

Химическая физика, 2020, T. 39, № 1, стр. 83-87
Моделирование взаимодействия катионов щелочных металлов с фрагментом графена в среде бензола
Г. П. Михайлов *
Уфимский государственный авиационный технический университет
Уфа, Россия
* E-mail: gpmihailov@mail.ru
Поступила в редакцию 09.01.2019
После доработки 05.06.2019
Принята к публикации 20.06.2019
Аннотация
Методом теории функционала плотности в приближении M06-2X/6-31G(d,p) проведено моделирование взаимодействия катионов щелочных металлов (M = Li+, Na+, K+) с фрагментом графена C36H16. Рассчитаны энергии связывания, физико-химические параметры ион-графеновых комплексов в вакууме и бензоле. Проведен анализ зависимости энергии связывания комплексов M+…C36H16 от расстояния между катионом и плоскостью фрагмента графена. Показано влияние природы катиона и неспецифической сольватации на прочность катион-π-взаимодействия.
ВВЕДЕНИЕ
Изучение взаимодействия катиона лития с графеновой поверхностью актуально при решении проблем, которые связаны с поиском новых материалов, способных накапливать ионы лития (например, для литиевых батарей). Фрагмент графена может быть использован как сенсор: например, даже 30–50 атомов углерода, собранные в графеновую структуру (или в так называемую графеновую квантовую точку) [1, 2]. Важно, как графеновые квантовые точки взаимодействуют с различными химическими элементами и ионами. Различные факторы влияют на взаимодействие ионов с поверхностью графена: природа катиона, сольватация и геометрия взаимодействия [3]. Работ, посвященных исследованию влияния сольватации на взаимодействие катионов с поверхностью графена, практически нет [4]. Анализ влияния неспецифической сольватации важен для электрохимических исследований с использованием графеновых поверхностей. Цель работы – расчет энергии связывания и анализ термодинамических параметров неспецифической сольватации комплексов катионов щелочных металлов Li+, Na+, K+ с фрагментом графена C36H16 в вакууме и среде бензола.
МЕТОДИКА РАСЧЕТОВ
Расчеты оптимальной структуры комплексов M+…C36H16 (M = Li+, Na+, K+) методом теории функционала плотности в приближении M06-2X/6-31G(d,p) проведены с использованием программного пакета GAUSSIAN 09 [5]. Выбор функционала M06-2X обусловлен его эффективностью в применении к исследованию термохимии, кинетики и нековалентных взаимодействий (силы Ван-дер-Ваальса, взаимодействия, связанные с переносом заряда, и диполь-дипольные взаимодействия) [6]. Нахождение стационарной точки при оптимизации геометрических параметров комплексов контролировалось тем, чтобы все рассчитанные колебательные частоты оказывались действительными. Учет неспецифической сольватации для графеновых комплексов в среде бензола проводился в рамках континуальной модели непрерывного поляризуемого диэлектрика (IEFРСМ) [7, 8]. Энергия связывания катиона с фрагментом графена рассчитывалась как разность полных энергий графенового комплекса и суммарной энергии его компонент.
Перенос электронного заряда из π-системы C36H16 в катион
Графен – это один слой sp2-углерода, состоящий из конденсированных шестичленных колец. Каждый атом графеновой плоскости ковалентно связан с тремя соседними. Неспаренные электроны атома углерода определяют особые электронные свойства графена [9]. Моделирование проводилось для фрагмента графена c атомами водорода, насыщающими оборванные связи на границах. Это необходимо для того, чтобы избежать влияния краевых дефектов на результаты моделирования. В качестве модели выбран простейший фрагмент графена с границами типа “зигзаг” и “кресло”, состоящий из 36 атомов углерода (рис. 1). Катион располагался перпендикулярно плоскости графена на расстоянии h, значение которого соответствует оптимальной структуре комплексов и зависит от природы иона (табл. 1). Ранее [10] методом теории функционала плотности в приближении M06-2X/6-311G(d,p) выполнен расчет энергии связывания, ΔE, комплексов катиона лития с молекулами бензола и полиароматических углеводородов (ПАУ) типа Li+${{{\text{C}}}_{{6{{n}^{2}}}}}$H6n (n = 2−5). Энергия связывания увеличивается в ряду C6H6 < < C24H12 < C54H18 < C96H24 < C150H30 и стремится к насыщению. Значения ΔE и диаметров d молекул ПАУ, равных среднему арифметическому размеров молекул вдоль осей X и Y, сильно взаимосвязаны с коэффициентом корреляции, равным 0.995. Энергия связывания Li+–C36H16, равная 209.5 кДж/моль (приближение M06-2X/6-311G(d,p)), хорошо соответствует уравнению регрессии ΔE от d, что показывает корректность выбранной модели фрагмента графена. При образовании комплексов M+…C36H16 (M = Li+, Na+, K+) длины связей не изменяются, за исключением незначительного увеличения на 0.3–0.6% длин связей C–C, C=C центрального кольца в контакте с M+. У иона лития 2s-оболочка свободна, а 1s-оболочка стабильна настолько, что исключает переход электрона на фрагмент графена и возникновение ионной связи.
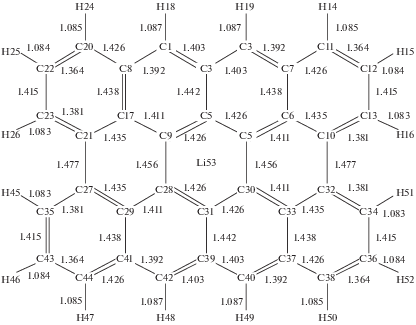
Таблица 1.
Значения заряда катионов q(M+), переноса электронного заряда Δq(C36H16), расстояния h между ионом M+ и C36H16, дипольного момента μ, ионного потенциала φион катиона, квадрупольного момента Θzz и средней поляризуемости αср ион-графеновых комплексов
| Комплекс | q(M+), a.e. | Δq(C36H16), a.e. | h, Å | µ, D | φион, а.е./нм | –Θzz, D ∙ Å | αср, a.e. |
|---|---|---|---|---|---|---|---|
| Li+–C36H16 | 0.527 (0.676) | 0.473 (0.324) | 1.84 (1.97) | 4.7 (6.1) | 17 | 199.8 (195.9) | 501.6 (642.8) |
| Na+–C36H16 | 0.707 (0.818) | 0.293 (0.182) | 1.69 (2.48) | 6.7 (8.5) | 10 | 194.7 (188.2) | 498.5 (638.2) |
| K+–C36H16 | 0.882 (0.911) | 0.118 (0.089) | 2.68 (2.83) | 8.6 (10.0) | 8 | 193.8 (188.1) | 500.4 (639.5) |
Взаимодействие катиона с графеном приводит к значительному переносу электронного заряда Δq(C36H16) из π-системы C36H16 в катион. Значения переноса электронного заряда по Малликену рассчитывались путем вычитания остаточного заряда на катионе в комплексе из начального заряда, составляющего +1 а.е. Значения Δq(C36H16) коррелируют с величиной ионного потенциала катиона φион = q/r, где r – радиус катиона. Заряды на всех атомах углерода (за исключением кольца, наиболее близкого к катиону) при образовании комплексов не изменяют знак, уменьшаются примерно на 2–8%, и характер изменения слабо зависит от природы катиона. Суммарный заряд на атомах водорода увеличивается примерно на 20–25% и не зависит от природы катиона. Значительно изменяются заряды атомов углерода кольца, наиболее близкого к катиону (табл. 2). Квадрупольный момент отражает распределение заряда по всей молекулярной системе. Компонента Θzz характеризует относительное распределение заряда вдоль оси Z перпендикулярно плоскости C36H16 (табл. 1). Отрицательные значения Θzz указывают на миграцию отрицательного заряда в результате катион-π-взаимодействия связей π(C–C) с 1s(Li+)-электроном.
Таблица 2.
Заряды на атомах, q, a.e., шестичленного кольца, входящего в состав C36H16 и комплексов M+…C36H16
| Атом | C36H16 | Li+…C36H16 | Na+…C36H16 | K+…C36H16 |
|---|---|---|---|---|
| C4 | –0.0389 (–0.0410) | –0.0606 (–0.0726) | –0.0728 | –0.0905 |
| C31 | –0.0389 (–0.0410) | –0.0612 (–0.0784) | –0.0728 | –0.0905 |
| C5 | 0.0305 (0.0291) | –0.0151 (–0.0180) | –0.0229 | –0.0315 |
| C9 | 0.0305 (0.0291) | –0.0149 (–0.0090) | –0.0229 | –0.0315 |
| C28 | 0.0305 (0.0291) | –0.0142 (0.0046) | –0.0229 | –0.0315 |
| C30 | 0.0305 (0.0291) | –0.0143 (–0.0046) | –0.0229 | –0.0315 |
| Σq | 0.0442 (0.0344) | –0.1803 (–0.1780) | –0.2372 | –0.3070 |
Энергия взаимодействия ион-графеновых комплексов
Энергия связывания ион-графеновых комплексов зависит от природы катиона и увеличивается в ряду K+ < Na+ < Li+ (табл. 1). Исследована зависимость энергии связывания от расстояния между катионом и плоскостью кольца вдоль оси Z, перпендикулярной плоскости C36H16 (рис. 2). В области действия сил притяжения потенциала взаимодействия M+…C36H16 при r > h отношение ΔE(r)/ΔE(r = h) хорошо описывается функцией A/rn (n = 2.1, коэффициент корреляции R = 0.998), где A = 6.0 для Li+, A = 7.8 для Na+ и A = 9.9 для K+. Следует отметить, что функция A/rn характерна для аппроксимации на больших расстояниях потенциала катион-π-взаимодействия катионов Na+ (n = 1.9), K+ (n = 2.0) с молекулой бензола (расчет в приближении M06/6-31G(d,p)) [11]. Смещение катиона M+ на r = 2h приводит к уменьшению ΔE примерно на 60% (Li+) и 70% (Na+, K+). Эти результаты показывают, что даже при неоптимальных расстояниях катион-π-взаимодействие вносит существенный вклад в энергию связывания комплексов. Фрагмент C36H16 обладает значительным квадрупольным моментом Θzz = = −212.6 Д ∙ Å (в среде бензола Θzz = −212.6 Д ∙ Å), но модель ион–квадруполь не позволяет полностью количественно описать катион-π-взаимодействие при r > h, так как предполагает, что заряды точечные, а зависимость энергии взаимодействия пропорциональна r−3 [12]. Дипольный момент изолированного фрагмента C36H16 близок к нулю (μ = 6 ∙ 10−4 Д). В комплексах M+…C36H16 значительный дипольный момент индуцируется из электронной плотности π-системы фрагмента графена за счет катиона (табл. 1). Значительные величины индуцированного дипольного момента, средней статической поляризуемости (αср) и заряд катионов (табл. 1) предполагают важность взаимодействия типа ион – индуцированный диполь (ΔEинд ~ q2αср/r4). Кроме этого, между катионом M+ и отрицательным зарядом, распределенным по поверхности молекул ароматических углеводородов, бензола и его производных, существует сильное электростатическое взаимодействие [13]. Зависимость ΔE(r) ~ r–2 на больших расстояниях может быть объяснена суперпозицией вкладов электростатической энергии взаимодействия и энергии взаимодействия типа ион – индуцированный диполь. Важность этих вкладов в энергию взаимодействия катионов (M = Li+, Na+, K+) с молекулами бензола, толуола, этилбензола и тетраэтилбензола показана посредством квантовохимических расчетов в приближении MP2/6-311G(d,p) [14].
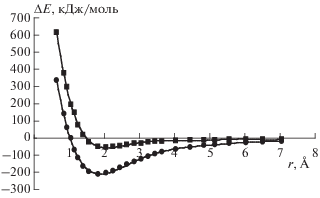
Влияние неспецифической сольватации
Одним из факторов, влияющих на прочность катион-π-взаимодействия с фрагментом графена, является неспецифическая сольватация. Энергия связывания ион-графеновых комплексов в среде бензола увеличивается в ряду K+ < Na+ < Li+, но менее зависит от природы катиона по сравнению с взаимодействием в вакууме (табл. 3). В области действия сил притяжения потенциала взаимодействия Li+…C36H16 при r > h энергия связывания ΔE(r) ~ 1/rn (n = 2.1 (вакуум), n = 1.7 (модель IEFPCM)). Свободная энергия сольватации комплексов (ΔGsolv) изменяется в ряду K+ < Na+ ≈ Li+ и по сравнению с изменением энергии связывания ΔE менее зависит от природы катиона (одноточечный “sp-расчет” с использованием параметра ExternalIteration [9]). Увеличение дипольного момента ион-графеновых комплексов в среде бензола обусловлено частичной компенсацией зарядов поляризованным континуумом. Влияние растворителя на растворенное вещество в методах поляризуемого континуума описывается эффектами “электростатических” и “неэлектростатических” взаимодействий. Вклад последних содержит дисперсионную (Edisp) и кавитационную (Ecav) составляющие, а также энергию отталкивания электронных оболочек молекул растворенного вещества и растворителя (Erep). Среди “неэлектростатических” (Enonel) членов энергии сольватации наибольшие значения дают дисперсионная и кавитационные составляющие. Они близки по модулю и имеют противоположные знаки, так что отчасти компенсируют друг друга. Влияние растворителя определяется главным образом “электростатическими” (Eel) взаимодействиями “растворенное вещество–растворитель”. С другой стороны, энергия связывания в среде бензола без учета неэлектростатических взаимодействий изменяется в интервале 44.9–47.8 кДж/моль и практически не зависит от природы катиона. Наибольший вклад в энергию сольватации обеспечивается катионами натрия (–126.9 кДж/моль), лития (–117.0 кДж/моль) и калия (–111.3 кДж/моль). Вклад от атомов фрагмента графена составляет 11–19% от свободной энергии сольватации.
Таблица 3.
Значения энергии (в кДж/моль) связывания (∆E) и составляющих энергии сольватации (ΔGsolv) ион-графеновых комплексов
| Комплекс | –∆E | –ΔGsolv | –Edisp | Erep | Ecav | Enonel | –Eel |
|---|---|---|---|---|---|---|---|
| Li+–C36H16 | 208.7 (54.6) | 144.7 | 150.8 | 14.0 | 186.1 | 49.4 | 194.1 |
| Na+–C36H16 | 157.5 (46.5) | 145.0 | 151.1 | 14.1 | 189.3 | 52.3 | 197.3 |
| K+–C36H16 | 121.0 (39.9) | 124.9 | 153.3 | 14.2 | 193.2 | 54.2 | 179.1 |
ЗАКЛЮЧЕНИЕ
При взаимодействии катионов щелочных металлов с фрагментом графена наиболее значительно изменяются заряды атомов углерода шестичленного кольца, наиболее близкого к катиону. Рост величины переноса заряда приводит к увеличению энергии связывания и хорошо коррелирует с ионным потенциалом катиона. Значительные величины индуцированного дипольного момента, средней поляризуемости и переноса заряда предполагают важность взаимодействия типа ион – индуцированный диполь при расстояниях, значительно превышающих расстояние между катионом и фрагментом графена, соответствующее оптимальной структуре комплекса. Погружение графена в континуум бензола существенно уменьшает энергию связывания и величину перераспределения заряда по сравнению с вакуумом. Наибольший вклад в свободную энергию сольватации ион-графеновых комплексов обеспечивается катионами.
Работа выполнена при финансовой поддержке Минобрнауки России в рамках реализации проекта № 16.1969.2017/4.6.
Список литературы
Gong X., Bi Y., Zhao Y. et al. // RSC Adv. 2014. V. 4. P. 24653.
Ting S.L., Ee S.J., Ananthanarayanan A. et al. // Electrochim. Acta. 2015. V. 172. P. 7.
Hill J.M., Chan Y. // Micro and Nano Lett. 2010. V. 5. № 5. P. 247.
Georgakilas V., Otyepka M., Athanasios B. et al. // Chem. Rev. 2012. V. 112. № 11. P. 6156.
Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J., Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers E., Kudin K.N., Staroverov V.N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas Ö., Foresman J.B., Ortiz J.V., Cioslowski J., Fox D.J. Gaussian 09. Rev. C. 1. Wallingford CT: Gaussian, Inc., 2009.
Zhao Y., Truhlar D.G. // Theor. Chem. Acc. 2008. V. 120. № 1–3. P. 215.
Cances E., Mennucci B. // J. Chem. Phys. 1998. V. 109. № 1. P. 249.
Tomasi J., Mennucci B., Cances E. // J. Mol. Struct. (THEOCHEM). 1999. № 464. P. 211.
Chen Zh., Lin Y.M., Rooks M.J. et al. // Phys. E. 2007. V. 40. № 2. P. 228.
Михайлов Г.П. // ЖОрХ. 2018. Т. 54. Вып. 11. С. 1723.
Davis M.R., Dougherty D.A. // Phys. Chem. Chem. Phys. 2015. V. 17. P. 29262.
Dougherty D.A. // Science. 1996. V. 271. № 5246. P. 163.
Mahadevi A.S., Sastry G.N. // Chem. Rev. 2013. V. 113. № 3. P. 2100.
Seiji Tsuzuki, Masaru Yoshida, Tadafumi Uchimaru et al. // J. Phys. Chem. A. 2001. V. 105. № 4. P. 769.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика