

Химическая физика, 2019, T. 38, № 4, стр. 45-52
Термические превращения 2,4-бис(N,N-диметиламино)-6-тринитрометил-1,3,5-триазина
В. В. Захаров 1, Н. В. Чуканов 1, *, Г. В. Шилов 1, Г. В. Малков 1, А. В. Шастин 1, Б. Л. Корсунский 1, **
1 Институт проблем химической физики Российской академии наук
Московская обл., Черноголовка, Россия
* E-mail: chukanov@icp.ac.ru
** E-mail: kors@polymer.chph.ras.ru
Поступила в редакцию 31.08.2018
После доработки 05.09.2018
Принята к публикации 20.09.2018
Аннотация
С целью прогнозирования термической стабильности энергоемких производных триазина, в том числе совместимости тринитрометильных заместителей с диалкиламиногруппами, термические превращения 2,4-бис(N,N-диметиламино)-6-тринитрометил-1,3,5-триазина (I) изучены в температурном интервале 170–623 K с применением методов дифференциального термического анализа, масс-спектрометрии, монокристальной и порошковой рентгеновской дифрактометрии. Соединение I характеризуется сильной анизотропией температурного расширения и при 365 K претерпевает полиморфное превращение α-I → β-I, которому предшествует скачкообразное падение плотности. Полиморфное превращение происходит с разрушением кристаллов и сопровождается частичным механохимическим разложением I. Плавление I при температуре около 396 K приводит к резкому ускорению разложения. Измерены тепловые эффекты полиморфного превращения, плавления и разложения I. По данным дифференциальной сканирующей калориметрии проведена оценка активационных параметров разложения I в расплаве. Определен состав образующихся газообразных продуктов и предложен механизм разложения I, согласно которому в лимитирующей стадии процесса происходит окисление диметиламиногруппы нитрогруппой.
ВВЕДЕНИЕ
Тринитрометильные производные полиазотистых гетероциклических соединений являются потенциальными компонентами новых мощных энергоемких композиций [1–6]. В последнее время был опубликован ряд работ по исследованию кинетики и механизмов термического разложения тринитрометильных производных триазина [7–10]. Однако имеющиеся данные о термической стабильности таких соединений и совместимости тринитрометильной группы с другими функциональными группами недостаточны. С целью восполнения этого пробела был синтезирован 2,4-бис(N,N-диметиламино)-6-тринитрометил-1,3,5-триазин (I) – модельное соединение, содержащее, наряду с тринитрометильной группой, диметиламиногруппы.
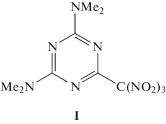
Предварительные исследования показали, что при температурах ниже точки плавления разложение I происходит с очень низкими скоростями, а, плавление сопровождается резким возрастанием скорости разложения, что, с одной стороны, делает практически невозможным проведение корректных изотермических кинетических измерений в расплаве, а с другой стороны, свидетельствует о плохой совместимости тринитрометильной группы с диметиламиногруппой, с точки зрения термической стабильности. Однако при изучении поведения соединения I при температурах ниже точки плавления нами был обнаружен ряд аномальных явлений. Настоящая работа посвящена исследованию термических превращений I.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Соединение I синтезировано нуклеофильным замещением тринитрометильной группы в 2-диметиламино-4,6-бис(тринитрометил)-1,3,5-триазине под действием диметиламина по известной методике [11]. Температуры плавления, ЯМР 1Н спектры I соответствовали литературным данным [11]. Чистота I подтверждена данными элементного анализа. Найдено (мас.%): C 29.03; H 3.78; N 33.52; C7H11N7O6. Вычислено (мас.%): C 29.07; H 3.83; N 33.90.
Термическое разложение I изучено в неизотермическом режиме методами дифференциально-сканирующей калометрии (ДСК) и термогравиметрии (ТГ) с применением синхронного термического анализатора NETZSCH STA 409C Luxx (Германия), сопряженного с квадрупольным масс-спектрометром QMS 403C Aeolos (диапазон температур 298–623 K, продувка аргоном со скоростью 40 мл/мин, скорость нагрева от 1 до 10 K/мин, масса навесок 3.2–7.3 мг). Интегрирование эндо- и экзотермических пиков на кривых ДСК и тем самым определение величин тепловых эффектов проводилось с использованием программного обеспечения NETZSCH Proteus® для термического анализа, лицензированного и установленного на синхронном термическом анализаторе. Масс-спектрометрический анализ газообразных продуктов термического разложения I проводился при энергии ионизирующих электронов, равной 70 эВ.
ДСК-эксперименты в режиме термоциклирования выполнены на дифференциальном сканирующем калориметре DSC821e (Mettler Toledo, Швейцария) для образца с массой 9 мг в диапазоне температур от 60 до 105 °С со скоростью нагревания и охлаждения 10 °С/мин; промежуточная выдержка при 60 °С в течение 3 мин; частота фиксации данных составляла 1 Гц. Для постановки экспериментов использовались стандартные алюминиевые тигли емкостью 40 мкл (№ 00027331, Mettler Toledo, Швейцария), вещество предварительно измельчалось в агатовой ступке. Первичную обработку термограмм проводили с помощью программного обеспечения STAR 9.10 (Mettler Toledo, Швейцария).
Исследования поликристаллических образцов 2,4-бис(N,N-диметиламино)-6-тринитрометил-1,3,5-триазина (I) методом порошковой рентгеновской дифрактометрии выполнены с использованием дифрактометра ARL X’TRA (THERMO FISHER SCIENTIFIC, Швейцария). Геометрия съемки θ-θ, Cu(Kα)-излучение, твердотельный детектор, шаг сканирования 0.02°, время измерения в точке 1 с. Исследования проводились при температурах 298, 358 и 378 K. Скорость изменения температуры составляла 5 K/мин. Образцы для дифрактометрических исследований готовили в виде смесей порошка соединения I с небольшим количеством силиконового масла. Монокристальные дифрактометрические исследования выполнены на дифрактометре Agilent Xcalibur c детектором Eos CCD в температурном интервале 170–350 K.
ЭКСПЕРИМЕНТАЛЬНЫЕ РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Изменения, происходящие в соединении I при варьировании температуры от 170 до 378 K, изучены с применением дифрактометрических методов. Данные, полученные на монокристалле, свидетельствуют об отсутствии полиморфных переходов при температурах ниже 350 K. Дифрактометрические данные соответствуют моноклинной α‑модификации I. Вплоть до температуры в 350 K кристалл сохраняет свою целостность, однако при более высоких температурах происходит его разрушение. В табл. 1 и на рис. 1, 2 приведены температурные зависимости параметров элементарной ячейки α-I, определенные из данных монокристальных исследований. Как видно из этих данных, температурное расширение α-I характеризуется сильной анизотропией, причем параметр b практически не изменяется с ростом температуры. Еще одной характерной особенностью α-I является резкое возрастание параметра c и менее резкий скачок параметра a при 350 K (рис. 1, 2). Возможно, резкое увеличение объема элементарной ячейки связано с разгрузкой внутренних напряжений в кристалле. С ростом температуры плотность кристалла уменьшается, но может не достигать равновесного значения из-за внутренних напряжений, создаваемых структурными дефектами. Тем не менее, при достаточно высокой температуре (особенно накануне фазового превращения) происходит мобилизация дефектов, что приводит к быстрому достижению равновесной при данной температуре плотности.
Таблица 1.
Температурные зависимости параметров элементарной ячейки α-I (по данным монокристальной дифрактометрии)
| T, K | a, Å | b, Å | c, Å | β, ° | V, Å3 |
|---|---|---|---|---|---|
| 170 | 8.846 | 12.625 | 13.303 | 108.57 | 1408.39 |
| 200 | 8.884 | 12.630 | 13.317 | 108.40 | 1417.89 |
| 230 | 8.926 | 12.629 | 13.331 | 108.22 | 1427.39 |
| 250 | 8.954 | 12.626 | 13.344 | 108.09 | 1434.08 |
| 270 | 8.982 | 12.624 | 13.359 | 107.93 | 1441.20 |
| 290 | 9.009 | 12.623 | 13.376 | 107.72 | 1449.03 |
| 300 | 9.024 | 12.624 | 13.390 | 107.58 | 1453.79 |
| 310 | 9.032 | 12.622 | 13.400 | 107.44 | 1457.37 |
| 320 | 9.048 | 12.613 | 13.417 | 107.36 | 1461.47 |
| 330 | 9.062 | 12.612 | 13.420 | 107.26 | 1464.75 |
| 340 | 9.075 | 12.610 | 13.435 | 107.14 | 1469.16 |
| 350 | 9.099 | 12.627 | 13.492 | 107.01 | 1482.45 |
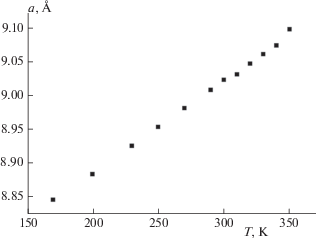
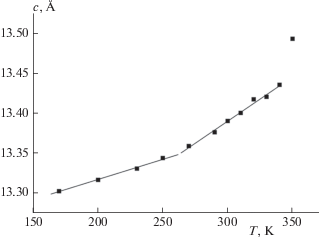
На графике температурной зависимости параметра c в области около 270 K наблюдается излом, свидетельствующий о возможном фазовом переходе второго рода. Можно предположить, что этот переход связан с изменением пространственной группы кристалла при сохранении моноклинной симметрии и параметров элементарной ячейки.
Анализ порошковых дифрактометрических данных показал, что при температуре 358 K большая часть рефлексов α-I сохраняется, но происходит существенное перераспределение их интенсивностей (рис. 3), что может являться следствием разгрузки микронапряжений в кристаллах (см. выше) и/или текстурирования поликристаллического образца. При 378 K рефлексы α-I не наблюдаются, и порошковая рентгенограмма соответствует новой, высокотемпературной модификации I (β-I) (см. рис. 4 и табл. 2). Полиморфный переход α-I → β-I происходит с резким увеличением объема и сопровождается выбросом части вещества из кюветы. Процессы, протекающие в кристалле при нагревании до 378 K, носят обратимый характер: после охлаждения до комнатной температуры порошковая дифрактограмма α-I практически полностью восстанавливается в течение 1 ч.
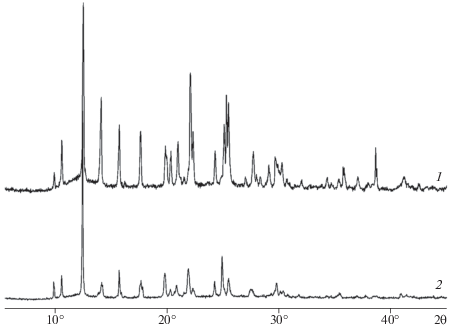
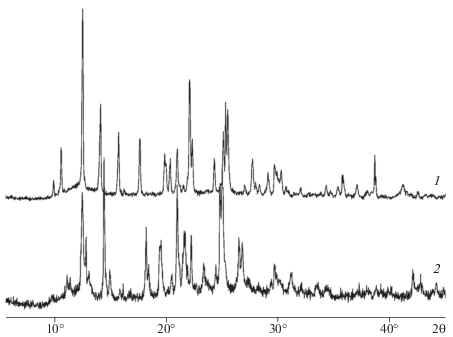
Таблица 2.
Порошковые рентгенографические данные для α-I и β-I
| 298 K (α-I) | 378 K (β-I) | |||
|---|---|---|---|---|
| I, % | d, Å | hkl | I, % | d, Å |
| 14 | 8.927 | 011 | 4 | 8.125 |
| 43 | 8.363 | –101 | 8 | 7.965 |
| 100 | 7.075 | 110 | 8 | 7.762 |
| 91 | 6.276 | 020 | 85 | 7.087 |
| 63 | 5.633 | 111 | 46 | 6.916 |
| 56 | 5.029 | –121 | 10 | 6.773 |
| 37 | 4.471 | –022 | 100 | 6.146 |
| 32 | 4.362 | –122 | 10 | 5.933 |
| 41 | 4.233 | –211 | 5 | 5.601 |
| 11 | 4.130 | –113 | 48 | 4.876 |
| 97 | 4.026 | 013 | 52 | 4.551 |
| 55 | 3.982 | 031 | 8 | 4.329 |
| 32 | 3.663 | –221 | 92 | 4.235 |
| 63 | 3.546 | 220 | 53 | 4.100 |
| 86 | 3.519 | 023 | 47 | 3.996 |
| 85 | 3.492 | 131 | 18 | 3.811 |
| 9 | 3.302 | 113 | 16 | 3.642 |
| 29 | 3.217 | 221 | 80 | 3.587 |
| 10 | 3.186 | 212 | 83 | 3.560 |
| 23 | 3.068 | –231 | 39 | 3.363 |
| 30 | 3.011 | 033 | 37 | 3.326 |
| 23 | 2.951 | –232 | 16 | 3.006 |
| 8 | 2.906 | 024 | 12 | 2.864 |
| 10 | 2.791 | –303 | 5 | 2.786 |
| 6 | 2.650 | –115 | 7 | 2.673 |
| 15 | 2.586 | 232 | 7 | 2.613 |
| 7 | 2.538 | 223 | ||
| 24 | 2.508 | –215 | ||
| 13 | 2.425 | –332 | ||
| 46 | 2.328 | 214 | ||
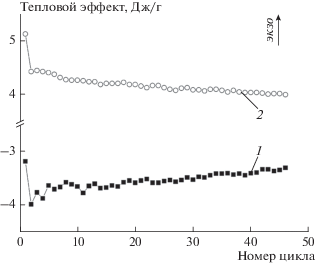
Термические превращения соединения I в неизотермическом режиме были изучены с помощью синхронного ДСК–ТГ анализа. На рис. 5 показаны кривые ДСК и ТГ анализа, полученные при плавлении и термическом разложении I при скорости нагревания 1.0 K/мин; на рис. 6 – кривые ДСК в диапазоне температур 298–623 K, полученные при различных скоростях нагревания.
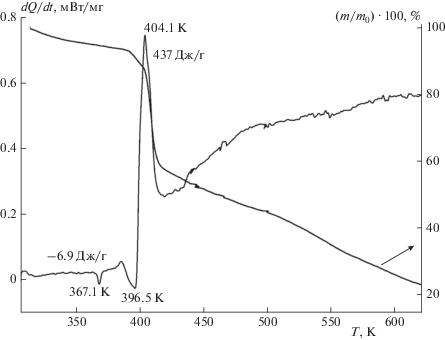
Рис. 6.
Кривые ДСК, полученные при различных скоростях нагревания I (K/мин): 1 (1), 2 (2), 5 (3), и 10 (4).
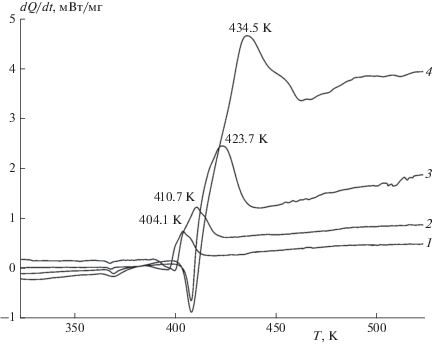
На кривых ДСК в диапазоне температур 366–371 K наблюдается эндотермический пик (–2.2 ± ± 0.2 кДж/моль), который соответствует полиморфному превращению α-I → β-I. В зависимости от скорости нагревания образцов I в диапазоне температур 396–409 K на кривых ДСК наблюдается также эндотермический пик, вызванный плавлением вещества, быстро переходящий в экзотермический вследствие высокой скорости термического разложения I в расплаве. Как видно из рис. 6, при увеличении скорости сканирования по температуре пики, соответствующие плавлению и максимальной скорости тепловыделения, смещаются в область более высоких температур.
Обратимость полиморфного превращения α-I → β-I, установленная на основании изучения порошковых дифрактограмм, была подтверждена методом ДСК при нагревании соединения I до 378 K и последующем его охлаждении. Кривые ДСК для полиморфных переходов α-I ↔ β-I в режиме термоциклирования приведены на рис. 7, 8. В ходе повторяющихся циклов нагрева–охлаждения изменяются не только формы кривых ДСК, но и положения пиков и величины тепловых эффектов (рис. 9, 10).
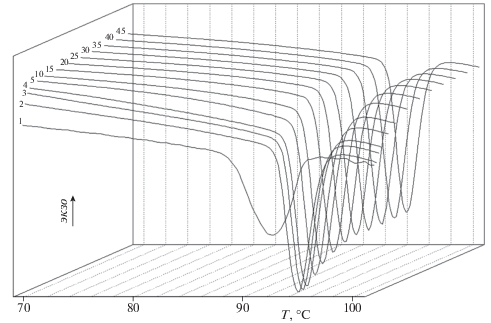
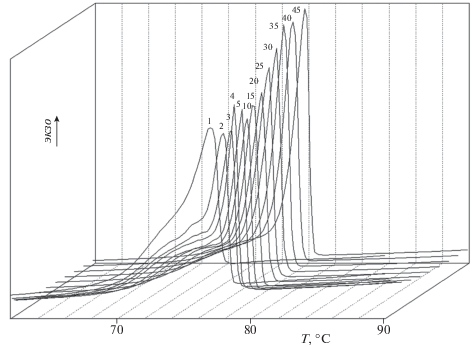
Рис. 9.
Зависимости положения пика на кривой ДСК от номера цикла для полиморфных переходов α-I → β-I (1) и β-I → α-I (2).
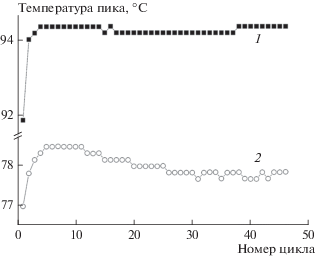
Рис. 10.
Зависимости теплового потока от номера цикла для полиморфных переходов α-I → β-I (1) и β-I → α-I (2).
Ступенчатый ход зависимостей на рис. 9 обусловлен дискретным шагом по температуре, равным 0.17 K. Важно отметить тот факт, что тепловые эффекты прямого и обратного полиморфных переходов уменьшаются с ростом номера цикла (за исключением второго цикла), причем в каждом цикле измеряемая абсолютная величина эндотермического эффекта при прямом переходе всегда ниже величины экзотермического эффекта при обратном переходе. Наиболее вероятное объяснение этих фактов заключается в том, что при прямом фазовом переходе, сопровождающемся увеличением объема, параллельно с процессом α-I → β-I происходит частичное разложение соединения I, имеющее механохимическую природу. Ранее мы уже сталкивались с подобным явлением при изучении полиморфного перехода ε → γ в кристаллах 2,4,6,8,10,12-гексанитро-2,4,6,8,10,12-гексаазаизовюрцитана [12]. Аномально высокие величины тепловых эффектов в первом цикле, скорее всего, обусловлены разложением термически нестабильной микропримеси, содержание которой в соединении I может составлять десятые доли процента.
Теплота разложения I в исследованном диапазоне температур 298–623 K найдена равной (139.1 ± ± 13.5) кДж/моль. Конечная потеря массы вещества в изученных условиях достигает 77%.
Для определения кинетических параметров термического разложения I было использовано уравнение Киссинджера (1) [13]:
(1)
$\ln \left( {\frac{\text{v}}{{T_{{max}}^{2}}}} \right) = - \frac{E}{{R{{T}_{{max}}}}} + \ln \left( {\frac{{AR}}{E}} \right),$При масс-спектрометрическом изучении газообразных продуктов разложения I, проведенном синхронно с исследованием тепловых эффектов, были зафиксированы пики при m/e = 15 $\left( {{\text{CH}}_{{\text{3}}}^{{\text{ + }}}} \right),$ 18 (H2O+), 30 (NO+), 31 $\left( {{\text{OCH}}_{{\text{3}}}^{{\text{ + }}}} \right),$ 44 (N2O+), 45 (НN2O+) и 46 $\left( {{\text{NO}}_{{\text{2}}}^{{\text{ + }}}} \right),$ что соответствует образованию метил- и/или метоксисодержащего летучего соединения, NO, N2O, H2O и NO2 в качестве основных газообразных продуктов термического разложения I. Следует отметить, что максимальные значения всех этих пиков были зарегистрированы при одном и том же значении температуры (404.1 K), которое совпадало с температурой, при которой скорость тепловыделения максимальна при той же скорости нагревания.
Кинетика термического разложения соединений, содержащих тринитрометильную группу, изучена очень подробно [14–16]. Характерной чертой этой реакции являются аномально высокие значения предэкспоненциального фактора. В то время как большинство мономолекулярных реакций характеризуется предэкспоненциальными множителями 1013–1014 с–1, тринитрометильные соединения вследствие высокой энтропии активации обычно распадаются с предэкспоненциальными факторами, находящимися в диапазоне 1015–1017 с–1. Приведенная выше величина 107.3 с–1 явно аномально низкая. Можно предположить, что эта величина является эффективной и описывает не элементарную (одностадийную), а сложную реакцию.
Мы полагаем, что сложность реакции обусловлена присутствием в молекуле субстрата диметиламиногрупп. Действительно, если в молекуле соединения I аминогруппы заменить на метоксильные, то разложение будет происходить так же, как и для типичных тринитрометильных соединений, а именно в соответствии с кинетическим уравнением первого порядка и предэкспоненциальным фактором, равным 1015.4 с–1 [16]. По-видимому, в исследованной реакции диметиламиногруппа не просто выполняет роль заместителя, проявляющего индуктивный, резонансный или какой-либо еще эффект. Скорее всего, она принимает непосредственное участие в одной из лимитирующих стадий процесса. Будучи, как все амины, восстановителем, она может окисляться нитрогруппой, входящей в состав тринитрометильного заместителя. Об этом, в частности, свидетельствует приведенный выше состав газообразных продуктов реакции: продукты, содержащие метильную группу, воду и окислы азота, выделяются практически синхронно. В рамках обсуждаемого механизма становится понятным и резкое ускорение реакции при переходе от твердой фазы к расплаву. Для эффективного окисления диметиламиногрупп нитрогруппами необходима должная взаимная ориентация этих групп. Такая ориентация затруднена в кристалле и значительно легче достигается в жидкости.
ВЫВОДЫ
Полученные результаты свидетельствуют о плохой кинетической совместимости тринитрометильных производных триазинов с диалкиламиногруппой. Обнаружен эффект аномального роста коэффициентов температурного уширения, предшествуюший полиморфному переходу в I и приводящий к дополнительному снижению термической стабильности вследствие возрастания свободного объема в кристалле.
Список литературы
Нечипоренко Г.Н., Лемперт Д.Б., Согласнова С.И. // Хим. физика. 2005. Т. 24. № 3. С. 74.
Shastin A.V., Godovikova T.I., Golova S.P. et al. // Mendeleev Commun. 1995. V. 5. P. 17.
Dharavath S., Zhang J., Imler G.H. et al. // J. Mater. Chem. A. 2017. № 10; https://doi.org/doi: 10.1039/C7TA00730B
Yuangang Xu, Cheng Shen, Qiuhan Lin et al. // J. Mater. Chem. A. 2016. V. 4. P. 17791.
Haiges R., Christe K.O. // Inorg. Chemistry. 2013. V. 52. № 12. P. 7249–7260.
Wang Yuan, Qi Cai, Song Jian-Wei et al. // J. Mol. Model. 2013. V. 19. P. 1079–1087.
Неделько В.В., Шастин А.В., Захаров В.В. // Матер. VII Всеросс. конф. “Энергетические конденсированные системы”. Черноголовка Дзержинский, 17–19 декабря 2014 г. Черноголовка: ИПХФ РАН, 2014. С. 31.
Неделько В.В., Шастин А.В., Конькова Т.С. и др. // Горение и взрыв. 2015. Т. 8. № 2. С. 160.
Неделько В.В., Захаров В.В., Корсунский Б.Л. и др. // Хим. физика. 2015. Т. 34. № 12. С. 39.
Kon’kova T.S., Miroshnichenko E.A., Nedel’ko V.V. et al. // Abstr. 18th Seminar on New Trends in Research of Energ. Mater. Pardubice, Czech Republic. 2015. P. 638.
Бахарев В.В., Гидаспов А.А., Якунина Н.Г., Булычев Ю.Н. // Хим.-фарм. журн. 2008. Т. 45. № 5. С. 11.
Chukanov N.V., Zakharov V.V., Korsounskii B.L. et al. // Cent. Eur. J. Energ. Mater. 2016. V. 13. № 2. P. 483.
Kissinger H.E. // Anal. Chem. 1957. V. 29. № 11. P. 1702.
Назин Г.М., Манелис Г.Б. // Успехи химии. 1994. Т. 63. № 4. С. 327.
Haзин Г.M. // Ibid. 1972. T. 41. № 9. C. 1537.
Неделько В.В., Захаров В.В., Корсунский Б.Л. и др. // Хим. физика. 2015. Т. 34. № 12. С. 39.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика