких концентрациях молекул Zn-ТФП благодаря повышенной скорости диффузии воды в объеме полимера. При таком воздействии происходит релаксация напряжений в неравновесной надмолекулярной структуре волокна, которая приводит к дополнительному уплотнению аморфных межкристаллитных областей в результате их укладки в кристаллиты и паракристаллиты. Радикал теряет взможность проникнуть в такие области. При более длительном времени экспозиции в водной среде всех типов образцов, независимо от концентрации Zn-ТФП, молекулярная подвижность ПГБ в волокнах увеличивается, что обусловлено уплотнением части аморфной фазы и, как результат, сорбцией радикала в наиболее рыхлых областях.
Также был рассчитан параметр ${{I_{ + }^{{\left( 1 \right)}}} \mathord{\left/ {\vphantom {{I_{ + }^{{\left( 1 \right)}}} {I_{ + }^{{\left( 2 \right)}}}}} \right. \kern-0em} {I_{ + }^{{\left( 2 \right)}}}},$ характеризующий отношение доли плотных областей к доле рыхлых. На основании полученных данных можно сделать вывод, что с ростом времени экспозиции в водной среде доля плотных областей, в которых сорбируется радикал, уменьшается. Полученные данные хорошо коррелируют с данными по молекулярной подвижности.
Воздействие озонолиза на динамику вращения радикала в волокнах композиции ПГБ/Zn-ТФП
При использовании биомедицинских материалов одновременно с механическим и температурным воздействиями на их структуру и сегментарную подвижность оказывает влияние озон. Здесь следует указать два источника его появления в атмосфере. Во-первых, озон образуется при работе мощных электрических приборов, обеспечивающих жизнедеятельность пациентов как в процессе хирургических операций, так и при терапии или мониторинге в стационаре. Во-вторых, озон в некоторых частных случаях продолжает использоваться при стерилизации медицинских изделий. Несмотря на повседневный контакт данного агрессивного соединения с полимерами, его влияние на их морфологические и динамические характеристики остается малоисследованной областью полимерного материаловедения.
В работе изучали влияние озонного воздействия на смесевые композиции при температуре 25 °С (рис. 12). Озонолиз сложным образом влияет на структуру полимера. Имеет место два параллельных процесса: 1) сшивка макромолекул, а также их распрямление, что обуславливает рост жесткости и, как следствие, замедление молекулярной подвижности; 2) рост подвижности из-за деструкции цепей и перехода их в более свернутую конформацию. Природа роста жесткости цепей заключается в том, что часть сегментов с высокой степенью выпрямленности β (β = h/L = $\sqrt {{2 \mathord{\left/ {\vphantom {2 k}} \right. \kern-0em} k}} $) после окисления стремятся принять предельно выпрямленную конфигурацию. Как показали результаты ранней работы [24], самопроизвольная доориентация при озонировании имеет место в трех случаях: 1) петли макромолекул на границе кристаллит–аморфная фаза окисляются с наиболее высокой скоростью вследствие высоких напряжений и при больших значениях β распрямляются; 2) макромолеклы, жесткость которых возрастает вследствие образования полярных кислородсодержащих групп в боковых цепях, образуют физические межмолекулярные сшивки, при этом параметр k уменьшается и, как результат, растет величина β, что приводит к распрямлению цепей; 3) макромолекулярные цепи в аморфных областях с достаточно высокой степенью выпрямленности при озонолизе имеют тенденцию к распрямлению. Эти три составляющие обуславливают рост τ. Параллельно имеют место процессы деструкции макромолекул, их переход в более свернутую конформацию и, как результат, рост молекулярной подвижности. На первых стадиях окисления преобладают процессы, обуславливающие замедление подвижности цепей, при более длительном озонолизе (более 100 мин) – процессы разрушения структуры волокна.
Рис. 12.
Зависимость времени корреляции τ от длительности озонного воздействия при содержании порфирина 0 (1), 1 (2), 3 (3) и 5% (4).
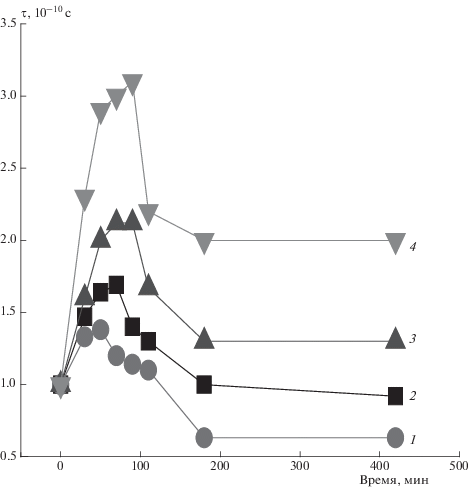
Из рис. 12 видно, что чем выше концентрация порфирина, тем выше эффект замедления подвижности радикала, а следовательно, молекулярной подвижности в волокне. Наблюдаемые изменения мы объясняем ростом степени кристалличности волокон за счет возникновения паракристаллических структур по мере увеличения концентрации Zn-ТФП, когда увеличивается доля трехмерных кристаллитов (растет доля складок на границе кристаллит–аморфная фаза), а также доля паракристаллических структур, что приводит все к более эффективной физической сшивке цепей при озонолизе. Аналогичные результаты были получены при озонолизе полиэтилена, полипропилена и смесей ПГБ с полиуретаном [25–27].
Для подтверждения изложенных выше механизмов, приводящих к замедлению подвижности радикала, а следовательно, молекулярной подвижности в аморфной фазе, проводились исследования методом ИК-спектроскопии данных образцов. В табл. 3 представлены данные по изменению площади пиков при 1179 (аморфная составляющая) и 1279 см–1 (кристаллическая составляющая), нормированных на площадь полосы при 1720 см–1 для исходных (1) и после озонолиза (1') волокон в течение 30 мин озонного воздействия. Представленные данные свидетельствуют о росте паракристаллических структур, во-первых, с увеличением концентрации порфириновых комплексов в волокне и, во-вторых, в процессе озонолиза. Представленные данные также подтверждают вывод о росте паракристаллических структур при озонолизе волокон при небольших временах воздействия.
Таблица 3.
Значения интегралов пиков
| № образца | Отношения оптических полос | |
|---|---|---|
| 1276/1720 см–1 | 1179/1720 см–1 | |
| 1 (исходный) | 0.39 | 0.73 |
| 1' (окисленный) | 0.41 | 0.7 |
| 2 (исходный) | 0.49 | 0.7 |
| 2' (окисленный) | 0.52 | 0.65 |
| 3 (исходный) | 0.52 | 0.61 |
| 3' (окисленный) | 0.55 | 0.54 |
| 4 (исходный) | 0.54 | 0.60 |
| 4' (окисленный) | 0.57 | 0.38 |
Таким образом, озонокислородное воздействие обуславливает рост времени корреляции зонда в волокнах, а следовательно, замедление молекулярной подвижности на ранних стадиях окисления (до 100 мин), что вызвано как межмолекулярной физической сшивкой макромолекул, так и процессами доориентации цепей. При более глубоких степенях окисления преобладают процессы деструкции цепей, что обеспечивает рост подвижности зонда, а следовательно, и молекулярной подвижности в аморфной фазе волокон.
ЗАКЛЮЧЕНИЕ
Введение малых концентраций порфирина (1, 3, 5%) приводит к росту степени кристалличности ПГБ, и как отклик на изменение кристаллической фазы нами было зарегистрировано изменение динамики вращения зонда, что свидетельствует об изменении молекулярной подвижности в аморфных областях полимера. Отжиг при 140 °C приводит к резкому увеличению доли кристаллической фазы в волокнах ПГБ и ПГБ + 1% Zn-ТФП. Введение 3% порфирина обуславливает резкое снижение степени кристалличности в волокнах, что вызвано деформацией частицами Zn-ТФП граничных кристаллических областей и кристаллов с дефектами. При 5%-ном содержании комплекса ΔH имеет более высокие значения, чем в ПГБ + 3% Zn-ТФП, что объясняется, на наш взгляд, сегрегацией частиц порфирина в частицы большего размера и, как следствие, достаточно низкой проницаемостью в расплавленные кристаллические области. Озонолиз (до 100 мин) волокон изучаемой смеси указывает на резкий рост времени корреляции, что объясняется ростом доли выпрямленных цепей и усилением межмолекулярного взаимодействия вследствие накопления кислородсодержащих групп. При более глубоком окислении подвижность радикала, а следовательно, и молекулярная подвижность возрастают, что объясняется интенсивной деструкцией цепей.
Взаимное влияние кристаллических и аморфных областей в биоразлагаемых высококристаллических полимерах и их композициях остается достаточно сложной и малоизученной проблемой современного полимерного материаловедения. Исследования состояния полимерной матрицы и роль Zn-ТФП впервые позволили интерпретировать на молекулярном уровне воздействие ряда агрессивных факторов (таких как температура и озон) на структурно-динамические характеристики волокон смесей ПГБ/Zn-ТФП.
Полученные в работе результаты позволяют рекомендовать данные материалы для биомедицинского применения в качестве матриц с антибактериальным эффектом. При этом стойкость к окислению рассматриваемых материалов в результате воздействия факторов, возникающих при некоторых видах стерилизации, достаточно высока.
В данной работе измерения кристалличности проводили методом ДСК на приборе DSC204 F1 фирмы “Netzsch”, Германия (Центр коллективного пользования Института биохимической физики РАН “Новые материалы и технологии”).
Список литературы
Соловьева A.Б., Беляев В.Е., Глаголев Н.Н. и др. // ЖФХ. 2005. Т. 79. № 4. С. 740.
Чудинов А.В., Румянцева В.Д., Лобанов А.В. и др. // Биоорган. химия. 2004. Т. 30. № 1. С. 99.
Kong L., Ziegler G.R. // Biomacromolecules. 2012. V. 13. P. 2247.
Schiffman J.D., Schauer C.A. // Polym. Rev. 2008. V. 48. № 2. P. 317; doi: 10.1080/15583720802022182
Xie G., Wang Y., Han X. et al. // Ind. Eng. Chem. Res. 2016. V. 55. № 26. P. 7116; https://doi:org/10.1021/ acs.iecr.6b00958
Kulkarni A., Bambole V.A., Mahanwar P.A. // Polym. Plas. Technol. and Eng. 2010. V. 49. № 5. P. 427.
Wang Y., Pan J., Han X. et al. // Biomaterials. 2008. V. 29. № 23. P. 3393; https://doi:org/10.1016/ j.biomaterials.2008.04.042
Mergaert J., Webb A., Anderson C. et al. // Appl. Environ. Microbiol. 1993. V. 59. № 10. P. 3233.
Loh X.J., Goh S.H., Li J. // Biomaterials. 2007. V. 28. № 28. P. 4113.
Жаркова И.И., Староверова О.В., Воинова В.В. и др. // Биомед. химия. 2014. Т. 60. Вып. 5. С. 553.
Карпова С.Г., Ольхов А.А., Иорданский А.Л. и др. // Хим. физика. 2016. Т. 35. № 8. С. 59.
Hu Y., Sato H., Zhang J. et al. // Polymer. 2008. V. 49. № 19. P. 4204; https://doi:org/10.1016/j.polymer.2008. 07.031
Ольхов А.А., Иорданский А.Л., Староверова О.В. и др. // Хим. волокна. 2015. № 5. С. 8.
Карпова С.Г., Ольхов А.А., Шилкина Н.Г. и др. // Высокомолекуляр. соединения. А. 2017. Т. 59. № 1. С. 53; doi: 10.7868/S2308112017010060
Иорданский А.Л., Ольхов А.А., Карпова С. Г. и др. // Том же № 3. С. 273; doi: 10.7868/S2308112017030075
Карпова С.Г., Ольхов А.А., Бакиров А.В. и др. // Хим. физика. 2018. Т. 37. № 2. С. 64.
Adler A.D., Longo F.R., Finarelli J.D. et al. // J. Org. Chem. 1967. V. 32. P. 476.
Филатов Ю.Н. Электроформование волокнистых материалов (ЭФВ-процесс). М.: “Нефть и Газ”, 1997.
Budil D.E., Lee S., Saxena S., Freed J.H. // J. Magn. Res. A. 1996. V. 120. P. 155.
Тимофеев В.П., Мишарин А.Ю., Ткачев Я.В. // Биофизика. 2011. Т. 56. № 3. С. 420.
Бучаченко А.Л., Вассерман А.М. Стабильные радикалы. М.: Химия, 1973.
Opfermann J. // Rechentechnik – Datenverarbeitung. 1985. V. 23. № 3. P. 26.
Vyazovkin S., Koga N., Schick C.V. Handbook of Thermal Analysis and Calorimetry, Applications to Polymers and Plastics. Amsterdam; Boston; London: Elsevier, 2002.
Озерин А.Н. Дис. … канд. хим. наук. М.: НИФХИ им. Л.Я. Карпова, 1977.
Карпова С.Г., Иорданский А.Л., Кленина Н.С. и др. // Хим. физика. 2013. Т. 32. № 6. С. 15.
Карпова С.Г., Попов А.А., Заиков Г.Е. // Высоко-молекуляр. соединения. 1991. Т. 33. № 5. С. 931.
Карпова С.Г., Иорданский А.Л., Попов А.А. // Хим. физика. 2012. Т. 31. № 1. С. 79.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика