

Химическая физика, 2019, T. 38, № 7, стр. 3-10
Идентификация химических соединений по спектрам рассеянного излучения в диапазоне длин волн 5.3–12.8 мкм с применением перестраиваемого квантово-каскадного лазера
И. С. Голяк 1, А. Н. Морозов 1, С. И. Светличный 2, А. С. Табалина 1, И. Л. Фуфурин 1, *
1 Московский государственный технический университет им. Н.Э. Баумана
Москва, Россия
2 Филиал Института энергетических проблем химической физики им. В.Л. Тальрозе
Российской академии наук
Черноголовка, Россия
* E-mail: igfil@mail.ru
Поступила в редакцию 15.02.2019
После доработки 14.03.2019
Принята к публикации 20.03.2019
Аннотация
Рассмотрены методика и экспериментальная лабораторная установка, позволяющая регистрировать спектры отражения и диффузного рассеяния инфракрасного излучения веществ, находящихся на различных поверхностях в твердом и жидком агрегатных состояниях. В качестве источника зондирующего излучения использовался квантово-каскадный лазер, перестраиваемый в диапазоне длин волн 5.3–12.8 мкм, при спектральном разрешении 2 см–1 и средней мощности 15 мВт. Экспериментальная установка позволяла регистрировать спектры диффузно рассеянного излучения исследуемых веществ. Предложены методики идентификации веществ по спектрам рассеянного излучения. Показано, что применение интегрального преобразования Крамерса–Кронига повышает степень селективности спектров и, как следствие, надежность идентификации.
ВВЕДЕНИЕ
Инфракрасная спектроскопия является общепринятым в отечественной и мировой практике оптическим методом исследования состава и структуры химических соединений [1]. Как правило, в рутинных лабораторных исследованиях для исследования веществ в различных агрегатных состояниях применяется инфракрасная фурье-спектроскопия [2, 3]. Особенное распространение данный метод получил для анализа химических соединений в газообразной фазе, в том числе для идентификации веществ в открытой атмосфере пассивным способом (инфракрасная фурье-спетрорадиометрия) [4–7]. Для исследования веществ в твердой и жидкой фазах в настоящее время, как правило, применяют метод спектроскопии комбинационного рассеяния света (раман-спектроскопия) [8–11]. Также в мировой практике существует направление исследования состава и структуры веществ с использованием терагерцового излучения [12], а также с помощью методов фотолюминесценции [13].
В 1994 г. в лаборатории Bell Labs (США) [14] впервые был представлен квантово-каскадный лазер (ККЛ) на гетероструктурах, на так называемой сверхрешетке. В настоящее время наблюдается существенный прогресс в разработке перестраиваемых инфракрасных квантово-каскадных лазеров и уже созданы ККЛ с диапазоном перестройки более 1000 см–1 и пиковой мощностью до 150 мВт. Применение подобных лазеров позволяет получать достаточно информативные спектры рассеяния веществ и, как следствие, успешно идентифицировать химические соединения [15, 16].
Разработка методики идентификации твердых и жидких веществ по рассеянному излучению с помощью перестраиваемого ККЛ является актуальным исследованием, поскольку в отличие от абсорбционной спектроскопии не требует пробоподготовки и может применяться для внелабораторных исследований [15, 17]. Поскольку плотность потока энергии рассеянного излучения на несколько порядков выше энергии комбинационного рассеяния света, для получения спектров рассеяния достаточно лазера средней мощностью около 10 мВт с поперечным размером лазерного пучка в несколько миллиметров. В этом случае нет риска физического повреждения или детонации исследуемого образца.
ЭКСПЕРИМЕНТАЛЬНАЯ УСТАНОВКА
Для регистрации рассеянного инфракрасного излучения от исследуемых образцов химических соединений в твердом и жидком агрегатных состояниях на различных подстилающих поверхностях создана экспериментальная установка на основе ККЛ, перестраиваемого в диапазоне длин волн 5.3–12.8 мкм, и кадмий-ртуть-теллуриевого (КРТ) фотоприемника, охлаждаемого каскадом ячеек Пельтье. Излучение ККЛ падает нормально на поверхность, на которой находится исследуемое вещество. Юстировка фотоприемника с собирающим объективом осуществляется таким образом, чтобы собирать максимум рассеянного излучения. Фотоприемник располагался под углом 30°–45° к нормали к подложке, чтобы минимизировать зеркально отраженное излучение и максимально собирать рассеянное излучение.
В качестве подложки (идеальная подложка) был выбран абсорбционный ИК-фильтр марки ИКС-970, изготовленный из стекол системы SiO2–ZnO–Na2O, окрашенных в массе сульфидами, селенидами и сульфоселенидами сурьмы. Подобная подложка обладает слабым френелевским отражением и слабым рассеянием и является хорошей подстилающей поверхностью для проведения тестовых лабораторных исследований. Для данного типа поверхностей коэффициент преломления n ~ 1.5 и поправка на френелевское отражение составляет порядка 3.8% [18]. Однако с учетом вариаций мощности лазера до 5% ференелевским отражением можно пренебречь при обработке экспериментальных результатов, полученных на описанной выше установке.
В качестве квантово-каскадного лазера и фотоприемника использовались элементы системы LaserTune (Block Engineering, США). Технические характеристики инфракрасного перестраиваемого ККЛ и КРТ-фотоприемника приведены в табл. 1 и 2.
Таблица 1.
Технические характеристики квантово-каскадного лазера
| Характеристика | Значение |
|---|---|
| Диапазон перестройки, мкм | 5.3–12.8 |
| Ширина линии, cм–1, не более | 2 |
| Максимальная импульсная мощность, мВт | 150 |
| Средняя мощность, мВт | 0.5–15.0 |
| Нестабильность по мощности, не более | 10% |
| Длительность импульса, нс | 30–300 |
| Частота повторений импульса, МГц | до 3 |
| Качество луча | одна поперечная мода |
| Диаметр луча, мм | 2 × 4, коллимированный |
| Расходимость луча, мрад | 5 |
| Нестабильность направления луча, мрад, не хуже | 1 |
| Поляризация | вертикально поляризованный |
Таблица 2.
Технические характеристики КРТ фотоприемника
| Характеристика | Значение |
|---|---|
| Материал фоточувствительного элемента | Cd–Hg–Te |
| Размер чувствительной площадки, мм | 1 × 1 |
| Спектральный диапазон, мкм | 6–12 |
| Воль/ваттная характеристика, В/Вт | 800 |
| Рабочий диапазон частот регистрации | 10 Гц – 20 МГц |
| Характерная рабочая температура, К | 223 |
| Пиковая пороговая мощность насыщения (при 8 мкм и 20 МГц), мВт | 5 |
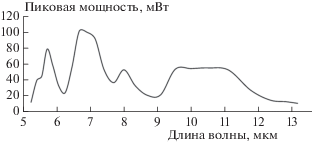
На рис. 1 приведена типичная спектральная характеристика пиковой мощности излучения ККЛ, используемого в экспериментальной установке. Фотоприемник оснащался объективом, рассчитанным таким образом, чтобы в пятне размером 0.023 мм было сконцентрировано 85% энергии. Квантово-каскадный лазер и фотоприемник аппаратно синхронизованы друг с другом в импульсном режиме, что позволяет регистрировать инфракрасные спектры в рабочем диапазоне 5.3–12.8 мкм с разрешением в 2 см–1, частотой следования порядка 200 кГц, шириной импульса порядка 50 нс и коэффициентом заполнения, составляющим 4–5%.
ЭКСПЕРИМЕНТАЛЬНЫЕ РЕЗУЛЬТАТЫ
С помощью описанной экспериментальной установки были зарегистрированы инфракрасные спектры рассеяния веществ в твердой и жидкой фазах. Для анализа и последующей процедуры идентификации веществ интерес представлял нормированный спектр рассеяния. Для его получения регистрировалось отраженное излучение от пластины с золотым покрытием, представляющее собой опорный сигнал с интенсивностью I0(ν). Опорный спектр с большой точностью соответствует спектру генерируемого лазерного излучения, поскольку спектральный коэффициент отражения золота составляет не менее 95% в рабочем спектральном диапазоне, а нестабильность мощности лазера, используемого в установке, составляет не более 5%.
Оптимальной схемой регистрации опорного сигнала in vivo является использование референтного канала фотоприемника, куда с помощью светоделителя направляется часть генерируемого излучения [17]. Такая схема работы позволяет нивелировать влияние нестабильности мощности лазера на нормированный спектр рассеяния исследуемого вещества. Подобная схема может стать усовершенствованием для предложенной в настоящей работе экспериментальной установки.
После регистрации опорного спектра на подложку помещалось исследуемое вещество и регистрировался спектр рассеянного излучения. Для повышения отношения сигнал/шум использовалось многократное измерение интенсивностей спектров I(ν) и находилось их среднее значение. Чтобы уменьшить влияние индикатрисы рассеяния на интенсивность спектральных линий, использовался прием “пространственного усреднения”, когда в процессе регистрации рассеянного излучения и его накопления подложку перемещали с амплитудой порядка нескольких миллиметров. Такой прием позволял усреднить излучение по достаточно широкому сектору диаграммы направленности рассеянного излучения и повысить воспроизводимость экспериментальных результатов.
Для анализа и идентификации веществ использовался нормированный спектр рассеяния R(ν), вычисляемый как R(ν) = I(ν)/I0(ν). Нормированный спектр рассеяния содержит селективные линии, характерные для колебательно-вращательных переходов молекул вещества [1], которые могут быть использованы для процедуры идентификации веществ [4, 19, 20].
Анализ спектров рассеяния порошка триптофана (β-(β-индолил)-α-аминопропионовая кислота) на идеальной подложке позволил зарегистрировать набор спектров рассеяния с использованием приема пространственного усреднения по диаграмме направленности. Экспериментально показано, что в данном случае среднее значение кросс-корреляции спектров рассеяния кристаллов триптофана с характерными размерами 0.5–1.5 мм составляет порядка 0.95, что позволяет утверждать о высокой воспроизводимости спектров рассеяния. Последнее обстоятельство, в свою очередь, позволяет утверждать о возможности идентификации химических соединений предложенным методом.
ИДЕНТИФИКАЦИЯ ХИМИЧЕСКИХ СОДИНЕНИЙ ПО ИНФРАКРАСНЫМ СПЕКТРАМ РАССЕЯНИЯ
Для идентификации веществ применялся известный в инфракрасной спектроскопии метод анализа, состоящий в последовательном сравнении экспериментального спектра со спектрами известных веществ, находящимися в базе данных. База данных представляет собой набор эталонных спектров рассеяния веществ с высоким отношением сигнал/шум. Эталонные спектры для базы данных создавались путем накопления и усреднения результатов по N = 100 экспериментам для каждого тестового вещества, что позволило применить метод синхронной фильтрации спектров и в $\sqrt N $ раз повысить отношение сигнал/шум в эталонных спектрах.
В состав базы данных вошли спектры порошков ацетилсалициловой кислоты (средний размер кристалла – 1 мм), лактозы (0.1 мм), фруктозы (0.5 мм), глюкозы (0.3 мм), аскорбиновой кислоты (0.2–0.3 мм) и триптофана (0.5–1.5 мм), а также жидкостей – глицерина, диэтилфталата, диметилсульфоксида, подсолнечного масла.
В качестве параметра степени схожести спектров выступал линейный коэффициент корреляции Пирсона [21], вычисляемый как
Скалярные произведения и средние значения интенсивности вычислялись с учетом весовой функции вида
Критерием идентификации i-го вещества из базы данных по инфракрасному спектру рассеяния является условие вида korri > thrпор, где korri – коэффициент корреляция экспериментального спектра R(ν) рассеяния и эталонного спектра Ri(ν) из базы данных; thrпор – порог обнаружения веществ по величине коэффициента корреляции.
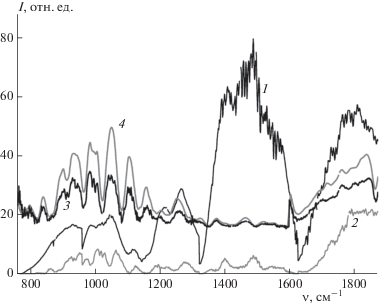
На рис. 2 приведен пример идентификации кристаллов порошка (L-тирозин) на идеальной нерассеивающей подложке. На инфракрасном спектре отчетливо видны селективные полосы в диапазоне 860–1300 см–1. Полоса в районе 1640 см–1 находится на границе стыковки спектров излучения двух лазерных чипов ККЛ и относится к артефактам обработки. Второй участок селективных линий находится в диапазоне 1670–1820 см–1.
Рис. 2.
Этапы обработки ИК-спектров рассеяния с последующей идентификации на примере L-тирозина: кривая 1 – спектр опорного сигнала, 2 – спектр рассеянного излучения, 3 – экспериментальный нормированный спектр рассеяния, 4 – эталонный нормированный спектр рассеяния. Коэффициент корреляции экспериментального и эталонного спектров равен 0.94.
Спектры рассеяния по своей физической природе обладают слабой селективностью. Более того, наблюдается зависимость интенсивности измеряемого спектра рассеяния от диаграммы направленности регистрации рассеянного излучения. Кроме того, зеркально отраженное излучение может вносить фазовые искажения в спектр исследуемого вещества. Одним из способов повышения селективности инфракрасных спектров является применение интегрального преобразования Крамерса–Кронига [17, 22, 23], позволяющего спектр рассеяния пересчитать в спектр поглощения (экстинкции).
ПРИМЕНЕНИЕ ПРЕОБРАЗОВАНИЯ КРАМЕРСА–КРОНИГА К СПЕКТРАМ РАССЕЯНИЯ ИНФРАКРАСНОГО ИЗЛУЧЕНИЯ
Соотношение Крамерса–Кронига представляет собой интегральную связь между действительной и мнимой частями любой комплексной функции, аналитичной в верхней полуплоскости [24]. Для оптических характеристик среды соотношение Крамерса–Кронига, согласно принципу причинности, устанавливает однозначную связь между показателями преломления и поглощения среды. Робинсон в 1953 году одним из первых показал возможность получения спектров поглощения вещества путем численного преобразования спектров рассеянного света [25]. Таким образом, в области спектроскопии рассеянного излучения соотношение Крамерса–Кронига является эффективным средством получения спектральных коэффициентов преломления и поглощения из спектра рассеянного излучения, что особенно актуально в тех случаях, когда экспериментально могут быть зарегистрированы только спектры рассеяния.
В электродинамике важным применением преобразования Крамерса–Кронига являются дисперсионные соотношения. Рассмотрим комплексный вид диэлектрической проницаемости среды:
где действительная и мнимая части диэлектрической проницаемости определяют показатель преломления n(ν) и показатель поглощения (экстинкции) k(ν) данной среды. Эти показатели не являются независимыми, и, следовательно, появляется принципиальная возможность по спектру одной из оптических постоянных вычислять спектр другой, не прибегая к непосредственным измерениям последней. Именно в этом и состоит суть соотношения Крамерса–Кронига. Справедливость этого соотношения неоднократно проверялась экспериментально при различной температуре и для различных сред в разных агрегатных состояниях – кристаллы, жидкости, растворы [17, 26, 27].Комплексный показатель рассеяния среды имеет следующий вид:
где $R(\nu ) = {{\eta }^{2}}(\nu )$ определяется как мощность рассеянного излучения; а φ(ν) – соответствующая фаза.Далее для величин (1) и (2) с учетом уравнений Френеля и соотношений Крамерса–Кронига для действительной и мнимой частей диэлектрической проницаемости вида (1) можно установить связь между спектрами рассеяния и поглощения среды [25–27]. Для рассеянного излучения спектры мощности и фазы связаны следующим образом:
(3)
$\varphi (\nu ) = - \frac{{2\nu }}{\pi }P\int\limits_0^\infty {\frac{{\ln \eta (\nu {\text{'}})}}{{{{\nu }^{{'2}}} - {{\nu }^{2}}}}} d\nu {\text{',}}$После восстановления фазы рассеянного излучения вида (3) вычисляется спектр поглощения (экстинкции) среды согласно уравнению
(4)
$k(\nu ) = \frac{{2\eta (\nu )\sin \varphi (\nu )}}{{1 - 2\eta (\nu )\cos \varphi (\nu ) + R(\nu )}}.$Спектр поглощения содержит спектральные линии, характерные для исследуемого вещества, и может быть применен для спектрального анализа и идентификации веществ.
Приведенные выше соотношения выполняются для случая близкого к нормальному падения возбуждающего излучения, а также при условии, что исследуемый образец находится в вакууме или воздушной среде. Интеграл вида (3) определен на интервале [0, ∞] при условии, что наблюдаемый спектр рассеяния R(ν) измеряется в ограниченном интервале волновых чисел ν. В этом смысле критичной для восстановления фазы φ(ν) становится экстраполяция спектра рассеяния. Методы экстраполяции изложены в работе [26] и их подробное рассмотрение выходит за рамки настоящей статьи. Более того, вычисление интеграла от функции с сингулярностью вызывает определенные математические сложности. В работе [28] изложены основные подходы к решению проблемы. В настоящей статье применялся метод двойного преобразования Фурье, аналитический вид которого с сохранением авторской нотации [29] представлен как
(5)
$\begin{gathered} i\varphi (\nu ) = - \frac{1}{{2\pi }}\int\limits_{ - \infty }^0 {dt} {{e}^{{ - i\nu t}}}\int\limits_{ - \infty }^\infty {d\nu \ln \eta (\nu ){{e}^{{i\nu t}}}} + \\ + \,\,\frac{1}{{2\pi }}\int\limits_0^\infty {dt} {{e}^{{i\nu t}}}\int\limits_{ - \infty }^\infty {d\nu \ln \eta (\nu ){{e}^{{i\nu t}}}} . \\ \end{gathered} $Преобразование Крамерса–Кронига с вычислением фазы рассеянного излучения в виде двойного преобразования Фурье, как правило, называют преобразованием Петерсона–Кнайта (P–K-преобразование) [29]. Именно в виде P–K-преобразования в настоящей работе вычислялись спектры поглощения из спектров рассеяния исследуемого образца.
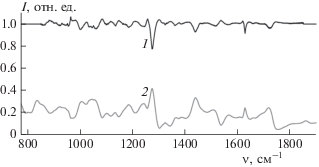
Далее приведены примеры вычислений спектров поглощения из спектров рассеяния, полученных на установке, описанной выше. На рис. 3 для иллюстрации метода P–K приведен пример преобразования спектра рассеяния в спектр поглощения для жидкого вещества на примере капли диэтилфталата (DEP). Диэтилфталат по ряду физико-химических свойств является аналогом вещества VX, чем и обусловлен интерес к исследованию DEP, связанный с обнаружением его следовых количеств на различных поверхностях.
Рис. 3.
Пример спектра поглощения диэтилфталата (1), численно полученного из спектра рассеяния (2) с помощью P–K-преобразования.
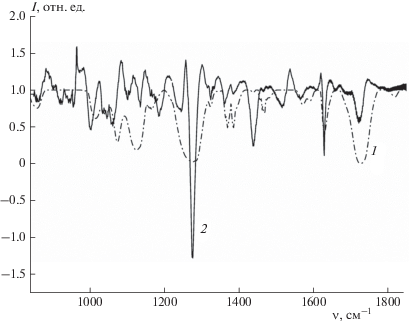
Для проверки корректности предлагаемого метода преобразования спектров P–K на созданной экспериментальной установке измерены спектры поглощения жидких образцов методом абсорбционной инфракрасной спектроскопии и выполнено сопоставление экпериментальных спектров, рассчитанных согласно (4) и (5), и полученных экспериментально. Расчетный и экспериментальный спектры поглощения приведены на рис. 4. Визуально наблюдается совпадение положения основных полос поглощения. Корреляция двух спектров на всем диапазоне составляет значение 0.5. Поскольку рассеянное излучение зависит от многих параметров, а также в расчетном методе имеют место особенности, связанные с необходимостью экстраполяции фазы в выражении (2) на бесконечный спектральный диапазон, в приведенных спектрах наблюдается искажение форм полос.
Рис. 4.
Экспериментальный (1) и рассчитанный методом P–K (2) спектры пропускания DEP. Корреляция для спектров поглощения DEP, полученных экспериментальным и численным способами, составляет 0.50.
Для практического применения разработанной методики с целью идентификации и распознавания веществ по спектрам рассеяния создана эталонная база данных спектров рассеяния, матрица кросс-корреляций которой приведена на рис. 5а. Для вычисления приведенной на рис. 5а матрицы использовались нормированные спектры рассеяния R(ν) следующих веществ: 1 – лактозы, 2 – диметилсульфоксида, 3 – аскорбиновой кислоты, 4 – фруктозы, 5 – диэтилфталата, 6 – триптофана, 7 – глюкозы, 8 – глицерина, 9 – подсолнечного масла, 10 – аспирина. Как видно из этого рисунка, для ряда веществ, например диметилсульфоксида, аскорбиновой кислоты, фруктозы, наблюдаются значения кросс-корреляции выше 0.6, что говорит о высокой вероятности их ложного обнаружения при невысоком отношении сигнал/шум в регистрируемых спектрах рассеяния.
Рис. 5.
Матрицы кросс-корреляции эталонных спектров рассеяния до (а) и после (б) применения P–K-преобразования.
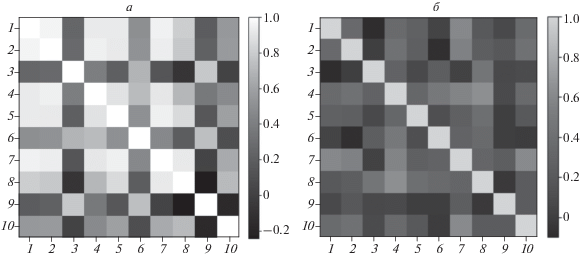
Эталонные спектры из базы данных спектров рассеяния были преобразованны, согласно (3)–(5), в спектры поглощения. После чего для базы данных спектров поглощения, полученных путем преобразования P–K из спектров рассеяния, вычислена матрица кросс-корреляций, приведенная на рис. 5б. Для ее вычисления использовались спектры рассеяния следующих веществ: 1 − аскорбиновой кислоты, 2 – ацетилсалициловой кислоты, 3 – диэтилфталата, 4 − диметилсульфоксида, 5 – фруктозы, 6 – глицерина, 7 − глюкозы, 8 – подсолнечного масла, 9 – триптофана, 10 – лактозы.
Из рис. 5 легко видеть, что база данных спектров поглощения, полученная методом P–K (рис. 5б), существенно ближе к диагональному виду, нежели матрица, приведенная на рис. 5а. Отсюда можно сделать вывод, что применение метода P–K для восстановления спектров должно существенно повысить селективность спектров, а значит, надежность обнаружения и идентификации веществ.
Далее в табл. 3 и 4 приведены экспериментальные данные по вероятностям правильного обнаружения веществ с помощью созданной экспериментальной установки с применением базы данных спектров поглощения, полученных методом P–K. Из этих таблиц видно, что применение созданной экспериментальной установки и методики анализа веществ на идеальной нерассеивающей подложке позволяет обнаруживать и идентифицировать широкий набор веществ в различных агрегатных состояниях с достаточно высокой вероятностью. Необходимо отметить, что для алюминия вероятность правильного обнаружения существенно ниже, чем для дерева и керамики. Этот факт объясняется высоким значением коэффициента отражения металлов в рабочем спектральном диапазоне. Идентификация веществ на различных естественных поверхностях представляет собой отдельную задачу и требует дополнительных исследований.
ЗАКЛЮЧЕНИЕ
Создана экспериментальная установка, состоящая из квантово-каскадного лазера с диапазоном перестройки 5.3–12.8 мкм при спектральном разрешении 2 см–1 и фотоприемника на основе соединения кадмий–ртуть–теллур, охлаждаемого холодильником Пельтье. Установка предназначена для регистрации спектров отражения, рассеяния и пропускания исследуемых веществ.
Показано, что применение интегрального преобразования Крамерса–Кронига с использованием уравнений Френеля позволяет вычислить спектр поглощения исследуемого вещества из спектра рассеяния. Такой метод дает возможность с высокой точностью получать спектры поглощения твердых и жидких веществ без предварительной пробоподготовки. На основе анализа матриц кросс-корреляции базы данных эталонных спектров показано, что эталонные спектры поглощения веществ обладают большей селективностью по сравнению со спектрами рассеяния, и, как следствие, применение преобразования Крамерса–Кронига позволяет повысить надежность обнаружения веществ по рассеянному излучению.
Приведены оценки значений вероятности правильного обнаружения веществ на идеальной подложке и на реальных подстилающих поверхностях. Для возможности широкого применения предлагаемого метода идентификации веществ на практике необходима разработка алгоритмов анализа влияния подстилающей поверхности на анализируемое излучение.
Исследование выполнено при финансовой поддержке Российским фондом фундаментальных исследований в рамках научного проекта № 18-29-02024.
Список литературы
Мальцев А.А. Молекулярная спектроскопия. М.: Из-во МГУ, 1980.
Вагин В.А., Гершун М.А., Жижин Г.Н., Тарасов К.И. Светосильные спектральные приборы / Под ред. Тарасова К.И. М.: Наука, 1988.
Морозов А.Н., Светличный С.И. Основы фурье-спектрорадиометрии. 2-е изд., испр. и доп. М.: Наука, 2014.
Кочиков И.В., Морозов А.Н., Фуфурин И.Л. // Компьютерная оптика. 2012. Т. 36. № 4. С. 554.
Морозов А.Н., Светличный С.И., Фуфурин И.Л. // Докл. АН. 2015. Т. 464. № 2. С. 156.
Harig R., Matz G. // Field Analyt. Chem. and Technol. 2001. V. 5. № 1. P. 75.
Голяк Иг.С., Голяк Ил.С., Карфидов А.О., Королёв П.А, Морозов А.Н., Миронов А.И., Строков М.А., Табалин С.Е., Фуфурин И.Л. // Приборы и техника эксперимента. 2014. № 6. С. 119.
Portnov A., Rosenwaks S., Bar I. // Appl. Phys. Lett. 2008. V. 93. № 4. P. 41115.
Gorelik V.S., Zlobina L.I., Sverbil P.P., Fadyushin A.B., Chervyakov A.V. // J. Rus. Laser Res. 2005. V. 26. № 3. P. 211.
Винтайкин И.Б., Васильев Н.С., Голяк Иг.С., Голяк Ил.С., Есаков А.А., Морозов А.Н., Светличный С.И., Табалин С.Е., Фуфурин И.Л. // Изв. РАН. Энергетика. 2016. № 6. С. 144.
Васильев Н.С., Винтайкин И.Б., Голяк Иг.С., Голяк Ил.С., Кочиков И.В., Фуфурин И.Л. // Компьют. оптика. 2017. Т. 41. № 5. С. 626.
Зайцев К.И., Гавдуш А.А., Карасик В.Е., Юрченко С.О. // Оптика и спектроскопия. 2015. Т. 118. № 4. С. 582.
Голяк И.С., Есаков А.А., Васильев Н.С., Морозов А.Н. // Там же. 2013. Т. 115. № 6. С. 990.
Capasso F., Tredicucci A., Gmachl C. et al. // IEEE J. 1999. V. 5. № 3. P. 792.
Deutsch E.R., Kotidis P., Zhu N. et al. // Proc. SPIE 9106, Advanced Environmental, Chemical, and Biological Sensing Technologies XI. Baltimore, Maryland: SPIE, 2014. P. 91060A-1; https://doi.org/10.1117/12.2058544
Ghorbani R., Schmidt F.M. Real-time breath gas analysis of CO and CO2 using an EC-QCL // Applied Physics B. 2017. V. 123. № 5. P. 144; https://doi.org/10.1007/s00340-017-6715-x
US Patent № US 2012/0033220 A1. Pub Feb. 9, 2012.
ГОСТ 9411-91. Стекло оптическое цветное. Технические условия (с Поправкой). М.: Издательство стандартов, 1992.
Морозов А.Н., Кочиков И.В., Новгородская А.В., Сологуб А.А., Фуфурин И.Л. // Компьют. оптика. 2015. Т. 39. № 4. С. 614.
Васильев Н.С., Голяк Ил.С., Морозов А.Н. // Оптика и спектроскопия. 2015. Т. 118. № 1. С. 157.
Pearson K. Notes on regression and inheritance in the case of two parents // Proc. Roy. Soc. London. 1895. V. 58. P. 240.
Grosse P., Offermann V. // Appl. Phys. A. 1991. V. 52. № 2. P. 138.
Phillip H.R., Taft E.A. // Phys. Rev. 1964. V. 136. № 5A. P. A1445.
Kronig R. de L., Gorter C.J. // Physica (Utr.). 1936. V. 3. P. 1009.
Robinson T.S., Price W.C. // Proc. Phys. Soc. (London). 1959. V. 66B. P. 969.
Musfeldt J.L., Tanner D.B., Paine A.J. // J. Opt. Soc. Amer. A. 1993. V. 10. № 12. P. 2648.
Hansen W.N., Abdou W.A. // Ibid. 1977. V. 67. № 11. P. 1537.
Koji Ohta, Hatsuo Ishida // Appl. Spectrosc. 1988. V. 42. P. 952.
Peterson C.W., Knight B.W. // J. Opt. Soc. America. 1973. V. 63. № 10. P. 1238.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика