

Химическая физика, 2019, T. 38, № 9, стр. 3-13
Полиуретаны без изоцианатов и изоцианаты без фосгена – новое направление “зеленой” химии: механизм, катализ, управление реакционной способностью
М. В. Забалов 1, *, М. А. Левина 1, Р. П. Тигер 1
1 Институт химической физики им. Н.Н. Семёнова Российской академии наук
Москва, Россия
* E-mail: zabalov@mail.ru
Поступила в редакцию 15.03.2019
После доработки 15.03.2019
Принята к публикации 20.03.2019
Аннотация
Представлен краткий обзор работ в области кинетики, катализа и механизма новых реакций “зеленой” химии полиуретанов, выполненных в Институте химической физики им. Н.Н. Семёнова РАН в последние полтора десятилетия. Основное внимание уделено процессам образования гидроксиуретанов без применения изоцианатов, основанным на взаимодействии циклокарбонатов с аминами, бесфосгенному получению изоцианатов путем термораспада ацилазидов и перспективам использования возобновляемого растительного сырья для производства новых полиуретанов.
ВВЕДЕНИЕ
Полиуретаны занимают весьма внушительный сегмент мирового рынка полимерных материалов, и их производство, возрастая ежегодно на 2.5%, приближается в настоящее время к 20 млн т/год. Между тем классическая технология производства полиуретанов из нефтяного сырья, разработанная более 80 лет назад и основанная на реакции NCO-групп ди- или полиизоцианатов с ОН‑группами ди- или полиолов, в экологическом отношении далеко не безупречна. Это связано в первую очередь с высокой токсичностью самих изоцианатов, которые получают путем реакций первичных аминов с фосгеном – исключительно опасным отравляющим веществом. В известной мере по этой причине производство полиуретанов постепенно начало перемещаться на территории развивающихся стран, где требования к промышленной экологии не столь жестки, как в Европе или Северной Америке. Одновременно с этим в странах-производителях полиуретанов стали развиваться работы по поиску альтернативных путей синтеза как изоцианатов без использования фосгена, так и самих полиуретанов без применения изоцианатов и с использованием ненефтяного сырья. Постепенно из этих исследований возникло новое направление в этой области – “зеленая” химия полиуретанов. Об актуальности работ по созданию неизоцианатных уретанов и перспективах развития этого направления полимерной химии свидетельствует ряд обзорных статей, опубликованных в последние годы [1–10].
Полтора десятилетия назад в Институте химической физики им. Н.Н. Семёнова РАН были начаты исследования, целью которых является разработка способов управления процессами получения неизоцианатных уретанов и бесфосгенных изоцианатов на основе сведений о механизме, кинетике и катализе соответствующих реакций, а также возможностей использования в химии новых уретанов возобновляемого растительного сырья. В настоящей статье, написанной в связи с 60-летием Отдела полимеров и композиционных материалов ИХФ РАН, кратко изложены основные результаты работ в данной области химической физики.
РЕАКЦИИ ЦИКЛОКАРБОНАТОВ С АМИНАМИ КАК АЛЬТЕРНАТИВНЫЙ ПУТЬ К НЕИЗОЦИАНАТНЫМ ПОЛИУРЕТАНАМ
Реакции первичных аминов с циклокарбонатами считаются сегодня наиболее перспективным путем синтеза новых уретанов. Они протекают с раскрытием циклокарбонатного цикла без выделения побочных продуктов, так же как и классические реакции уретанообразования:
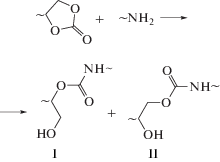
Полиуретаны, образующиеся в этой реакции, содержат гидроксильные группы – первичные – I и вторичные – II в равных или несколько отличающихся соотношениях в зависимости от строения реагентов и условий проведения процесса. Наличие ОН-групп в гидроксиуретанах способствует их гидролитической и вообще химической стабильности за счет образования внутри- и межмолекулярных водородных связей и открывает пути для соответствующей модификации полимеров. Для “зеленой” химии полиуретанов важно, что в процессах не принимают участие высокотоксичные изоцианаты, а исходные олигомеры можно получать из эпоксидсодержащих предшественников за счет реакции с диоксидом углерода, безупречной в экологическом отношении, в том числе из возобновляемого сырья – растительных масел путем их окисления до эпоксидсодержащих триглицеридов с последующей каталитической карбонизацией под действием СО2 [11].
Реакция присоединения аминов к циклокарбонатам известна с середины прошлого века, но именно в связи с перспективами ее использования в “зеленой” химии полиуретанов механизм и количественные закономерности образования гидроксиуретанов стали интенсивно изучаться лишь в последние годы [12–23]. В наших экспериментальных и теоретических работах [14–19, 23] было впервые установлено, что аминолиз циклокарбонатов протекает по двум параллельным каналам с участием одной и двух молекул амина. Путем квантовохимических расчетов методом функционала плотности показано, что присоединение первичных аминов к циклокарбонатам может протекать по одностадийному (Схема 1 ) и многостадийному, включающему образование аминоспирта в качестве интермедиата (Схема 2 ), механизмам.
Схема 1
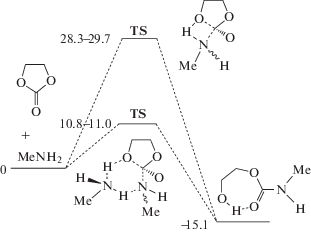
В обоих случаях при участии в реакции двух молекул амина расчетная величина энергии активации оказывается почти втрое ниже, чем энергия активации простого бимолекулярного взаимодействия. Этот эффект обусловлен каталитическим содействием второй молекулы амина акту переноса протона к циклокарбонату в шестичленном переходном состоянии (TS), существенно менее напряженном, чем четырехчленное состояние.
Схема 2
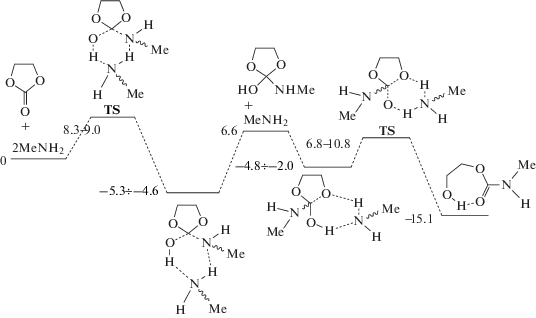
Цифры на всех схемах соответствуют барьерам превращения различных изомеров (в ккал/моль), отсчитанным от суммы энергии реагентов, разнесенных друг от друга на бесконечность. При многостадийном превращении максимальный барьер реакции с двумя молекулами амина составляет 8.3–10.8 ккал/моль, что много ниже энергии активации реакции с участием одной молекулы.
Кинетические закономерности процесса проще всего исследовать в избытке амина в режиме псевдопервого порядка. Протекание реакции по двум параллельным каналам с участием одной (константа скорости k1) или двух (константа скорости k2) молекул BuNH2 приводит к выполнению следующей зависимости наблюдаемой константы скорости псевдопервого порядка, kнабл, от концентрации амина в избытке последнего в растворе:
(1)
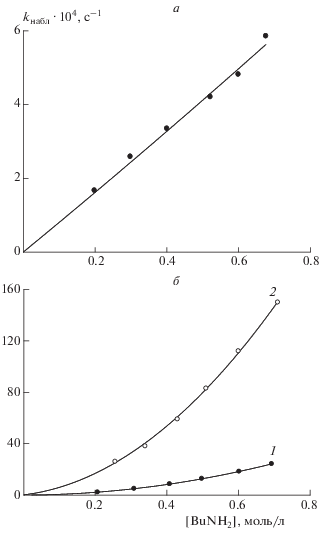
${{k}_{{{\text{набл}}}}} = {{k}_{1}}\left[ {{\text{BuN}}{{{\text{H}}}_{{\text{2}}}}} \right] + {{k}_{2}}{{\left[ {{\text{BuN}}{{{\text{H}}}_{{\text{2}}}}} \right]}^{2}}.$Такого типа зависимости неоднократно наблюдались нами при изучении модельных и целевых реакций в различных растворителях в отсутствие катализаторов. На рис. 1 представлены такие зависимости на примере аминолиза модельного этиленкарбоната и его олигомерных аналогов в различных средах. В соответствии с предсказаниями теории экспериментальные результаты также дают трехкратное снижение энергии активации реакции с участием двух молекул амина по сравнению с реакцией с одной молекулой, хотя сами значения энергий активации и отличаются от расчетных. Так, например, для взаимодействия этиленкарбоната с н-бутиламином в диоксане Е1 = 14.0 ккал/моль, Е2 = 3.7 ккал/моль, в н-бутаноле Е1 = 7.0 ккал/моль, Е2 = 2.5 ккал/моль [16]. Снижение активационного барьера и ускорение реакции в спиртовой среде, осуществляющей каталитическое содействие акту переноса протона от амина к циклокарбонату, побудило авторов данного обзора исследовать аминолиз в присутствии карбоновых кислот – хорошо известных бифункциональных катализаторов ацильного переноса. Было показано [17], что кинетические закономерности катализируемой уксусной кислотой реакции этиленкарбоната с н-бутиламином в диоксане те же, что и некаталитической реакции (рис. 2). В обоих случаях имеют место нелинейные зависимости наблюдаемой константы скорости от концентрации амина при [AcOH] = const и концентрации катализатора при [BuNH2] = const, свидетельствующие об участии одной или двух молекул амина в элементарных актах превращения. Соответствующие величины энергий активации каталитического процесса составляют (5.3 ± 0.2) и (1.1 ± 0.2) ккал/моль, что меньше значений, характерных для некаталитической реакции (см. выше). В той же работе в результате квантовохимического исследования механизма катализа уксусной кислотой обнаружено протекание процесса по одностадийному и многостадийному путям и получены данные о строении соответствующих предреакционных комплексов и переходных состояний. Барьеры превращений по наиболее выгодным одностадийному и многостадийному механизмам различаются незначительно и составляют ~12 ккал/моль, что существенно ниже расчетных значений энергии активации некаталитической реакции. Решающую роль в снижении активационного барьера играет структура карбоновой кислоты как бифункционального катализатора, которая благодаря мезомерии карбоксильной группы способна объединять разные части реагирующих молекул и осуществлять тем самым эффективный перенос протона между ними.
Рис. 1.
Типичные кинетические закономерности н-бутиламинолиза этиленкарбоната в н-бутаноле (а) при температурах: 1 – 20, 2 – 28, 3 – 40, 4 – 55 °С [16] и в ДМСО (б) при 55 °С [23]: 1 – этиленкарбонат, 2 – 4-(2-этилгексилоксиметил)циклокарбонат, 3 – трициклокарбонат полиоксипропилентриола, 4 – олигомер на основе триглицеридов соевого масла средней функциональности ~4.5.

Рис. 2.
Типичные зависимости kнабл каталитических реакций н-бутиламинолиза этиленкарбоната от концентрации амина: а – катализ при [ТБД] = const = = 3.5 ⋅ 10–3 моль/л в диметилсульфоксиде при 55 °С [24]; б – катализ при [AcOH] = const = 1.0 ⋅ 10–2 моль/л в диоксане при 55 °С [17]; 1 – некаталитическая, 2 – каталитическая реакция.
Самым активным катализатором реакций аминолиза циклокарбонатов является амин класса гуанидинов – 1,5,7-триазабицикло[4.4.0]децен-5 (ТБД):
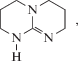
механизм действия которого и кинетические закономерности модельных реакций в его присутствии изучены нами экспериментально и теоретически [24]. Отличительная особенность реакции в присутствии ТБД – линейные зависимости kнабл от концентраций катализатора и амина. Последнее свидетельствует об отсутствии канала с участием двух молекул амина в каталитической реакции. Сравнительные закономерности катализа под действием уксусной кислоты и ТБД приведены на рис. 2. Катализ под действием ТБД принципиально отличен от катализа той же реакции уксусной кислотой, при котором происходит ускорение обоих каналов, хотя и в том и в другом случае ускоряющее действие катализатора направлено преимущественно на мономерный канал.
О чрезвычайно высокой активности ТБД можно судить путем сопоставления соотношения констант скорости каталитической и некаталитической реакций в присутствии одинаковых количеств ТБД и уксусной кислоты. Так, например, при одинаковых концентрациях [ТБД] = [AcOH] = = 9.8 ⋅ 10–3 моль/л и [BuNH2] = 0.4 моль/л соотношение kкат/k0 ~ 56 при катализе ТВД и лишь ~6 при катализе уксусной кислотой. Экспериментальное значение эффективной энергии активации каталитической реакции в присутствии ТБД при [BuNH2] = 0.4 моль/л и [ТБД] = 3.5 ⋅ 10–3 моль/л составляет 2.3 ккал/моль, что примерно в 2.5 раза ниже, чем энергия активации некаталитического процесса (5.4 ккал/моль) при той же концентрации амина в диметилсульфоксиде (ДМСО). На Схемах 3 и 4 приведены результаты квантовохимического исследования синхронного и стадийного путей аминолиза на примере катализируемой ТБД реакции этиленкарбоната с метиламином, протекающей по мономерному каналу [24].
В качестве исходного состояния (RC) принята энергия каталитического комплекса, включающего этиленкарбонат, метиламин и ТБД состава 1 : 1 : 1. Видно, что в энергетическом отношении многостадийный путь примерно вдвое выгоднее одностадийного. В работе [24] подробно описаны строение и энергетические характеристики изомерных (а–d) переходных состояний TS различных стадий реакции. Для механизма каталитического действия ТБД характерно, что образование катионоподобной структуры протонированного катализатора происходит во всех переходных состояниях, где ТБД участвует в переносе протона. Образование таких структур не является энергозатратным, что связано с делокализацией заряда в катионе ТБД. Акцептирование и отрыв протона в ТБД типичны для катализаторов бифункционального типа. В совокупности с низкими стерическими препятствиями для подхода реагентов и гибкостью структуры эти факторы обеспечивают ТБД высокую каталитическую активность.
Наряду с решением целевых задач “зеленой” химии полиуретанов реакция аминолиза циклокарбонатов использовалась нами для разработки методологии теоретических расчетов механизмов химических реакций в растворах в приближении супермолекулы. Это отдельное направление химической физики жидкофазных процессов, позволяющее исследовать и количественно интерпретировать роль растворителя в элементарных актах превращения.
Схема 3
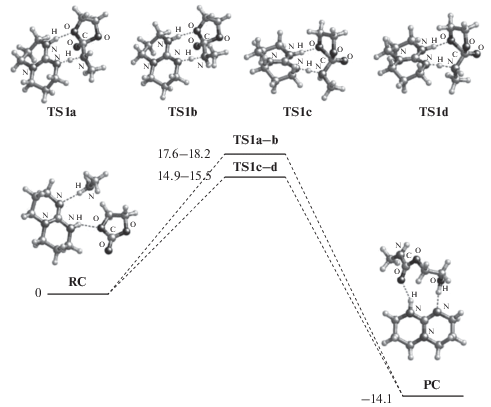
Схема 4
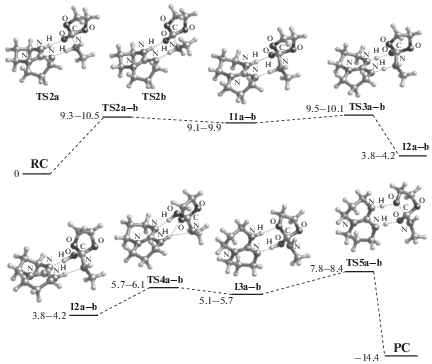
Схема 5
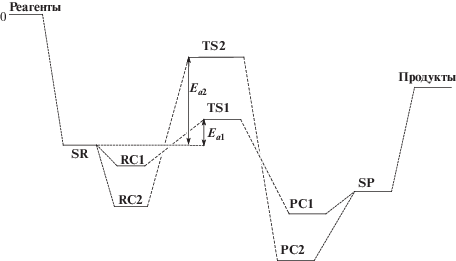
В работах [18, 19] на примере расчетов реакции этиленкарбоната с метиламином в среде диоксана и метанола в приближении супермолекулы мы впервые предложили использовать в качестве уровня отсчета энергию сольватированных, не взаимодействующих между собой реагентов (SR). На Схеме 5 приведена энергетическая диаграмма, указывающая на положение SR, TS, предреакционных комплексов (RC), комплексно-связанных продуктов (PC) и сольватированных продуктов (SP) на координате реакции, протекающей по двум параллельным каналам с энергиями активации Еа1 и Еа2.
Основным критерием правильного построения структур SR был учет всех специфических локальных взаимодействий молекул реагентов с молекулами растворителя. Важно, что энергии взаимодействия реагентов со средой в SR и TS примерно одинаковы и при вычитании одной из другой не влияют на расчетное значение энергии активации. Для процессов в ассоциированной за счет водородных связей среде введены представления об образовании циклов переноса протона (с1) и циклов стабилизации (с2) переходных состояний, размеры которых влияют на величины активационных барьеров. В соответствии со Схемой 6 реакция начинается с образования RC из SR. Первое переходное состояние (TSA) отвечает за присоединение амина к карбонильной группе, второе (TSB) – за раскрытие пятичленного цикла и отрыв уходящей группы. Интермедиат (I) является цвиттер-ионным и может образоваться только при достаточной его стабилизации молекулами растворителя. Минимум для интермедиата I на поверхности потенциальной энергии (ППЭ) относительно TSA, как правило, неглубокий.
Схема 6
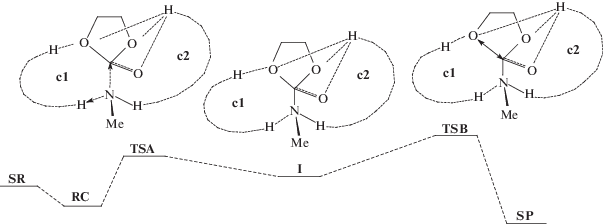
Установлены наиболее выгодные структуры для стабилизации переходных состояний и снижения активационных барьеров за счет образования циклов с участием водородных связей. Образование восьмичленных циклов переноса протона и восьми- или десятичленных циклов стабилизации (на Схеме 6 представлены штриховыми линиями) предпочтительно для рассматриваемой реакции. Наиболее характерные значения барьера реакции по данному механизму в метаноле составляют 15.5–20 ккал/моль [18].
ТРИГЛИЦЕРИДЫ РАСТИТЕЛЬНЫХ МАСЕЛ КАК ВОЗОБНОВЛЯЕМОЕ СЫРЬЕ ДЛЯ НОВЫХ УРЕТАНОВ
С позиций современной экологии представляется желательным при разработке новых процессов “зеленой” химии использовать ненефтяное возобновляемое сырье. В этом отношении новые полиуретаны хороши тем, что для синтеза исходных олигомеров для них могут быть использованы триглицериды ненасыщенных кислот, являющиеся основными компонентами растительных масел. Последние путем окисления легко переводятся в эпоксидированные аналоги, а затем по реакции с СО2 – в соответствующие олигомеры с циклокарбонатными функциональными группами в цепи. Условия синтеза таких олигомеров из растительного сырья описаны в литературе, содержание ненасыщенных и насыщенных кислот в большинстве растительных масел также известно. Однако для регулируемого синтеза полиуретанов принципиальное значение имеют не столько число и содержание ненасыщенных связей в исходных маслах, сколько состав соответствующих триглицеридов после окисления и карбонизации и их функциональность по эпоксидным и циклокарбонатным группам. От этого зависят молекулярная структура и свойства конечного, сшитого за счет реакции с амином, продукта. Таких данных для триглицеридов конкретных растительных масел в литературе нет.
Нами разработан масс-спектрометрический метод анализа функционального состава триглицеридов с эпоксидными и циклокарбонатными группами в цепях и осуществлен синтез циклокарбонатсодержащих производных соевого масла путем каталитической (катализатор – Bu4NBr) карбонизации эпоксидсодержащих предшественников под действием СO2 при 140 °С и давлении 1 МПа [25]. На рис. 3 приведен фрагмент масс-спектра триглицерида, показывающий, что для него характерно наличие цепей с различным числом циклокарбонатных групп. Из анализа масс-спектров продуктов карбонизации с различной степенью превращения следует, что в них содержатся в качестве основных компонентов триглицериды, имеющие олеиновые (O), линолевые (L), линоленовые (Ln), стеариновые (S) и пальмитиновые (Р) цепочки, такие как LLO, LLL, OLO, OOO, LLS, SOO, PLO, PLL, POO, OLnO и другие (всего 25 типов триглицеридов). Главным отличием циклокарбонатсодержащих триглицеридов подсолнечного масла, исследованных нами вслед за производными соевого масла, является отсутствие в них Ln-фрагментов. Естественно, что от состава олигомеров будут зависеть физико-механические свойства конечных полиуретанов, а также кинетика их образования, так как реакционная способность циклокарбонатных групп О-, L- и Ln-фрагментов может быть разной. Это, в частности, следует из результатов недавних квантовохимических расчетов реакции метиламина с циклокарбонатами а, b и c:
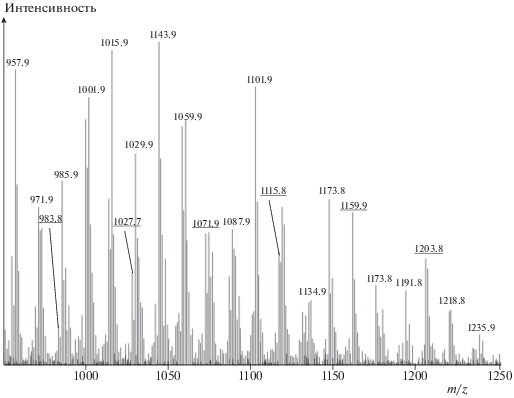
Рис. 3.
Пример фрагмента масс-спектра циклокарбонатсодержащего триглицерида, демонстрирующий процесс карбонизации его эпокси-аналога с исходной молекулярной массой 983.3 Да [25]. Подчеркнутые массы пиков соответствуют структурам с одной (1027.7), двумя (1071.9), тремя (1115.8), четырьмя (1159.9) и пятью (1203.8) циклокарбонатными группами.
моделирующими линолевый – а и олеиновый – b и c фрагменты циклокарбонатсодержащих цепей триглицерида. Так, минимальные значения активационных барьеров превращения циклокарбонатов а, b и c составляют 32.3, 33.1 и 34.3 ккал/моль, соответственно, а барьер превращения второй циклокарбонатной группы в а – много ниже: 25.9 ккал/моль. Отсюда следует, что реакционная способность линолевого радикала триглицерида с двумя расположенными рядом циклокарбонатными группами должна быть выше, чем олеинового. Находит ли это отражение в кинетике реакции уретанообразования с участием таких олигомеров, должны показать дальнейшие кинетические исследования модельных и целевых реакций.
АЦИЛАЗИДЫ КАК СКРЫТЫЕ ИЗОЦИАНАТЫ
Исследования механизмов реакций, лежащих в основе бесфосгенных способов получения изоцианатов, в хронологическом отношении были начаты нами раньше, чем реакций образования неизоцианатных уретанов. Первая работа на эту тему была опубликована в 2004 г. в виде обзора существующих на тот момент экспериментальных и теоретических исследований превращения ацилазидов в изоцианаты путем термической перегруппировки Курциуса [10]:
Эта классическая реакция органической химии протекает при 60–80 °С со 100%-ным выходом изоцианатов, в отличие от фотохимической перегруппировки, при которой образуются различные побочные продукты. Несмотря на то, что сама реакция термической перегруппировки ацилазидов по Курциусу известна давно и используется в органическом синтезе, ее механизм долгое время установлен не был. Основная проблема механизма этой реакции сводилась к вопросу о том, возникает ли в ходе термической перегруппировки Курциуса промежуточный бирадикал – ацилнитрен (стадийной механизм) или же реакция протекает в один акт, при котором образование изоцианата осуществляется синхронно с распадом азидной группировки и выделением молекулы азота (синхронный или концертный механизм). Решению этого вопроса и ряда кинетических проблем, а также связи реакционной способности ацилазидов с их строением посвящены наши последующие работы в данной области [26–30].
Вопрос о стадийности или синхронности перегруппировки Курциуса был решен, когда в результате квантовохимических расчетов строения НСОN3 (1), CH3CON3 (2), PhCON3 (3) стало ясно, что ацилазиды могут находиться в виде син- и анти-конформеров по отношению к связи C–N. Сам факт существования ацилазидов в различном конформационном состоянии ранее не был известен, но, как выяснилось, именно он определяет возможность реализации принципиально разных путей их термической перегруппировки в изоцианаты [29]. Син-конформеры по их энергии образования на 1–7 ккал/моль устойчивее соответствующих анти-аналогов, энергии активации син-анти-изомеризации ацилазидов 1–3 составляют соответственно 9.4, 7.0, 9.2 ккал/моль, обратной реакции – 8.5, 6.1, 2.5 ккал/моль. Как видно из Схемы 7 , син-конформеры ацилазидов 1–3 перегруппировываются в изоцианаты по концертному механизму, а анти-конформеры – по стадийному, через образование синглетного ацилнитрена в качестве интермедиата, причем энергетический барьер по первому пути – через TS1 – ниже, чем по второму– через TS2 и ТS3.
Схема 7
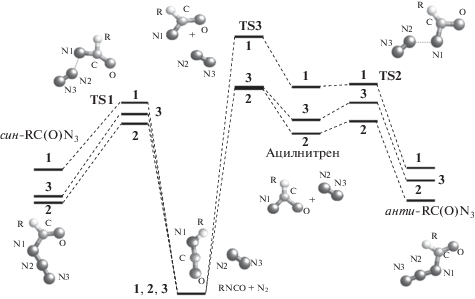
В кинетическом отношении перегруппировка Курциуса чрезвычайно проста: описывается кинетическим законом первого порядка, константа скорости слабо чувствительна к изменению среды, экспериментальные значения энергии активации составляют 25–32 ккал/моль при величинах предэкспонента 1012–1014 с–1, типичных для мономолекулярных реакций термического разложения органических соединений. Расчетные значения активационных барьеров перегруппировки син-конформеров 1–3 равны соответственно 28.0, 32.9 и 34.5 ккал/моль, не сильно отличаясь от экспериментальных величин.
Среди других проблем механизма реакции решался вопрос о причинах аномально высокой реакционной способности орто-замещенных бензоилазида в перегруппировке Курциуса. С этой целью изучено строение шести арилацилазидов общей формулы
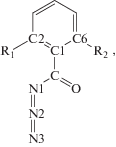
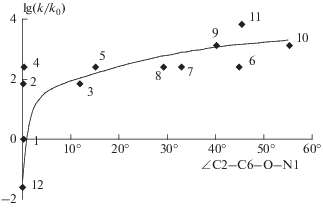
где R1 = Н, R2 = H; R1 = Н, R2 = CH3; R1 = Н, R2 = = изо-C3H7; R1 = Н, R2 = трет-C4H9; R1 = CH3, R2 = CH3; R1 = Н, R2 = OH; и рассчитаны фрагменты ППЭ их синхронной перегруппировки в изоцианаты [27]. Показано, что решающим фактором ускорения перегруппировки является дестабилизация сопряженной структуры арилацилазида за счет стерических эффектов орто-заместителей. Образование внутримолекулярной водородной связи, как это имеет место в случае орто-гидроксизамещенного бензоилазида, стабилизирует сопряженную структуру и приводит к увеличению энергетического барьера реакции. Дана количественная интерпретация “орто-эффекта” на основе установленной связи между реакционной способностью орто-алкилзамещенных бензоилазида и величиной двугранного угла ∠C2–C6–O–N1, характеризующего степень вывода ацилазидной группы из плоскости бензольного кольца (рис. 4).
Рис. 4.
Зависимость реакционной способности арилацилазидов от величины двугранного угла ∠С2–С6–О–N1 в син-конформерах как параметра, характеризующего степень вывода ацилазидной группы из плоскости бензольного кольца под влиянием орто-заместителей: цифры 1–12 относятся к различным устойчивым изомерам орто-замещенных фенилацилазидов 1–6 [27].
В теоретических работах [28, 29] было изучено электронное и геометрическое строение донорно-акцепторных комплексов син-бензоилазида, его 2-метил- и 2,6-диметилзамещенных с льюисовыми кислотами – BF3, AlCl3, SbCl5, а в экспериментальной работе [30] – каталитическая активность некоторых из этих кислот в перегруппировке Курциуса. Комплексы образуются по атомам кислорода и азота ацилазидной группы и имеют состав 1 : 1 или 1 : 2 в зависимости от строения кислоты Льюиса (комплексы по атому кислорода более устойчивы). В результате комплексообразования происходит вывод ацилазидной группировки из плоскости бензольного кольца, что приводит к снижению энергии активации их перегруппировки по сравнению с некаталитической реакцией. Установлены корреляции между энергией активации перегруппировки комплексов и величиной двугранного угла ∠C2–C6–O–N1 в них, характеризующего степень нарушения сопряжения ацилазидной группы с бензольным кольцом под влиянием комплексообразования с кислотой Льюиса. Получены сведения о строении переходных состояний перегруппировки комплексов, в которых осуществляется отрыв молекулы азота с синхронной перестройкой остальных атомов, ведущей к образованию конечного продукта. Таким образом, комплексообразование с кислотами Льюиса может служить эффективным способом регулирования реакционной способности ацилазидов.
ЗАКЛЮЧЕНИЕ
Проведенный комплекс исследований механизмов образования новых уретанов и бесфосгенных изоцианатов может служить количественной основой разработки соответствующих процессов “зеленой” химии, так же как и сведения о распределении по функциональности циклокарбонатсодержащих триглицеридов растительных масел – возобновляемого сырья для полиуретанов. Малоизученной в количественном отношении остается пока реакция фиксации СO2 эпоксидами как метод получения циклокарбонатов. До сих пор экспериментаторам не удалось найти катализаторы и условия проведения реакций при низких температурах и давлениях СО2, и здесь квантовая химия является весьма перспективным методом исследования. Нами в настоящее время ведутся расчеты методом функционала плотности с использованием в качестве катализаторов тетраалкиламмониевых солей, льюисовых кислот и оснований с разнообразными добавками, ионных жидкостей. Показано, что в результате взаимодействия нуклеофильного фрагмента катализатора с эпоксидом образуется активный интермедиат – алкоголят-анион, способный присоединяться практически к любым доступным в ближайшем окружении молекулам, в том числе и к CO2:
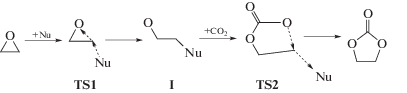
Для снижения температуры и давления при проведении такой реакции необходимо присутствие в системе доноров протонов, которые за счет образования разветвленных цепей водородных связей стабилизировали бы анионный центр интермедиата и способствовали снижению энергии переходного состояния. Перспективными в этом смысле катализаторами могут быть соли третичных и вторичных аминов, а также ионные жидкости с добавлением или без добавления других амфотерных или слабокислых доноров протонов, таких как вода. Есть основания надеяться на успех исследований такого типа систем и их дальнейшее практическое применение.
Работа выполнена в рамках госзадания (тема V 45.5, 0082-2014-0015, № АААА-А17-117032750201-9) и при финансовой поддержке Российским фондом фундаментальных исследований (проект № 17-03-00146).
Список литературы
Cornille A., Auvergne R., Figovsky O., Boutevin B., Caillol S. // Eur. Polym. J. 2017. V. 87. P. 535.
Rokicki G., Parzuchowski P.G., Mazurek M. // Polym. Adv. Technol. 2015. V. 26. № 7. P. 707.
Maisonneuve L., Lamarzelle O., Rix E., Grau E., Cramail H. // Chem. Rev. 2015. V. 115. № 22. P. 12407.
Blattmann H., Fleischer M, Bähr M., Mülhaupt R. // Macromol. Rapid Comm. 2014. V. 35. № 14. P. 1238.
Figovsky O., Shapovalov L., Leykin A., Birukova R., Pota-shnikova R. // PU Magazine Int. 2013. V. 10. № 4. P. 256.
Nohra B., Candy L., Blanco J.-F. et al. // Macromolecules. 2013. V. 46. № 10. P. 3771.
Figovsky O., Shapovalov L., Leykin A., Birukova R., Pota-shnikova R. // Intern. Lett. Chem. Phys. and Astron. 2012. V. 3. P. 52.
Guan J., Song Y., Lin Y. et al. // Ind. Eng. Chem. Res. 2011. V. 50. № 11. P. 6517.
Blazek K., Datta J. // Crit. Rev. Env. Sci. and Technol. 2019. V. 49; https://doi.org/10.1080/10643389.2018.15377
Тигер Р.П. // Высокомолекуляр. соединения. 2004. Т. 46. № 5. С. 931.
Garrison T.F., Kessler M.R. // Bio-based plant oil polymers and composites. / Eds. Madbouly S.A., Zhang C., Kessler M.R. Amsterdam: Elsevier Inc., 2016. P. 37.
Гарипов Р.М., Сысоев В.А., Михеев В.В. и др. // Докл. АН. 2003. Т. 393. № 1. С. 61.
Lambert R.H., Henderson T.J. // Polymer. 2013. V. 54. № 21. P. 5568.
Забалов М.В., Тигер Р.П., Берлин А.А. // Докл. АН. 2011. Т. 441. № 4. С. 480.
Забалов М.В., Тигер Р.П., Берлин А.А. // Изв. АН. Сер. хим. 2012. № 3. С. 518.
Левина M.A., Крашенинников В.Г., Забалов М.В., Тигер Р.П. // Высокомолекуляр. соединения. 2014. Т. 56. № 2. С. 139.
Забалов М.В., Левина М.А., Крашенинников В.Г., Тигер Р.П. // Изв. АН. Сер. хим. 2014. № 5. С. 1740.
Zabalov M.V., Tiger R.P. // Theor. Chem. Acc. 2017. V. 136. Article 95.
Забалов М.В., Тигер Р.П. // Изв. АН. Сер. хим. 2016. № 3. С. 631.
Tomita H., Sanda F., Endo N. // J. Polym. Sci., Part A: Polym. Chem. 2001. V. 39. № 21. P. 3678.
Lamarzelle O., Durand P.-L., Wirotius A.-L. et al. // Polym. Chem. 2016. V. 7. № 7. P. 1439.
Cornille A., Blain M., Auvergne R. и дp. // Ibid. 2017. V. 8. № 3. P. 592.
Левина М.А., Забалов М.В., Крашенинников В.Г., Тигер Р.П. // Высокомолекуляр. соединения. 2018. Т. 60. № 5. С. 372.
Левина М.А., Забалов М.В., Крашенинников В.Г., Тигер Р.П. // Там же. 2017. Т. 59. № 5. С. 317.
Левина М.А., Милославский Д.Г., Придатченко М.Л. и др. // Там же. 2015. Т. 57. № 6. С. 413.
Забалов М.В., Тигер Р.П. // Изв. АН. Сер. хим. 2005. № 10. С. 2200.
Забалов М.В., Тигер Р.П. // Там же. 2007. № 1. С. 7.
Забалов М.В., Тигер Р.П. // Там же. 2012. № 9. С. 1678.
Zabalov M.V., Tiger R.P. // J. Mol. Struct.: THEOCHEM. 2010. V. 962. № 1–3. P. 15.
Тигер Р.П., Забалов М.В., Птицына Н.В. // Кинетика и катализ. 2009. Т. 50. № 3. С. 397.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика