

Химическая физика, 2020, T. 39, № 1, стр. 3-6
Влияние внутри- и межмолекулярных взаимодействий на возможность внутреннего вращения вокруг С=С-связи в молекулах FOX-7 и его производных
Б. Э. Крисюк *
Институт проблем химической физики Российской академии наук
Черноголовка, Россия
* E-mail: bkris@mail.ru
Поступила в редакцию 02.08.2018
После доработки 21.04.2019
Принята к публикации 20.05.2019
Аннотация
В работе методами квантовой химии с использованием метода DFT c функционалом PBE и базисным набором cc-pVDZ выполнены расчеты барьера внутреннего вращения в 1,1-диамино-2,2-динитроэтилене (ДАДНЭ или FOX-7) и 2-(нитрометилен)-1,3-диазациклогептане (НМДАЦГ). Расчет для изолированных молекул показал, что вращение в таких молекулах вокруг С=С-связи вполне возможно. Барьер вращения составляет 19 (ДАДНЭ) и 16 ккал/моль (НМДАЦГ) соответственно. Для исследования влияния на этот барьер межмолекулярного взаимодействия исследовали комплексы ДАДНЭ и НМДАЦГ с двумя молекулами нитрометана. Последние использованы в качестве моделей ДАДНЭ и НМДАЦГ. Расчет показал, что межмолекулярное взаимодействие снижает барьер внутреннего вращения. В комплексах молекул ДАДНЭ и НМДАЦГ с двумя молекулами нитрометана этот барьер составляет 12 и 10 ккал/моль соответственно. Кроме того, в комплексе НМДАЦГ с двумя молекулами нитрометана наиболее энергетически выгодным оказалось состояние, в котором плоскость нитрогупп развернута на 120° относительно плоскости NH–C–NH.
ВВЕДЕНИЕ
Среди интенсивно изучаемых в последнее время энергоемких соединений 1,1-диамино-2,2-динитроэтилен (ДАДНЭ или FOX-7) является одним из самых перспективных из-за его высокой термической стабильности и низкой чувствительности к удару и трению [1]. Несмотря на большое количество работ, посвященных изучению термолиза ДАДНЭ, детальный механизм первичной стадии до сих пор остается не ясным. В качестве первичной реакции термолиза предполагали отщепление NO2, и это предположение подтверждается в том числе последними результатами, полученными методом молекулярной динамики в конденсированной фазе [2, 3]. Однако этот результат не соответствует данным по энергии активации термолиза, равной 45.7 ккал/моль [4], а прочность C–NO2 выше 70 ккал/моль. Также предполагали нитро-нитрит-изомеризацию (энергия активации около 60 ккал/моль) [5, 6] и внутримолекулярный перенос атома водорода с аминогруппы на углерод и атома кислорода нитрогруппы на другой углерод [7]:
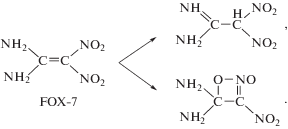
Наконец, недавно был предложен механизм с прямым внутримолекулярным переносом атома водорода с амино- на нитрогруппу и одновременным конформационным переходом [8], когда в продукте атом водорода развернут в сторону, противоположную аминогруппе:
Обратим внимание, что практически во всех механизмах первичной стадии термолиза происходит изменение гибридизации атома углерода и двойная связь становится одинарной. При этом происходит поворот плоскости нитрогрупп относительно плоскости аминогрупп. Циклические производные ДАДНЭ (2-(нитрометилен)-1,3-диазациклопентан (НМДАЦГ), а также циклогексан и циклогептан, т.е. 5-, 6- и 7-членные циклы) также представляют большой интерес как перспективные материалы. А в таких производных с 6- и 7-членными циклами уже в исходном состоянии в кристалле плоскость цикла развернута примерно на 90° относительно плоскости остальной молекулы [9]. Поэтому исследование возможности внутреннего вращения в ДАДНЭ и его производных важно как для объяснения структуры последних, так и для анализа первичной стадии термолиза, где это вращение необходимо для протекания реакции.
Следует также отметить, что в большинстве цитированных работ моделирование термолиза выполняли в газовой фазе, т.е. для изолированных молекул. Тогда как эксперименты в основном делали в жидкой и твердой фазах, где из-за высокой полярности ДАДНЭ велика роль не только внутри-, но и межмолекулярных взаимодействий. Поэтому в данной работе мы рассмотрели не только возможность внутреннего вращения в этих молекулах, но и влияние межмолекулярных взаимодействий на него. В настоящей работе мы выполнили исследование на примере ДАДНЭ и
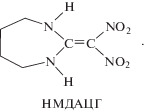
МЕТОДИКА РАСЧЕТА
Расчеты проводили на многопроцессорном кластере в вычислительном центре ИПХФ РАН и на восьмиядерном компьютере AMD с помощью программ GAUSSIAN 09 [10], GAMESS [11] и FireFly [12], частично основанной на [11]. Основные расчеты выполнены с использованием метода DFT c гибридным функционалом на базе PBE (PBE0) и базисным набором cc-pVDZ. Выбор этого уровня расчета обусловлен тем, что он достаточно хорошо воспроизводит результаты, полученные на гораздо более высоком уровне расчета в работе [5] (расхождение наших данных с результатами цитируемой работы не превышает 2 ккал/моль [6]), и в то же время не так ресурсозатратен. Многоконфигурационные расчеты методами MCSCF и MRMP2 выполнены с помощью пакета GAMESS [11] и с использованием геометрии, оптимизированной на уровне PBE/cc-pVDZ.
Внутреннее вращение исследовали сканированием торсионного угла между плоскостями амино- и нитрогрупп. Сканирование осуществляли с шагом в 5–10 град. Геометрические параметры, соответствующие вершине барьера вращения, дополнительно оптимизировали методами TS и QST. Истинность положения барьера контролировали путем сканирования внутренней координаты реакции в обе стороны от барьера с помощью процедуры IRC, реализованной в [10]. При сканировании угла вращения в комплексах использовали несколько стартовых точек, так как при очередном шаге могла измениться структура комплекса и мы попадали на другую ветвь кривой.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
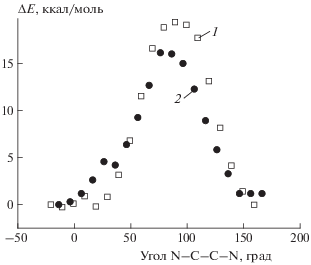
На рис. 1 представлены результаты сканирования торсионного угла в изолированных молекулах ДАДНЭ и НМДАЦГ. Видно, что в исходных молекулах из-за наличия внутримолекулярного сопряжения связь С=С сильно ослаблена и по сути не является двойной. Барьер вращения составляет всего 19 ккал/моль для ДАДНЭ и 16 ккал/моль для НМДАЦГ, содержащего семичленный цикл. Результаты рис. 1 получены методом PBE по сути в однодетерминантном приближении. Однако вращение вокруг двойной связи в общем случае связано с разрывом и затем образованием π-связи и в этом приближении может описываться некорректно. Чтобы убедиться, что для данной задачи метод PBE дает адекватные результаты, были выполнены расчеты на уровне MCSCF и MRMP2 (базис cc-pVDZ). Для молекулы ДАДНЭ величина барьера внутреннего вращения составила 14 (MCSCF) и 22 ккал/моль (MRMP2) соответственно. Первая величина получена без учета корреляции электронов и потому не точна, а вторая достаточно хорошо соответствует данным расчетов методом PBE. Следовательно, выбранный метод применим для данной задачи и поэтому остальные результаты получены на этом уровне расчета.
Рис. 1.
Изменение энергии молекул при повороте плоскости нитрогрупп в молекулах ДАДНЭ (1) и НМДАЦГ (2).
Оптимизация переходного состояния методами TS и QST качественно картины не изменяет (изменяется лишь значение энергии барьера в пределах долей ккал/моль). Также и сканирование внутренней координаты с помощью процедуры IRC от барьера в обе стороны приводит к исходному или конечному состояниям. Поэтому для экономии места эти результаты не приведены.
Отметим, что в молекуле, содержащей цикл, барьер вращения ниже. При этом значения величины барьера вращения сопоставимы с энергией межмолекулярного взаимодействия в подобных молекулах. Следовательно, зафиксированный в работе [9 ] поворот кольца в НМДАЦГ может быть объяснен низким барьером вращения.
Следует отметить, что данный результат подтверждает отмеченную ранее и экспериментально изученную закономерность снижения барьера внутреннего вращения для поляризованных С=С-связей (см., например, [13]). Так, в случае наличия электронодононых и электроноакцепторных заместителей у С=С-связи энергия активации вращения составляет 15–24 ккал/моль [13], что хорошо соответствует нашим результатам расчетов (рис. 1).
Однако в конденсированных фазах межмолекулярное взаимодействие аминогрупп может еще больше ослабить С=С-связь за счет смещения сопряженных π-электронов на аминогруппу. Для проверки этого предположения мы исследовали внутреннее вращение в комплексах с молекулами нитрометана (модель водородных связей в кристаллах ДАДНЭ и его производных). На рис. 2 показана структура комплекса исходной молекулы ДАДНЭ с двумя молекулами нитрометана. Все координаты оптимизированы. Расстояние между атомами водорода ДАДНЭ и атомами кислорода нитрометана составляет соответственно 2.07 и 3.16 Å в каждой из пар. В этом смысле комплекс является симметричным. Аналогичный комплекс образует и НМДАЦГ. Но в этой молекуле вместо NH2-групп присутствуют NH-группы, поэтому молекулы нитрометана ориентированы на единственный в каждой паре водород, а все расстояния N…H примерно одинаковы и составляют 3 Å.
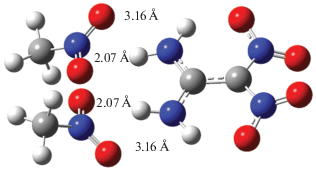
На рис. 3 представлены кривые изменения энергии таких комплексов при вращении плоскости нитрогрупп. Для комплекса ДАДНЭ наблюдается примерно та же картина, что и для изолированной молекулы (рис. 1), но барьер вращения существенно ниже и составляет 12 ккал/моль, что на 7 ккал/моль меньше, чем для свободной молекулы.
Рис. 3.
Изменение энергии комплексов исследуемых молекул при вращении плоскости нитрогрупп в ДАДНЭ (1) и НМДАЦГ (2).
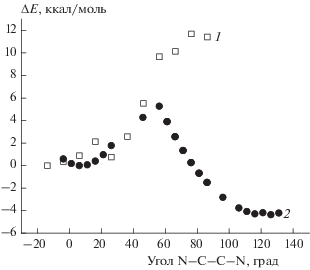
Иная картина наблюдается для НМДАЦГ. Здесь комплекс с углом поворота нитрогрупп, равным 110°–120°, оказывается энергетически более выгодным по сравнению с комплексом исходной молекулы. Это демонстрирует, что при наличии межмолекулярных связей основным состоянием для циклических замещенных ДАДНЭ могут стать состояния, в которых плоскость цикла развернута относительно плоскости нитрогрупп. Барьер вращения, отсчитываемый от более выгодного развернутого состояния, составляет всего 10 ккал/моль.
Таким образом, из-за внутримолекулярных взаимодействий в молекулах аминонитроэтиленов, таких как ДАДНЭ и его производные, барьер внутреннего вращения оказывается существенно понижен и составляет величину порядка 20 ккал/моль. В конденсированных фазах межмолекулярные водородные связи не только еще больше снижают барьер внутреннего вращения (до 10 ккал/моль) в ДАДНЭ и его циклических производных, но и могут сделать более энергетически выгодными состояния, в которых плоскость N–C–N в цикле повернута на значительный угол относительно плоскости нитрогрупп.
ВЫВОДЫ
1. Квантовохимические расчеты показывают, что в изолированных молекулах ДАДНЭ и НМДАЦГ возможно внутреннее вращение вокруг С=С-связи. Из-за внутримолекулярного сопряжения барьер такого вращения составляет 19 (ДАДНЭ) и 16 ккал/моль (НМДАЦГ) соответственно.
2. Межмолекулярное взаимодействие снижает этот барьер. В комплексах исследованных молекул с двумя молекулами нитрометана он составляет 12 и 10 ккал/моль соответственно.
3. В комплексе НМДАЦГ с двумя молекулами нитрометана наиболее энергетически выгодным оказалось состояние, в котором плоскость нитрогупп развернута на 120° относительно плоскости NH–C–NH.
Список литературы
Latypov N.V., Bergman J., Langlet A., Wellmar U., Bemm U. // Tetrahedron 1998. V. 54. P. 11525.
Liu Y., Li F., Sun H. // Teor. Chem. Acc. 2014. V. 133. P. 1557; https://doi.org/10.1007/s00214-014-1567-5
Jiang H., Jiao Q., Zhang C. // J. Phys. Chem. C. 2018. V. 122. P. 15125; https://doi.org/10.1021/acs.jpcc.8b03691
Burnham A.K., Weese R.K., Wang R., Kwok Q.S.M., Jones D.E.G. // Proc. 35th Intern. Annual Conf. of ICT. Karlsruhe, Germany. 2005. P. 70; https://www.osti.gov/biblio/862394
Gindulyte A., Massa L., Huang L., Karle J. // J. Phys. Chem. A. 1999. V. 103. P. 11045.
Khrapkovskii G.M., Nikolaeva E.V., Chachkov D.V., Shamov A.G. // Rus. J. General Chem. 2004. V. 74. № 6. P. 908.
Kiselev V.G., Gristan N.P. // J. Phys. Chem. 2014. V. 118. № 36. P. 8002.
Крисюк Б.Э., Веретин В.С. // Бутлеровские сообщ. 2017. Т. 49. № 2. С. 30.
Baun K., Bigelow S.S., Nguyen N.V., Archibald T.G. // J. Org. Chem. 1992. V. 57. № 1. P. 235.
Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J.A., Jr., Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers E., Kudin K.N., Staroverov V.N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas Ö., Foresman J.B., Ortiz J.V., Cioslowski J., Fox D.J. // Gaussian 09. Rev. C.01. Gaussian. Inc.: Wallingford CT, 2009.
Granovsky A.A. Firefly version 8.0.0; http://classic. chem.msu.su/gran/firefly/index.html
Schmidt M.W., Baldridge K.K., Boatz J.A. et al. // J. Comput. Chem. 1993. V. 14. P. 1347.
Потапов В.М. Стереохимия. М.: Химия, 1988.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика