

Химическая физика, 2019, T. 38, № 4, стр. 62-68
Механизм каталитической полимеризации 2-гидроксиэтилметакрилата под действием оксокомплекса ванадия(IV)
С. Н. Холуйская 1, *, А. А. Гриднев 1
1 Институт химической физики им. Н.Н. Семёнова, Российской академии наук
Москва, Россия
* E-mail: s_n_khol@mail.ru
Поступила в редакцию 12.04.2018
После доработки 31.10.2018
Принята к публикации 21.11.2018
Аннотация
Обнаружена высокая каталитическая активность комплекса оксованадия(IV) с диметилсульфоксидом в реакции полимеризации 2-гидроксиэтилметакрилата (ГЭМА), причем с высокой селективностью по мономеру. Комплекс оксованадия(IV) с диметилсульфоксидом не вызывает полимеризацию других мономеров, в том числе других метакрилатов. С помощью ЯМР на ядрах 13С установлена карбоцепная структура полимерного ГЭМА. Получаемый поли-ГЭМА имеет узкое, унимодальное молекулярно-массовое распределение. Установлено, что полимеризация имеет каталитический характер. Предложен координационный механизм с участием комплекса ванадия.
ВВЕДЕНИЕ
2-Гидроксиэтилметакрилат (ГЭМА) принадлежит к важному классу мономеров – (мет)акрилатам, содержащих группы с подвижными атомами водорода. На базе этих соединений получают уникальные по своим свойствам современные материалы, такие как сверхразветвленные [1, 2], стереоградиентные [3] и “умные” [4, 5] полимеры. Кроме того, гомо- и сополимеры ГЭМА, благодаря своей гидрофильности, нетоксичности и биосовместимости [6], стали объектом первостепенного значения в медицинском материаловедении. Полимеры ГЭМА, как правило, получают в процессе радикальной полимеризации, причем последние два десятилетия особое внимание уделяется разработке методов контролируемой полимеризации. Так, в 1999 году [7] впервые реализована контролируемая радикальная полимеризация ГЭМА с переносом атома (ATRP), катализированная соединениями одновалентной меди. Дальнейшее усовершенствование методики позволило проводить реакцию в протонных и апротонных растворителях (спирты, ДМСО, ТГФ) в присутствии воды [8–10]. Следующим шагом стал синтез гомо- и сополимеров ГЭМА с использованием активаторов, генерируемых и регенерируемых одноэлектронным переносом – AGET ATRP [11] и ARGET ATRP [12]. Однако указанные методы ограничивались синтезом полимеров ГЭМА (ПГЭМА) с невысоким молекулярным весом и, кроме того, по-прежнему требовали значительных концентраций металлсодержащих катализаторов, что негативно сказывалось на чистоте конечного продукта.
В 2013 году авторы работы [13] получили высокомолекулярный ПГЭМА методом “живой” полимеризации посредством электронного переноса (SET-LRP) при инициировании системой метил- α-бромфенилацетат/Cu(0). Реакция проходит с высокой скоростью в мягких условиях при низкой концентрации инициаторов, однако ее недостатками являются чувствительность к кислороду воздуха и наличие функциональных групп инициатора и атомов галогена в концевых группах макромолекул. Таким образом, из краткого обзора существующих данных видно, что продолжается постоянный поиск новых методов синтеза полимеров ГЭМА. В данной работе мы впервые использовали в качестве катализатора полимеризации ГЭМА соединение ванадия VO(DMSO)5(ClO4)2 (I).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Мономер ГЭМА (“ФГУП НИИ химии и технологии полимеров им. В.А. Каргина” г. Дзержинск) очищали от примесей дистилляцией в вакууме, отделяя фракцию с температурой кипения 67 °С при давлении 3.5 Торр. Катализатор I синтезировали по методу, изложенному в работе [14].
Реакцию полимеризации проводили в вакууме и на воздухе, а также в инертной атмосфере и кислороде, насыщая мономер и растворы катализатора инертным газом (аргоном класса чистоты “ос. ч.”) или О2 методом барботажа. При полимеризации в блоке катализатор растворяли непосредственно в мономере (интенсивное перемешивание в течение 5 мин). В остальных экспериментах I растворяли в этаноле или воде, затем смешивали с ГЭМА. Растворимый ПГЭМА получали в изопропаноле. К 4.5 мл спирта добавляли 0.4 мл ГЭМА, барботировали Ar в течение 15 мин, затем вводили шприцем предварительно насыщенный аргоном раствор I в ДМСО (40 мМ) и термостатировали при 50 °С. После термостатирования в течение 4 ч конверсия ГЭМА составила 100% по данным 1H-ЯМР (растворитель ДМСО-d6). Для исследования методом динамического светорассеяния реакционную смесь разбавили в 5 раз изопропанолом.
Гидродинамический диаметр молекул и распределение интенсивности рассеяния света частицами получены методом динамического светорассеяния на лазерном спектрометре-анализаторе Zetasizer Nano S (фирма Malvern Instruments, Великобритания) с углом детектора 173°.
Инфракрасные спектры регистрировали на ИК-фурье-спектрометре Bruker IFS 45 (Германия) с рабочим диапазоном 4700–400 см–1, детектором DTGS с разрешением 1–4 см–1. Регистрацию спектров проводили в таблетках KBr и между солевыми стеклами. Концентрацию ненасыщенных двойных связей определяли по интенсивности пика поглощения на частоте ν = 1635 см–1, относящейся к деформационным колебаниям С–Н связи группы –СН=СН2.
Твердотельные спектры ЯМР на ядрах 13С с вращением образца под магическим углом (ВМУ) получены на спектрометре Bruker AVANCE-II 400 (Германия), оснащенном системой ВМУ с частотами 400.1 и 100.4 МГц для ядер 1Н и 13С соответственно. Спектры регистрировались по методике переноса поляризации с высокомощной развязкой по протонам. В качестве внешнего стандарта шкалы химических сдвигов использовался кристаллический адамантан. Образец ПГЭМА получен полимеризацией в массе в течение 4 ч при температуре 50 °С и концентрации катализатора 1.0 мM.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Ранее мы обнаружили, что соединение I может катализировать некоторые реакции с участием спиртов. Во-первых, в присутствии I происходит одноэлектронное восстановление молекулярного О2 спиртом с образованием свободных радикалов [15, 16]. Во-вторых, I является эффективным катализатором алкоголиза эпоксидов и активированных двойных связей по реакции Михаэля [17]. В частности, было показано, что присоединение метанола к циклогексен-2-ону-1 происходит без изменения валентного состояния ванадия с образованием продукта с выходом 76%:
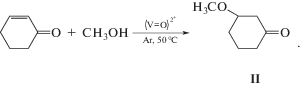
Обнаруженные реакции указывали на возможную активность катализатора I в полимеризации виниловых мономеров, в том числе, содержащих гидроксильные группы. Действительно, оказалось, что полимеризация ГЭМА происходит относительно быстро при комнатной температуре. В то же время, для таких мономеров, как метилметакрилат (ММА), акриловая кислота и N-винилпирролидон, катализатор неактивен. Важно отметить, что реакция нечувствительна к наличию кислорода: скорости полимеризации на воздухе и в аргоне близки.
Комплекс I вследствие своей ионной природы хорошо растворим в полярных растворителях, а также в ГЭМА, что позволяет проводить полимеризацию в массе. Вода и низшие спирты не препятствуют реакции, однако при содержании воды более 30 об.% происходит разделение фаз, которое не наблюдается, если в качестве растворителя применяются низшие спирты (этанол, изопропанол). Для блочной полимеризации ГЭМА использовали концентрации катализатора 0.2–5.0 мМ, т.е. менее 0.01 вес.%. При комнатной температуре реакция завершается за 22 ч, при температуре в 50 °С – в течение 4 ч. В результате полимеризации в массе образуется однородный, твердый, прозрачный, нерастворимый и химически стабильный материал. Он ограниченно набухает в полярных органических растворителях и воде; попытка его растворения при 80 °С в ДМФА в течение нескольких часов оказалась безуспешной, что доказывает сетчатую структуру полимера. Следует отметить, что образование сшивок при полимеризации ГЭМА в массе не говорит о специфике механизма реакции, а является системной особенностью мономера, которая обусловлена присутствием диметакрилата этиленгликоля.
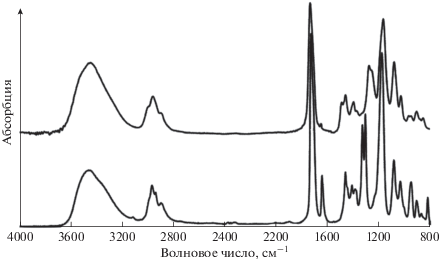
Окончанию реакции соответствует исчезновение в ИК-спектрах полос, относящихся к колебаниям двойной связи ГЭМА с ν = 1639 и 817 см–1 (рис. 1). При этом в полимере сохраняются интенсивные полосы поглощения карбонильной группы С=О (ν = 1722 и 1450 см–1), связей С–О (ν = 1175 и 1070 см–1) и гидроксильных групп в области 3200–3700 см–1. Мы не наблюдали различий в ИК-спектрах образцов, полученных на воздухе и в аргоне.
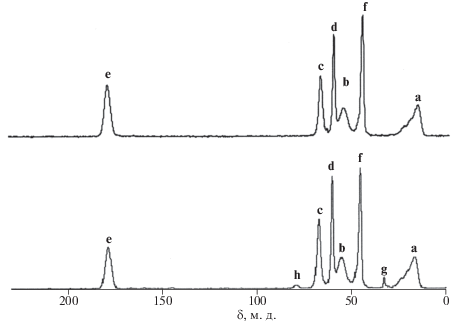
Рис. 1.
ИК-спектры мономера ГЭМА (нижний спектр) и ПГЭМА (верхний спектр), полученного в массе в присутствии I ([I]o = 1.0 мM, t = 22 °С, воздух).
В свете наших прежних экспериментов по присоединению спиртов к С=С связи (реакция (1)) нельзя было исключить образование линейного полиэфира:
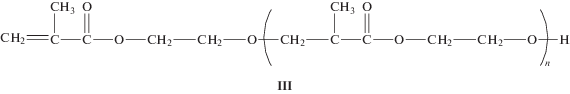
Макромолекулы такого строения могут образовываться, если ключевая стадия роста цепи представляет собой присоединение по Михаэлю алкоксильного остатка ГЭМА к двойной связи молекулы мономера. Образование полиэфира как основного продукта анионной полимеризации ГЭМА многократно описано в работах [18, 19]. При реализации данного направления в ИК-спектре должно было бы наблюдаться расходование полосы поглощения OH-групп, однако, как видно из рис. 1, интенсивность полос в области 3200–3700 см–1 при полимеризации не уменьшается.
Метод ЯМР подтвердил отсутствие гетероцепей в синтезированном ПГЭМА. На рис. 2 представлены твердотельные спектры двух образцов, синтезированных в присутствии О2 (воздух) и в вакууме. Оба спектра принципиально не различаются. Спектры содержат основные мультиплетные сигналы со значениями химического сдвига δ = 16.32, 45.20, 55.19, 60.23, 67.26 и 178.62 м. д., относящиеся к метильной группе a, четвертичному атому углерода f, метиленовой группе b основной цепи, углеродам этиленгликольной группы с и d и углероду карбонильной группы е соответственно. Эти данные полностью совпадают со спектром карбоцепного ГЭМА, приведенным в работе [19]. В бескислородной среде в спектре дополнительно проявляются слабые сигналы с δ = 32.83 (g) и 78.54 (h) м. д. (рис. 2, нижний спектр) и суммарной интегральной интенсивностью около 2%. Сигналы относятся к третичному атому углерода –CCH2CH(CH3) концевого фрагмента цепи и углероду фрагмента –CO2CH2CH2OCH2–. Авторы работы [1] наблюдали аналогичные сигналы в спектре сверхразветвленного поли-ГЭМА. Следовательно, синтезированный ПГЭМА имеет следующее строение:
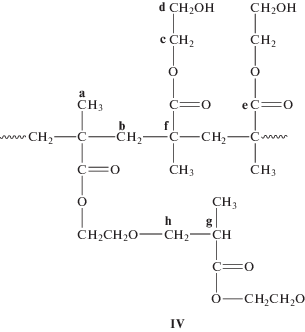
Рис. 2.
Спектры 13C-ЯМР полимера ПГЭМА, полученного на воздухе (верхний спектр) и в вакууме (нижний спектр) при полимеризации в массе в течение 4 ч, [I]o = 1.0 мM, t = 50 °С.
Таким образом, метод ЯМР показал, что основными структурными элементами синтезированного ПГЭМА являются карбоцепные макромолекулы и доля полиэфирных фрагментов пренебрежимо мала.
Полимеризация акрилатов может происходить по радикально-цепному, анионному и координационному механизмам, катионная полимеризация для этих соединений неизвестна. Радикальная полимеризация ГЭМА в присутствии I представлялась вполне возможной с учетом ранее полученных данных о генерации свободных радикалов при катализированном I окислении углеводородов. В литературе описана эмульсионная полимеризация ММА и стирола, инициированная радикальными частицами, возникающими при взаимодействии молекулярного O2 с комплексным соединением ванадия с циклопентадиенильными лигандами Cp2VCl2 [20]. Однако, как отмечалось выше, ММА и другие виниловые мономеры в присутствии I не полимеризуются ни на воздухе, ни в инертной атмосфере.
Наиболее наглядный способ идентификации радикального механизма состоит в изучении взаимодействия радикальных ловушек с активным центром роста цепи. К сожалению, ингибиторы радикальной полимеризации, такие как нитроксильные радикалы и гидрохинон, взаимодействуют с катализатором I, и поэтому было невозможно применить метод ингибиторов для выяснения природы активных центров.
Известно, что ряд соединений кобальта, а именно кобалоксимы, служат эффективными агентами переноса цепи на мономер в реакции радикально-цепной полимеризации метакрилатов [21]. Миллимолярной концентрации этих соединений достаточно, чтобы снизить молекулярный вес полиметакрилатов до нескольких сотен. Следовательно, кобалоксимы Co могут применяться для индикации радикально-цепного процесса. Так, их введение в концентрации 0.1 мM при радикальной полимеризации ГЭМА приводит к образованию вязкого раствора олигомеров. Наши эксперименты с кобалоксимами V и VI в концентрациях 0.3–0.6 мM показали, что они не влияют на полимеризацию ГЭМА в присутствии I (1.0 мM): скорость реакции практически не изменилась, а конечный продукт по-прежнему представлял собой твердый, стеклообразный, сшитый полимер.
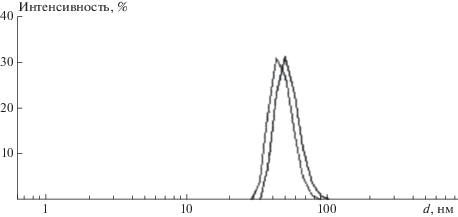
Полученные результаты показывают, что полимеризация ГЭМА под действием I не происходит ни по ионному, ни по свободно-радикальному механизмам. Для координационного механизма характерно узкое молекулярно-массовое распределение, поэтому мы исследовали синтезированный под действием I растворимый ПГЭМА методом динамического светорассеяния. Распределения частиц по размерам в единицах интенсивности показаны на рис. 3. Из рисунка видно, что наблюдается хорошая сходимость результатов двух измерений, проведенных для полученного образца; кроме того, как следует из анализа, форма кривых удовлетворительно описывается функцией Гаусса. Распределение имеет унимодальный характер с модой, соответствующей гидродинамическому радиусу ~25 нм, причем максимальная доля частиц, представленная данной фракцией, составляет 99.8%. Данные динамического светорассеяния свидетельствуют, что синтезированный под действием I ПГЭМА имеет более узкое молекулярно-массовое распределение в сравнении с образцами, полученными радикальной и анионной полимеризацией в присутствии обычно используемых инициаторов. Предположение о координационном характере реакции подтверждается исследованием взаимодействия катализатора и мономера методом ИК-спектроскопии. Введение эквимолекулярной концентрации (20 мM) комплекса I в раствор ГЭМА в ацетонитриле приводит к уширению полосы поглощения карбонильной группы с частотой ν = 1720 нм и появлению плеча с максимумом поглощения ν = 1711 нм, относящегося к ассоциированным с металлокомплеком карбонильным группам. Подобное изменение спектра ГЭМА, связанное с протонированием карбонильной группы, описано в работе [18].
Рис. 3.
Распределение интенсивности рассеяния света частицами ПГЭМА в зависимости от их гидродинамического диаметра d.
Таким образом, координационный механизм полимеризации ГЭМА под действием I представляется единственным механизмом, который может объяснить все полученные нами экспериментальные данные. Координационная полимеризация метакриловых мономеров, названная О. Вебстером полимеризацией с переносом группы (group-transfer polymerization, GTP), впервые описана в работе [22]. В качестве инициатора этой реакции использовали силилкетенацеталь. Инициирование заключается во внедрении молекулы мономера по O–Si связи енолята VII с образованием С–С связи и переносом триметилсилильной группы. Реакция роста протекает аналогичным образом и каждый акт присоединения мономера приводит к переносу Si-енольной группы и ее воспроизведению в качестве активного центра на конце растущей полиметакрилатной цепи. К признанным достоинствам GTP относятся технологически удобный режим реакции (температура 50–80 °С, широкий круг растворителей, включая полярные, инертность по отношению к кислороду) и возможность получения блок-сополимеров [23].
Детали механизма полимеризации с переносом группы все еще не выяснены полностью, однако очевидно, что именно енольная форма метакрилата способна участвовать в реакции продолжении цепи. Кроме того, поскольку ММА не полимеризуется в присутствии I, можно заключить, что гидроксильная группа ГЭМА играет важную роль при координации мономера катализатором. Мы предполагаем, что первая стадия полимеризации ГЭМА под действием I представляет собой образование аддукта енольной формы ГЭМА VIII, где группа Х вероятно представляет собой гидроксил и/или алкоксидную группу RO. Координация второй молекулы мономера приводит к перераспределению связей с образованием структуры IX, что способствует присоединению по активированной двойной связи енолята и формированию интермедиата X. В результате каждый акт роста цепи при полимеризации ГЭМА происходит путем присоединения молекулы мономера к концевому активному центру, который представляет собой циклическую алкокси-ванадий-енольную группу.
где X≡OH, RO.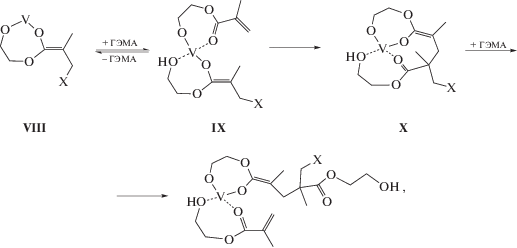
Полимеризация с переносом группы происходит в режиме “живых” цепей. Возможность “живой” полимеризации ГЭМА под действием I в настоящий момент изучается.
ЗАКЛЮЧЕНИЕ
Разработан метод получения полимерного материала на основе ГЭМА, отличающийся тем, что реакция полимеризации проводится в присутствии комплексного соединения оксованадия(IV) в мягких условиях (при комнатной температуре), нечувствительна к наличию кислорода, может осуществляться как в массе, так и в присутствии растворителей (вода, спирты). Преимуществами предлагаемого метода синтеза являются также полнота полимеризации: практически 100%-ное расходование мономера подтверждено методом ЯМР 13С и ИК-спектроскопией. Полученный полимерный продукт имеет карбоцепную структуру (данные ЯМР 13С) и узкое, унимодальное молекулярно-массовое распределение (данные светорассеяния).
Получены факты, свидетельствующие о нерадикальном характере полимеризации. Так, реакция нечувствительна к агентам переноса цепи (кобалоксимы) и имеет контролируемый характер. Кроме того, обнаружено, что комплекс оксованадия(IV) неактивен в полимеризации акрилатов без гидроксильной группы (ММА). Совокупность полученных результатов позволила выдвинуть гипотезу о координационной природе полимеризации ГЭМА с определяющей ролью гидроксильной группы. Предложен координационный механизм реакции полимеризации ГЭМА под действием соединения ванадия.
Работа выполнена в рамках темы № 45.11 госзадания ФАНО России № 0082-2014-0009 (№ гос. регистрации АААА-А17-117040610309-0), а также в рамках госзадания 0082-2014-0015 (№ АААА-А17-117032750201-9).
Список литературы
Jia Z., Yan D. // J. Polym. Sci., Part A: Polym. Chem. 2005. V. 43. P. 3502.
Chen Y., Shen Z., Barriau E. et al. // Biomacromolecules. 2006. V. 7. P. 919.
Miura Y., Shibata T., Satoh K. et al. // J. Amer. Chem. Soc. 2006. V. 128. P. 16026.
Xu F.-J., Kang E.-T., Neoh K.-G. // Biomaterials. 2006. V. 27. № 14. P. 2787.
Cayre O.J., Chagneux N., Biggs S. // Soft Matter. 2011. V. 7. № 6. P. 2211.
Hsieh K.-H., Young T.-H. // Polymeric Materials Encyclopedia / Ed. Salamon J.C. V. 5. Boca Raton: CRC Press, 1996. P. 3087.
Beers K.L., Boo S., Gaynor S.G. et al. // Macromolecules. 1999. V. 32. P. 5772.
Robinson K.L., Khan M.A., de Paz Banez M.V. et al. // Ibid. 2001. V. 34. P. 3155.
Teoh R.L., Guice K.B., Loo Y.-L. // Ibid. 2006. V. 39. P. 8609.
Bories-Azeau X., Armes S.P., van den Haak H.J.W. // Ibid. 2004. V. 37. P. 2348.
Oh J.K., Matyjaszewski K. // J. Polym. Sci., Part A: Polym. Chem. 2006. V. 44. P. 3787.
Paterson S.M., Brown D.H., Chirila T.V. et al. // Ibid. 2010. V. 48. P. 4084.
Nguyen N.H., Leng X., Percec V. // Polym. Chem. 2013. V. 4. P. 2760.
Selbin J., Holmes L.H. // J. Inorg. Nucl. Chem. 1962. V. 24. № 9. P. 1111.
Холуйская С.Н., Каспаров В.В., Рубайло В.Л. // Кинетика и катализ. 1991. Т. 32. № 5. С. 1140.
Холуйская С.Н., Рубайло В.Л. // Ibid. С. 1146.
Nikitin A.V., Kholuiskaja S.N., Rubailo V.L. // J. Chem. Research (S). 1994. № 9. P. 358.
Розенберг Б.А. // Высокомолекуляр. соединения. А. 2007. Т. 49. № 7. С. 1389.
Розенберг Б.А., Бойко Г.Н., Гурьева Л.Л. и др. // Ibid. 2004. Т. 46. № 3. С. 405.
Patra B.N., Bhattacharjee M. // J. Polym. Sci., Part A: Polym. Chem. 2006. V. 44. P. 2749.
Gridnev A.A., Ittel S.D. // Chem. Rev. 2001. V. 101. P. 3611.
Webster O.W., Hertler W.R., Sogah D.Y. et al. // J. Amer. Chem. Soc. 1983. V. 105. P. 5706.
Webster O.W. // Adv. Polym. Sci. 2004. V. 167. P. 1.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика