

Химическая физика, 2019, T. 38, № 6, стр. 15-21
Новые амфифильные производные каликс[4]арена с 4,5-дикарбокситриазолильными фрагментами – синтез и использование в мицеллярном катализе
Г. А. Фатыхова 1, Е. Г. Макаров 1, Д. А. Миронова 1, Э. Д. Султанова 1, В. А. Бурилов 1, *, С. Е. Соловьёва 1, 2, И. С. Антипин 1, 2
1 Казанский (Приволжский) федеральный университет
Казань, Россия
2 Институт органической и физической химии им. А.Е. Арбузова
Казанского научного центра Российской академии наук
Казань, Россия
* E-mail: ultrav@bk.ru
Поступила в редакцию 19.11.2018
После доработки 06.12.2018
Принята к публикации 21.01.2019
Аннотация
C использованием реакции азид-алкинового присоединения получены амфифильные производные каликсаренов с различной липофильностью, содержащие четыре 4,5-дикарбокситриазолильных фрагмента на верхнем ободе в стереоизомерной форме “конус”. Показано, что амфифильные макроциклы, замещенные четырьмя октильными и тетрадецильными группами на нижнем ободе, способны образовывать монодисперсные наноагрегаты в водных растворах. Установлено, что полученные макроциклы обладают флуоресценцией в синей области, а при росте концентрации наблюдается батофлорный сдвиг максимума эмиссии, согласующийся с образованием агрегатов. Полученные макроциклы были с успехом использованы в качестве мицеллярной среды для проведения реакции сочетания Сузуки, продемонстрировав количественные выходы продуктов сочетания йод- и бромаренов в водной среде при комнатной температуре.
ВВЕДЕНИЕ
В водной среде протекают важнейшие физиологические и природные процессы. Использование водной среды является актуальным и для проведения химических реакций [1]. Среди двенадцати принципов “зеленой” химии выбор соответствующего растворителя, способного снизить токсичность процесса, а также повысить его эффективность, является ключевым, поскольку на долю растворителя в химических реакциях приходится не менее 80% массовой доли [2]. Учитывая, что большинство органических молекул являются ограниченно или полностью водонерастворимыми, для возможности проведения их химических превращений в водных растворах можно использовать мицеллярный катализ, набирающий в последнее время большую популярность [3, 4]. Поверхностно-активные вещества (ПАВ) формируют в воде наноразмерные агрегаты, способные солюбилизировать водонерастворимые реагенты. Взаимодействие реагентов в малом объеме мицеллы способствует увеличению эффективности реакции. Однако для обеспечения высокой селективности и увеличения выходов продуктов реакции очень важен подбор ПАВ, способного создать подходящее микроокружение, поэтому разработка новых амфифильных веществ представляет собой актуальное направление мицеллярного катализа [5, 6].
В этой связи большими перспективами обладают производные каликс[4]аренов, отличающиеся от традиционных ПАВ способностью к образованию комплексов типа “гость–хозяин” [7]. Наличие нескольких реакционных центров, нетоксичность, многообразие стереоизомерных форм и доступность cделали молекулу каликсарена универсальной структурой для молекулярного распознавания, экстракции, создания стимул-чувствительных коллоидных систем, биомедицины и т.д. [8–11]. Благодаря наличию гидрофобной полости каликсарены нашли применение и в мицеллярном органокатализе [12, 13].
Ранее в нашей научной группе был разработан синтетический подход к производным каликсаренов, содержащих азидные фрагменты на верхнем ободе и алкильные фрагменты различной длины – на нижнем [14, 15]. Полученные азид-содержащие макроциклы могут быть использованы для синтеза любых функциональных, в том числе амфифильных, молекул с конъюгацией соответствующих функциональных фрагментов через реакцию азид-алкинового циклоприсоединения. Используя полученные ранее тетраазиды с бутильными и октильными заместителями [15], а также синтезированные тетраазиды с тетрадецильными фрагментами, в данной работе получены новые амфифильные макроциклы, содержащие четыре 4,5-дикарбокситриазолильных фрагмента, с использованием реакции азид-алкинового циклоприсоединения, а также исследованы их агрегационные характеристики. Полученные соединения были использованы в качестве мицеллярной среды для проведения некоторых органических реакций.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Каликсарены 11 и 12 (см. Схему 1 ) были получены по ранее отработанной методике [15]. Тетраазид 13 с тетрадецильными фрагментами был получен по аналогии с макроциклами 11 и 12 с использованием методики, включающей тетраалкилирование, ипсо-нитрование, восстановление и последующее диазотирование аминопроизводных 8, 9 с замещением диазогруппы на азид-анион. Полученные тетра-азидсодержащие макроциклы были введены в реакцию с ацетилендикарбоновой кислотой с получением окта-карбоксипроизводных 14–16, которые были выделены с практически количественными выходами.
Схема 1
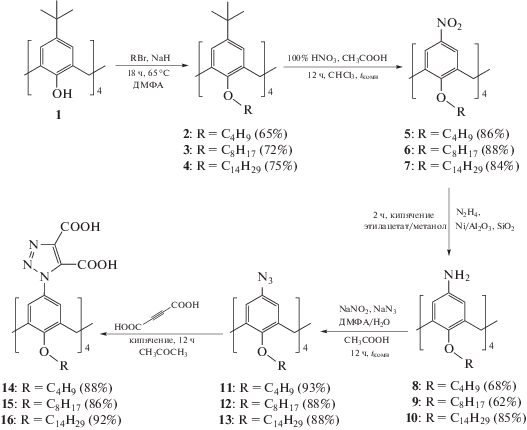
Структура полученных макроциклов 14–16 была установлена с использованием комплекса физических методов исследования, а состав – элементным анализом. В ЯМР-1Н-спектрах 14–16 в области 1–2 м. д. проявляются сигналы протонов –CH2- и –CH3-групп алкильных заместителей в виде неразрешенных мультиплетов. Мостиковые метиленовые группы резонируют при 3.2 и 4.5 м. д., уширенные сигналы ароматических протонов и OCH2-групп проявляются при 6.6 и 4.1 м. д. соответственно. Уширение сигналов в спектрах ЯМР-1Н является следствием обмена амфифильными молекулами между раствором и агрегатами [16]. В спектрах матрично-активированной лазерной ионизации (МАЛДИ) каликсаренов 14–16 были зафиксированы пики молекулярных ионов с отщеплением молекул CO2 и H2O. Так, в спектре МАЛДИ макроцикла 14 наблюдается серия пиков с m/z = 1226 [M–CO2 + 2H]+, 1182 [M–2CO2 + 2H]+, 1138 [M–3CO2 + 2H]+, 1094 [M–4CO2 + 2H]+, а также сигналы 1206 [M–CO2 – H2O]+, 1162 [M–2CO2 – H2O]+, 1118 [M–3CO2 – H2O]+. Наличие серии сигналов декарбоксилированных продуктов обусловлено легкостью декарбоксилирования в N‑гетероциклах, содержащих карбоксильную группу во 2-ом положении гетероцикла [17, 18]. Присутствие в молекулах 14–16 карбоксильных групп подтверждали методом ИК-спектроскопии. В области 1720–1730 см–1 проявляются валентные колебания карбонильной группы, а в области 3440–3455 см–1 – валентные колебания связанных водородными связями гидроксильных групп.
Каликсарены 14–16 оказались хорошо растворимыми в воде. Для измерения критической концентрации агрегации (ККА) макроциклов использовался метод солюбилизации пирена [19], для чего наблюдали изменения в соотношении интенсивностей первой и третьей полос испускания (при 373 и 384 нм) пирена при увеличении концентрации макроциклов. Было установлено (табл. 1), что значения ККА уменьшаются в ряду 14–16 сообразно увеличению гидрофобности молекул. Последняя согласуется с литературными данными, согласно которым увеличение гидрофобности молекул приводит к уменьшению значения ККА [20].
Таблица 1.
Значения ККА, размеры, индекс полидисперсности и поверхностный потенциал макроциклов 14–16
| Каликсарен | ККА, ммоль/л | d, нм | ИПД | ξ, мВ |
|---|---|---|---|---|
| 14 | 0.83 | 323 ± 39 | 0.459 ± 0.11 | –66 ± 3 |
| 15 | 0.13 | 19 ± 1 | 0.203 ± 0.01 | –45 ± 4 |
| 16 | 0.053 | 30 ± 3 | 0.301 ± 0.01 | –53 ± 6 |
Согласно данным динамического рассеяния света соединение 14 образует субмикронные агрегаты с размером порядка 300 нм и высоким индексом полидисперсности (ИПД), тогда как более гидрофобные макроциклы 15 и 16 образуют компактные наночастицы с размерами 19 и 30 нм соответственно (табл. 1). Согласно данным электрофоретического рассеяния света (ЭФРС) поверхностный заряд агрегатов составляет порядка –50÷–60 мВ, что свидетельствует об образовании устойчивых коллоидных систем (табл. 1). Такое распределение размеров можно объяснить следующим образом: гидрофобности макроцикла 14 недостаточно, чтобы преодолеть электростатическое отталкивание между анионными головными группами макроцикла и сформировать компактную мицеллу, в результате чего образуется полидисперсная смесь агрегатов различной морфологии. В случае же более липофильных макроциклов 15 и 16 образуются монодисперсные наночастицы.
Было также обнаружено, что макроциклы 14–16 проявляют интенсивную флуоресценцию в синей области спектра (рис. 1а).
Рис. 1.
Спектры флуоресценции соединения 16 при концентрациях 0.08 мкмоль/л – 2 ммоль/л (а); зависимость длины волны максимума эмиссии макроциклов 14 (◼), 15 (⚫) и 16 (▲) от десятичного логарифма концентрации макроцикла (б). Условия регистрации люминесценции: ТРИС, рН = 7.3, Т = 25 °С (λвозб = 400 нм).
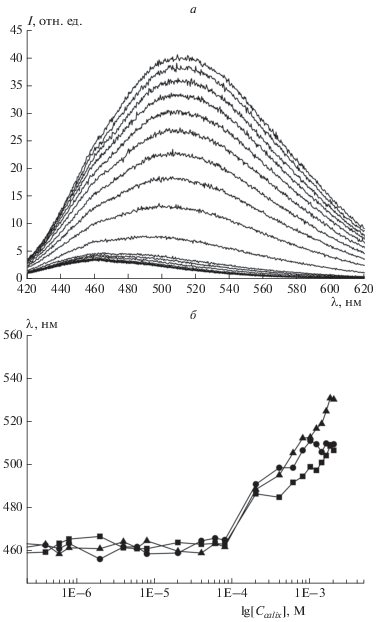
Оказалось, что с увеличением концентрации макроцикла наблюдается батофлорный сдвиг максимума эмиссии с 460 до 510 нм в случае макроциклов 14, 15 или до 530 нм в случае более липофильного макроцикла 16, что может быть связано с уменьшением полярности среды микроокружения флуорофора (сольватофлорный эффект) вследствие образования агрегатов. И, действительно, батофлорный сдвиг происходит в диапазоне концентраций, близких к значениям ККА (рис. 1б). Наличие собственной люминесценции открывает возможность использования макроциклов 14–16 в качестве флуоресцентных сенсоров для распознавания различных субстратов [21].
Способность полученных амфифильных макроциклов образовывать устойчивые ассоциаты позволила использовать их в качестве мицеллярной среды для реакций кросс-сочетания в водной среде. Реакция Сузуки (реакция сочетания арилгалогенидов с бороновыми кислотами, катализируемая палладием) является одним из наиболее универсальных и широко применяемых синтетических протоколов для селективного формирования углерод-углеродных связей, в частности для получения замещенных бифенилов [22]. Однако традиционно в данной реакции используют достаточно токсичные органические растворители (ДМФА, ТГФ, ацетонитрил и т.д.). Одним из решений данной проблемы является проведение реакции в условиях мицеллярного катализа, в котором реагенты могут быть солюбилизированы амфифильными соединениями [23]. На примере реакции сочетания п-йоднитробензола с фенилборной кислотой, катализируемой ацетатом палладия в водной среде, было изучено влияние длины алкильного заместителя в макроциклах 14 и 16 на выход целевого бифенила (Схема 2 ).
Схема 2
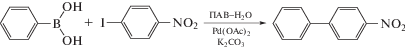
Для образования устойчивых коллоидных систем макроциклы использовались в концентрациях, на один порядок превышающих их ККА. Выход продукта устанавливали методом газовой хромато-масс-спектрометрии (ГХМС), отбирая пробы через 5 и 10 ч. Для выявления влияния структуры ПАВ на каталитическую активность также была проведена реакция в присутствии другого ПАВ – со стеарата натрия. Установлено, что проведение реакции в присутствии макроцикла 16 при комнатной температуре приводило к образованию основного продукта с выходом 99% (табл. 2). Аналогичная реакция с макроциклом 14 приводила к образованию лишь 11% целевого продукта, что может быть связано с образованием коллоидно-нестабильных полидисперсных агрегатов, неспособных эффективно солюбилизировать реагенты. В случае использования стеарата натрия, не имеющего гидрофобной полости, выход продукта составил 71%. Таким образом, наличие гидрофобной полости в структуре макроцикла 16, а также четырех липофильных заместителей приводит к увеличению его солюбилизирующей способности и, как следствие, повышает эффективность мицеллярного катализа.
Таблица 2.
Выход 4-нитро-1,1'-бифенила по данным ГХМС*
| ПАВ | Время, ч | Выход, % |
|---|---|---|
| – | 10 | 2 |
| 14 | 5 | 5 |
| 14 | 10 | 11 |
| 16 | 5 | 78 |
| 16 | 10 | 99 |
| Стеарат натрия | 10 | 71 |
Макроцикл 16 был использован в качестве мицеллярной среды в сочетании менее активных бром- и хлораренов с разными функциональными группами как донорной, так и акцепторной природы (Схема 3 , табл. 3).
Схема 3
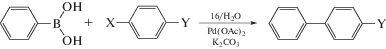
Таблица 3.
Выход 4-Y-1,1'-бифенилов по данным ГХМС*
| X | Y | Выход, % |
|---|---|---|
| Br | COCH3 | 99 |
| Br | NO2 | 91 |
| Br | OCH3 | 65 |
| Cl | NO2 | 20 |
Согласно полученным данным макроцикл 16 оказался одинаково эффективным как для йодаренов, так и для бромаренов с акцепторными группами. В случае бромарена с донорной метоксигруппой выход продукта снижался, что объясняется уменьшением скорости диссоциации С–Br при окислительном присоединении арилгалогенида к палладию (0) [22]. При использовании наименее активного п-хлорнитробензола выход снижался до 20%.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Физико-химические измерения
Эксперименты ЯМР выполнены на спектрометре Avance 400 Nanobay фирмы “Bruker”. В качестве внутреннего стандарта использовали CDCl3 (δ(Н) = = 7.26 м. д.) при температуре 25 °С. ИК-спектры регистрировали на спектрометре Bruker Vector-22 в интервале волновых чисел 400–4000 см–1 в таблетках KBr. Анализ методом тонкослойной хроматографии проводили на пластинах “Merck UV 254” с проявлением ультрафиолетовой лампой VL-6.LC (6W –254 nm tube). Элементный анализ полученных соединений был осуществлен с использованием анализатора PerkinElmer PE 2400 СHNS/О.
Масс-спектры MAЛДИ получены на масс-спектрометре UltraFlex III TOF/TOF в линейном режиме. В качестве матрицы использовали п‑нитроанилин или 2,5-дигидроксибензойню кислоту (Nd: VAG-лазер, λ = 355 нм). Фиксировались положительно заряженные ионы.
Эксперименты по динамическому и электрофоретическому рассеянию света проводились на приборе Zetasizer Nano ZS (“Malvern Instruments”, США) с источником излучения He–Ne-лампы с мощностью 4 мВт, длиной волны 633 нм и углом рассеяния света 173°. Данные обрабатывались программным обеспечением DTS (Dispersion Technology Software 5.00). Растворы фильтровали через фильтр Millex HV с размером пор 0.45 мкм перед измерением для удаления пыли. Для каждой системы проводилось минимум три эксперимента в одноразовых пластиковых ячейках DTS 0012 (“Sigma-Aldrich”, USA) при 298 K или капиллярных ячейках DTS 1070 при регистрации ЭФРС. Обработка статистических данных проводилась с использованием коэффициента t-Student, а погрешность определения размера частиц составляла <2%. Изучаемые образцы перед измерениями предварительно подвергали ультразвуковой обработке в течение 30 мин при 25 °С.
Спектры флуоресценции регистрировали в кварцевых кюветах с длиной оптического пути 10 мм на спектрофлуориметре Fluorolog FL-221 (“HORIBA Jobin Yvon”) в диапазоне 350–430 нм при длине волны возбуждения 335 нм с щелью в 2.5 нм для пирена и в диапазоне 420–620 нм при длине волны возбуждения 400 нм с щелью в 2.0 нм для каликсаренов 14–16. Все исследования проводили в буферном растворе (ТРИС, pH = 7.3) при 298 К.
Газовая хромато-масс-спектрометрия проводилась на хромато-масс-спектрометре GCMS-QP2010 Ultra фирмы “Shimadzu” на колонке HP-5MS (внутренним диаметром 0.25 мкм и длиной 30 м) со следующими параметрами: газ-носитель – гелий “А”, температура инжектора – 250 °С, скорость потока через колонку – 2 мл/мин, режим c 40-кратным делением потока; температурная программа термостата – градиентное повышение температуры от 70 до 140 °С с шагом 10 °С/мин, затем выдерживание температуры в течение 2 мин с последующим ее повышением от 140 до 250 °С с шагом 10 °С/мин. Диапазон сканируемых масс – (35÷400) m/z. Образцы для хроматографического анализа были приготовлены путем экстракции 1 мл реакционной смеси хлороформом (2 раза по 2 мл). Для количественного анализа замещенных бифенилов, полученных в серии тестовых реакций, использовался метод внутреннего стандарта c использованием ундекана, для чего предварительно строился калибровочный график с использованием индивидуальных образцов бифенилов. Внутренний стандарт в количестве 0.025 ммоль (5 мкл) вносился в реакционный сосуд до начала реакции.
Методы получения исследуемых соединений
Все реагенты были приобретены из каталогов Acros или Sigma-Aldrich и использовались без дополнительной очистки. Растворители марки х. ч. или ч. д. а. очищали в соответствии со стандартными методами [24]. Соединения 2, 3, 5, 6, 8, 9, 11, 12 [15] и 4, 7 [25] синтезированы по литературным методикам.
Соединение 10 – 5,11,17,23-тетра-амино-25,26,27,28-тетратетрадецилоксикаликс[4]арен (методика синтеза аналогична для синтеза соединений 8, 9 [15]) было получено с выходом 85% и без дополнительных очисток было введено в реакцию диазотирования.
Соединение 13 – 5,11,17,23-тетра-азид-25,26,27,28-тетра-тетрадецилоксикаликс[4]арен (методика синтеза аналогична для синтеза соединений 11, 12 [15]) светло-коричневое порошкообразное вещество (выход – 88%). Спектр ЯМР 1H (400 МГц, CDCl3), δ, м. д.: 0.89 (т, J = 6.7 Гц, 12H, –CH3), 1.31 (м, J = 33.8 Гц, 80H, –CH2–), 1.61 (м, 8H, –CH2–), 1.85 (м, 8H, –CH2–), 3.10 (д, J = = 13.7 Гц, 4H, Ar–CH2–Ar), 3.82 (т, J = 7.3 Гц, 8H, –O–CH2), 4.40 (д, J = 13.5 Гц, 4H, Ar–CH2–Ar), 6.34 (с, 8H, ArH). Спектр ЯМР 13С{Н} (100.6 МГц, CDCl3, 25 °С), δ, м. д.: 14.28, 22.86, 26.43, 29.57, 29.89, 29.97, 30.06, 30.12, 30.34, 31.27, 32.10, 118.65, 133.71, 136.43, 154.13. ИК-спектр, ν, см–1: 1236 (νas (Ar–O–CH2–)), 2109 (νas (N3)), 2852 (νs(–CH2–)), 2922 (νas(–CH2–)), 2955 (νas(–CH3)); MALDI-TOF, m/z: [M + H]+ = 1373; найдено: [M + Na]+ = 1395. Элементный анализ. Найдено (%): C – 73.80, H – 9.80, N – 12.11. Вычислено для C84H132N12О4 (%): C – 73.43, H – 9.68, N – 12.23; Тпл = 87 °С (с разложением); Rf = 0.14 (гексан/этилацетат 5/1).
Методика получения соединений 14–16. В колбу загрузили 0.1 ммоль тетраазида 11, 12 или 13, 0.23 г (2.0 ммоль) ацетилендикарбоновой кислоты, 5 мл ацетона. Реакционную смесь кипятили в течение 40 ч с обратным холодильником. Для выделения продукта ацетон удалили на роторном испарителе, к полученному осадку добавили 30 мл воды, осадок отфильтровали на фильтре Шотта. Полученный осадок сушили в вакуум-эксикаторе над P2O5 в течение 24 ч.
Соединение 14 – 5,11,17,23-тетра(4,5-карбокси-1,2,3-триазол-1-ил)-25,26,27,28-тетрабутоксикаликс[4]арен – коричневое порошкообразное вещество (выход – 88%).
Спектр ЯМР 1H (400 МГц, CDCl3), δ, м. д.: 1.05 (уш. с, 12H, –CH3), 1.56 (уш. м, 8H, –CH2–), 1.92 (уш. м, 8H, –CH2–), 3.28 (уш. с, 4H, ArCH2Ar), 4.20 (уш. с, 8H, –O–CH2–), 4.57 (уш. с, 4H, ArCH2Ar), 6.65 (уш. с, 8H, ArH). ИК-спектр, ν, см–1: 1216 (ν (C–O)), 1239 (νas(Ar–O–CH2–)), 1378 (δ(–CH3)), 1731 (ν (–C(O)), 2852 (νs(–CH2–)), 2871 (νs(–CH3)), 2919 (νas(–CH2–)), 2959 (νas(–CH3)), 3456 (ν (−ΟΗ)). MALDI-TOF, m/z: [M]+ = 1268; найдено: 1226 [M–CO2 + 2H]+, 1206 [M–CO2 – H2O]+, 1182 [M–2CO2 + 2H]+, 1162 [M–2CO2 – H2O]+, 1138 [M–3CO2 + 2H]+, 1118 [M–3CO2 – H2O]+, 1094 [M–4CO2 + 2H]+. Элементный анализ. Найдено (%): C – 56.92, H – 4.90, N – 13.06. Вычислено для C60H60N12О20 (%): C – 56.78, H – 4.77, N – 13.24; Тпл = 146 °С (с разложением).
Соединение 15 – 5,11,17,23-тетра(4,5-карбокси-1,2,3-триазол-1-ил)-25,26,27,28-тетраоктилокси-каликс[4]арен – коричневое порошкообразное вещество (выход – 86%). Спектр ЯМР 1H (400 МГц, CDCl3), δ, м. д.: 0.90 (уш. с, 12H, –CH3), 1.1–1.64 (уш. с, 40H, –CH2–), 1.92 (уш. с, 8H, –CH2–), 3.32 (уш. с, 4H, ArCH2Ar), 4.20 (уш. с, 8H, –O–CH2–), 4.57 (уш. с, 4H, ArCH2Ar), 6.65 (уш. с, 8H, ArH). ИК-спектр, ν, см–1: 1266 (νas(Ar–O–CH2–)), 1730 (ν(–C(O)), 2853 (νs(–CH2–)), 2922 (νas(–CH2–)), 2955 (νas(–CH3)), 3442 (ν(−ΟΗ)). MALDI-TOF, m/z: [M]+ = 1492; найдено: [M–3CO2 + Н]+ = 1273. Элементный анализ: найдено (%): C – 61.26, H – 6.34, N – 11.05. Вычислено для C76H92N12О20 (%): C – 61.12, H – 6.21, N – 11.25; Тпл = 143 °С (с разложением).
Соединение 16 – 5,11,17,23-тетра(4,5-карбокси-1H-1,2,3-триазол-1-ил)-25,26,27,28-тетратетрадецилокси-каликс[4]арен – коричневое порошкообразное вещество (выход – 92%). Спектр ЯМР 1H (400 МГц, CDCl3), δ, м. д.: 0.88 (уш. с, 12H, –CH3), 1.05–1.65 (уш. с, 88H, –CH2–), 1.94 (уш. с, 8H, –CH2–), 3.32 (уш. с, 4H, ArCH2Ar), 4.20 (уш. с, 8H, –O–CH2–), 4.57 (уш. с, 4H, ArCH2Ar), 6.65 (уш. с, 8H, ArH). ИК-спектр, ν, см–1: 1226 (νas(Ar–O–CH2–)), 1726 (ν(–C(O)), 2852 (νas(–CH2–)), 2922 (νas(–CH2–)), 2950 (νas(–CH3)), 3451 (ν (−OH)). MALDI-TOF, m/z: [M]+ = 1830; найдено: [M–CO2 + H]+ = 1787, [M–2CO2 – H]+ = = 1743, [M–3CO2 + H]+ = 1699. Элементный анализ: найдено (%): C – 65.79, H – 7.83, N – 9.05. Вычислено для C100H140N12О20 (%): С – 65.62, H – 7.71, N – 9.18; Тпл = 146 °С (с разложением).
ЗАКЛЮЧЕНИЕ
Впервые получены новые амфифильные производные каликс[4]арена с анионными карбоксильными головными группами на верхнем ободе и алкильными заместителями различной длины – на нижнем. Изучена агрегация амфифильных макроциклов в водных растворах и обнаружено, что полученные макроциклы обладают флуоресценцией в синей области и сольватофлорным эффектом, что представляет интерес с точки зрения создания флуоресцентных сенсорных систем. Полученный макроцикл 16 с тетрадецильными фрагментами был использован в качестве мицеллярной среды для проведения реакции сочетания Сузуки, продемонстрировав количественные выходы продуктов сочетания йод- и бромаренов в водной среде при комнатной температуре за 10 ч. Установлено, что макроцикл 16 благодаря наличию гидрофобной полости по эффективности мицеллярного катализа превосходит коммерчески доступный стеарат натрия.
Работа выполнена при финансовой поддержке Российским научным фондом (грант № 18-73-10033).
Список литературы
Anastas P.T., Kirchhoff M.M. // Acc. Chem. Res. 2002. V. 35. Issue 9. P. 686; https://doi.org/10.1021/ar010065m
Sheldon R.A. // Green Chem. 2007. V. 9. № 12. P. 1273; https://doi.org/10.1039/b713736m
La Sorella G., Strukul G., Scarso A. // Green Chem. 2015. V. 17. Issue 2. P. 644; https://doi.org/10.1039/ c4gc01368a
Lipshutz B.H., Ghorai S., Cortes-Clerget M. // Chem. Eur. J. 2018. V. 24. Issue 26. P. 6672; https://doi.org/10.1002/chem.201882662
Yang H., Jiao X., Li S. // Chem. Commun. 2012. V. 48. Issue 91. P.11217; https://doi.org/10.1039/c2cc36273b
Yeung D.K.J., Gao T., Huang J. et al. // Green Chem. 2013. V. 15. Issue 9. P. 2384; https://doi.org/10.1039/ c3gc41126e
Shinkai S., Mori S., Koreishi H. et al. // J. Amer. Chem. Soc. 1986. V. 108. P. 2409; https://doi.org/10.1021/ ja00269a045
Nimsea S.B., Kim T. // Chem. Soc. Rev. 2013. V. 42. P. 366; https://doi.org/10.1039/C2CS35233HO
Solovieva S.E., Burilov V.A., Antipin I.S. // Macroheterocycles. 2017. V. 10. № 2. P. 134; https://doi.org/10.6060/mhc170512a
Burilov V., Valiyakhmetova A., Mironova D. et al. // New J. Chem. 2018. V. 42. Issue 4. P. 2942; https://doi.org/10.1039/C7NJ04099G
Yakimova L.S., Gilmanova L.H., Evtugyn V.G. et al. // J. Nanopart. Res. 2017. V. 19. Issue 5. P. 173; https://doi.org/10.1007/s11051-017-3868-9
Mirgorodskaya A.B., Yackevich E.I., Kudryashova Y.R. et al. // Colloids Surf. B. 2014. V. 117. P. 497; https://doi.org/10.1016/j.colsurfb.2014.02.003
Sayin S., Yilmaz M. // Tetrahedron. 2016. V. 72. Issue 41. P. 6528; https://doi.org/10.1016/j.tet.2016.08.066
Фатыхова Г.А., Бурилов В.А., Докучаева М.Н., Соловьёва С.Е., Антипин И.С. // ДАН. 2018. Т. 479. № 6. С. 645; https://doi.org/10.7868/S0869565218120095
Burilov V.A., Fatikhova G.A., Dokuchaeva M.N. et al. // Beilstein J. Org. Chem. 2018. V. 14. P. 1980; https://doi.org/10.3762/bjoc.14.173
Huc I., Oda R. // Chem. Commun. 1999. Issue 20. P. 2025; https://doi.org/10.1039/A906141J
Moser R.J., Brown E.V. // J. Org. Chem. 1972. V. 37. Issue 4. P. 3938; https://doi.org/10.1021/jo00797a037
Lu P., Sanchez C., Cornella J. et al. // Org. Lett. 2009. V. 11. № 24. P. 5710; https://doi.org/10.1021/ol902482p
Ranganathan R., Vautier-Giongo C., Bales B.L. // J. Phys. Chem. B. 2003. V. 107. P. 10312; https://doi.org/10.1021/jp034346i
Basílio N., Garcia-Rio L. // Chem. Phys. Chem. 2012. V. 13. Issue 9. P. 2368; https://doi.org/10.1002/ cphc.201200175
Park C., Song H.J., Choi H.C. // Chem. Commun. 2009. Issue 32. P. 4791; https://doi.org/10.1039/B900328B
Suzuki A. // Angew. Chem. Intern. Ed. 2011. V. 50. P. 6722; https://doi.org/10.1002/anie.201101379
Chatterjee A., Ward T.R. // Catal. Lett. 2016. V. 146. P. 820; https://doi.org/10.1007/s10562-016-1707-8
Armarego W.L.F., Chai C.L. Purification of laboratory chemicals. New York: Elsevier, 2009.
Brake M., Böhmer V., Krämer P. et al. // Supramol. Chem. 1993. V. 5. P. 65; https://doi.org/10.1080/ 10610279308027509
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика