

Химическая физика, 2020, T. 39, № 2, стр. 18-29
Исследование кинетических особенностей бимодального процесса гелеобразования в вязкотекучих жидкостях
И. В. Кумпаненко 1, *, Н. А. Иванова 1, Е. О. Панин 1, Е. Г. Раевская 1, Н. Ю. Ковалева 1, А. В. Рощин 1
1 Институт химической физики им. Н.Н. Семёнова Российской академии наук
Москва, Россия
* E-mail: ivkumpan@chph.ras.ru
Поступила в редакцию 20.03.2019
После доработки 20.03.2019
Принята к публикации 22.04.2019
Аннотация
Изучены кинетические особенности бимодального процесса гелеобразования в вязкотекучих жидкостях, в частности в нефтепродуктах, методом вискозиметрии. Гелеобразование и загущение нефтепродуктов облегчают ликвидацию последствий их разлива. Измерены экспериментальные зависимости от времени вязкости смесей бензина, керосина, зимнего и летнего дизельных топлив с тетралкоксиборатом лития как гелеобразующим агентом. Обнаружено, что эти зависимости имеют сигмоидальную бимодальную форму, называемую в научных публикациях формой “доза–эффект”. Предложена математическая формула для описания этих бимодальных зависимостей. В соответствии с этой формулой процесс гелеобразования проходит через две стадии: 1) реакция трет-бутилата лития Li–O–C(CH3)3 и триалкоксибората B(OC8H17)3 с образованием композиции тетраалкоксибората лития; 2) реакция тетраалкоксибората лития и углеводородов вышеупомянутых нефтепродуктов с образованием геля. Формула содержит ряд параметров подгонки: например, tinf 1 и tinf 2 (ч) – времена начала процесса гелеобразования и образования стабильного геля, соответственно; h1 и h2 (ч–1) – константы скорости соответственно для 1-й и 2-й стадий процесса гелеобразования и т.п. Определены зависимости этих параметров от концентрации гелеобразующих агентов. На основе этих зависимостей получены выводы относительно кинетических особенностей бимодального процесса гелеобразования в вязкотекучих жидкостях.
1. ВВЕДЕНИЕ
В последние десятилетия наблюдается повышенное внимание к исследованиям процессов гелеобразования и загущения жидкостей в связи с их использованием в различных приложениях, таких как тканевая инженерия [1–4], доставка лекарственных средств к участку их действия в организме [5–8] и их контролируемое удаление [9, 10], сенсорные технологии [11–13], очистка воды от загрязняющих веществ [14, 15], разработка солнечных [16, 17] и топливных [18, 19] элементов в электротехнике и пр. Одной из важнейших причин возросшего интереса к исследованию процесса загущения вязкотекучих жидкостей является возникающая на практике потребность перевести нефть или продукты ее перегонки из жидкого (текучего) в гелеобразное, резиноподобное или твердое состояния, что значительно облегчает процедуру механического и/или ручного сбора нефти и нефтепродуктов (ННП) в случае чрезвычайной ситуации их разлива на грунте или водной поверхности. Наиболее многообещающим для этой цели представляется использование так называемых супрамолекулярных гелей, получающихся из низкомолекулярных гелеобразующих агентов. Отметим, что часто в научной литературе для обозначения таких гелей используется термин “низкомолекулярные гели” (low molecular weight gels (LMWGs)) [20–24].
Ранее [25, 26], рассматривая и сравнивая различные существующие способы остановки разливов ННП и ликвидации их последствий, мы отмечали, что с точки зрения экономии времени и средств одним из наиболее подходящих является метод структурирования разлитой жидкости с помощью низкомолекулярных гелеобразователей (ГО), ее загущения, отверждения и дальнейшего сбора. Важнейшей особенностью процесса загущения разлитых на водной поверхности ННП, которая впервые была отмечена в статье [20], является так называемая фазовая селективность гелеобразования (ФСГ) нефти в смеси нефть/вода. Сущность явления ФСГ состоит в образовании геля в одной из двух присутствующих в смеси фаз, а именно в гидрофобной фазе. Хотя в работе [20] указывается на образование геля в нефти, в последующих статьях [21–24] было показано, что данное явление характерно для смесей воды с нефтепродуктами и большинством углеводородных растворителей.
Отметим, что в большинстве описанных в научной литературе работ в качестве экспериментального метода регистрации факта гелеобразования используется так называемый тест на переворачивание сосуда (ТПС, (stable-to-inversion of the container method)) [27–30]. При выполнении этого теста ампулу, содержащую жидкость, в которой исследуется образование геля, переворачивают вверх дном, после чего ведется наблюдение за возможным вытеканием жидкости из ампулы. Если в течение нескольких минут после переворачивания ампулы жидкость в ней остается неподвижной, считается, что в ходе исследуемого процесса образовался стабильный гель. Важной количественной характеристикой, измеряемой в ходе ТПС, является минимальная концентрация гелеобразователя (МКГ, minimum gelator concentration) [29, 30], т.е. минимальная концентрация низкомолекулярной присадки, способствующей образованию стабильного геля.
В нашей предыдущей работе [25] для исследования структурирования и гелеобразования в нефтепродуктах, смешанных с водой, МКГ определялась близким к ТПС методом поворота сосуда с жидкостью на 40° (ПС40) относительно исходного положения. Очевидно, что методы ТПС и ПС40 недостаточно информационно-содержательны и требуется привлечение более совершенных методов. Одним из таких методов, на наш взгляд, является метод вискозиметрии.
В последние годы появились статьи, в которых приведены экспериментально измеренные зависимости вязкости жидкости, в которой протекает процесс гелеобразования (загущения), от времени, например в ходе затвердевания сульфоалюминатных цементов [31], индуцированного в потоке гелеобразования в микроволоконной суспензии [32]; реологических изменений в фенолформальдегидной смоле с добавками пиролизного масла, используемой в качестве консерванта для древесины [33]; изменений вязкости гидрогелей, используемых в фармацевтике [34]. Особый интерес представляет исследование изменения вязкости эмульсии нефти в воде при использовании различных эмульгаторов [35] – явления, лежащего в основе одного из эффективных способов сбора разливов ННП на водной поверхности.
Во всех перечисленных процессах [31–35] загущение (гелеобразование) происходит по совершенно различным физико-химическим механизмам. Однако во всех случаях наблюдается схожий вид зависимости вязкости от времени. Далее мы остановимся на этом вопросе подробнее.
В настоящей статье описывается применение вискозиметрического метода для исследования ФСГ и последующего загущения нефтепродуктов под действием гелеобразующих агентов в смесях с водой.
2. ИССЛЕДОВАНИЕ ПРОЦЕССА ГЕЛЕОБРАЗОВАНИЯ МЕТОДОМ ВИСКОЗИМЕТРИИ
Процесс образования геля в жидкости можно наблюдать, измеряя зависимость ее вязкости от времени. Как правило, эта зависимость имеет вид сигмоидальной кривой и в простейшем случае описывается логистической функцией. Например, зависимость динамической вязкости η (мПа ∙ с) от времени t (ч) может быть представлена в виде
(1)
$\eta (t) = {{\eta }_{0}} + \frac{{{{\eta }_{m}} - {{\eta }_{0}}}}{{1 + \exp \left\{ {h({{t}_{{inf}}} - t)} \right\}}},$На рис. 1 приведены кривые, рассчитанные по формуле (1). Цифрами 1 показаны точки перегиба на кривых. Опубликовано множество статей, в которых экспериментально измеренные зависимости вязкости от времени имеют вид, показанный на рис. 1 [36–38].
Рис. 1.
Зависимости динамической вязкости жидкости от времени в процессе образования в ней геля, рассчитанные с помощью логистической формулы (1) при η0 = 7, ηm = 2000, h = 0.2: а – tinf = 40, б – tinf = 20.
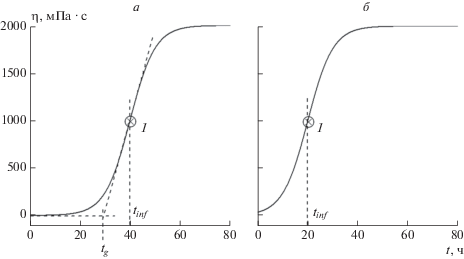
В условиях чрезвычайной ситуации разлива нефти и нефтепродуктов важнейшими характеристиками процесса гелеобразования являются время его начала, скорость его протекания и время образования стабильного геля. На рис. 1а показаны два способа определения времени начала процесса. Первый из них был предложен в статье [37], в которой время начала образования геля в жидкости, tg, определено как “время, необходимое для достижения точки перегиба на кривой зависимости вязкости от времени”. Это время может быть определено как абсцисса точки пересечения двух прямых: 1) касательной к кривой в точке перегиба и 2) асимптоты начального участка рассматриваемой кривой. На самом деле обозначенное как tg время не соответствует определению, приведенному выше в цитате из [37]. В настоящей работе мы предлагаем другой способ определения данного времени в строгом соответствии с приведенной выше цитатой, согласно которому время начала процесса гелеобразования соответствует абсциссе точки перегиба 1, обозначенной на рис. 1а и б как tinf.
Одно из преимуществ предлагаемого нами способа определения времени начала процесса гелеобразования продемонстрировано на рис. 1б, из которого видно, что не всегда можно установить положение горизонтальной асимптоты начального участка кривой. С другой стороны, абсциссы точек перегиба tinf кривой зависимости η = η(t) математически определяются как корни уравнения
что легко осуществить, если известны вид и параметры функции η = η(t).
Заметим, что зависимость вязкости от времени не всегда может быть описана простой логистической функцией вида (1): такой функцией описываются только сигмоидальные кривые, обладающие инверсионной симметрией точечной группы Ci с центром в точке перегиба 1 (рис. 1). Часто данные кривые демонстрируют отсутствие какой-бы то ни было симметрии, как это, например, показано на рис. 2. Кривая на рис. 2а имеет типичный вид, характерный для бимодальной зависимости, именуемой в научной литературе зависимостью “доза–эффект” или “доза–ответ” (dose-response) [39–41], содержащей две точки перегиба, каждая из которых расположена между ее отрезками с выпуклостями, направленными вниз (слева) и вверх (справа), и может быть описана функцией
(3)
$\begin{gathered} \eta (t) = {{\eta }_{0}} + ({{\eta }_{m}} - {{\eta }_{0}}) \times \\ \times \,\,\left[ {\frac{p}{{1 + \exp \left\{ {{{h}_{1}}({{t}_{{inf}}}_{1} - t)} \right\}}} - \frac{p}{{1 + \exp ({{h}_{1}}{{t}_{{inf}}}_{1})}}} \right. + \\ \left. { + \,\,\frac{{1 - p}}{{1 + \exp \left\{ {{{h}_{2}}({{t}_{{inf}}}_{2} - t)} \right\}}} - \frac{{1 - p}}{{1 + \exp ({{h}_{2}}{{t}_{{inf}}}_{2})}}} \right], \\ \end{gathered} $Рис. 2.
Зависимости динамической вязкости жидкости от времени в процессе образования в ней геля, рассчитанные с помощью формулы (3) при η0 = 7.22, ηm = 2020, tinf 1 = 24.752, h1 = 0.25, h2 = 0.125: а – p = 0.1, tinf 2 = 79.8354; б – p = 0.9, tinf 2 = 60.
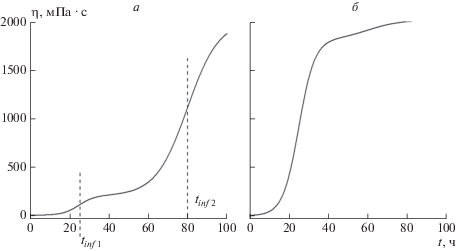
где tinf 1 и tinf 2 (ч) – значения t, соответствующие 1-й и 2-й точкам перегиба; h1 и h2 (ч–1) – параметры, характеризующие скорости реакций в процессах гелеобразования в разных временны́х интервалах; p – доля 1-й реакции в бимодальном процессе; η0 и ηm определены выше.
Эффект бимодальности в процессе гелеобразования можно объяснить изменением механизма реакции при переходе от первой стадии процесса, протекающего в низковязкостном растворе, ко второй стадии, когда процесс протекает в условиях среды с высокой вязкостью. Например, при добавлении к нефти полимерного ГО, имеющего сшивающие агенты на концах цепей [42, 43], на начальной стадии (t = 20–35 ч, рис. 2а) протекает процесс адсорбции ГО на нефти, который может замедлиться (t = 35–60 ч), а затем вновь ускориться (t = 60–100 ч). В иных условиях такая бимодальность может не наблюдаться (рис. 2б). К сожалению, причины бимодальности в процессе гелеобразования фундаментально до сих пор не исследовались.
Вискозиметрическое исследование бимодального процесса и экспериментальное определение для него зависимости динамической вязкости жидкости от времени дают возможность определить такие важнейшие характеристики процесса, как время его начала и время образования стабильного геля. По аналогии со сказанным выше относительно унимодального процесса, проиллюстрированного на рис. 1, мы предположили, что для бимодального процесса время его начала соответствует положению первой точки перегиба на временнóй оси – tinf 1, а время образования стабильного геля – положению на этой оси второй точки перегиба – tinf 2.
Для того чтобы апробировать предложенную формулу (3) для описания бимодальной зависимости вязкости от времени, мы аппроксимировали с ее помощью экспериментальную зависимость от времени вязкости смеси фенолформальдегидной смолы (ФФС) с пиролизным маслом и хлористой медью, используемой в качестве консерванта для древесины [34]. Изменение вязкости после смешивания указанных компонентов происходит в результате реакции перекрестного сшивания молекул ФФС, в которой пиролизное масло играет роль сшивающего агента, а ионы меди – катализатора (и одновременно – ингибитора роста грибковых образований в сохраняемой древесине).
На рис. 3 показаны упомянутая экспериментальная зависимость (кружки), полученная по данным, представленным на рис. 5 в статье [34] (образец R385Cu), и теоретическая зависимость (сплошная линия), построенная с помощью формулы (3). Параметры из этой формулы определялись методом подгонки с минимизацией среднеквадратичного отклонения теоретических значений от экспериментальных: η0 = 60.39, ηm = 4544.03, p = 0.106, tinf 1 = 247.90, tinf 2 = 422.64, h1 = 0.061, h2 = = 0.047. Как видно из рис. 3, качество подгонки хорошее: коэффициент детерминации R2 = 0.99991. Анализ величин параметров из формулы (3) показывает следующее.
Рис. 3.
Зависимость динамической вязкости фенолформальдегидной смолы с добавками пиролизного масла от времени.
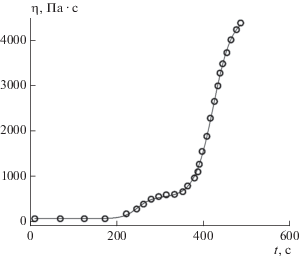
Исходная небольшая величина вязкости исследуемой смеси равна η0 = 60.39 Па ∙ с и сохраняется таковой в течение 200 с. Этого времени достаточно, чтобы нанести консервант на обрабатываемую поверхность древесины и дать ему возможность проникнуть внутрь на глубину ~100 мкм. В соответствии со сказанным выше через tinf 1 = 247.90 с начинается процесс образования геля, а через tinf 2 = 422.64 с образуется стабильный гель с динамической вязкостью ~2000 Па ∙ с. Конечная вязкость смеси наблюдается спустя >600 с и равна ηm = 4544.03 Па ∙ с.
Как отмечалось выше, бимодальность зависимости вязкости от времени можно объяснить изменением механизма реакции при переходе от первой стадии процесса, протекающего в низковязкостном растворе, ко второй стадии, когда процесс протекает в условиях среды с высокой вязкостью. При этом величина параметра p = 0.106 в уравнении (3) характеризует долю реакции первой стадии в бимодальном процессе, т.е. реакция первой стадии по времени занимает всего лишь около 10% от всего процесса загущения. Конечно, результаты такого анализа носят приблизительный характер, и в дальнейшем требуется провести более тщательное фундаментальное исследование данной проблемы. Параметры h1 = 0.061 с–1, h2 = 0.047 с–1, характеризующие скорости реакций в процессах перекрестного сшивания, показывают, что эти скорости отличаются друг от друга незначительно.
3. ВИСКОЗИМЕТРИЧЕСКОЕ ИССЛЕДОВАНИЕ ФАЗОВО-СЕЛЕКТИВНОГО ГЕЛЕОБРАЗОВАНИЯ НЕФТЕПРОДУКТОВ В СМЕСИ С ВОДОЙ
Описанный вискозиметрический метод был нами применен для исследования процесса гелеобразования в следующих продуктах перегонки нефти: бензин АИ-93 зимний и летний, керосин, дизельное топливо зимнее и летнее. В табл. 1 приведены значения вязкости и плотности ННП, как исследованных в настоящей работе, так и некоторых других для сравнения. Значения динамической вязкости ННП нам понадобятся в дальнейшем для корректировки расчета параметров из уравнения (3).
Таблица 1.
Вязкость и плотность ННП
| Нефтепродукт | Температура, °С | Плотность, г/см3 | Кинематическая вязкость ν, сСт | Динамическая вязкость η0, мПа ∙ с |
|---|---|---|---|---|
| Бензин АИ-93 зимний | 20 | 0.7552 | 0.5254 | 0.3968 |
| Бензин АИ-93 летний | 20 | 0.7550 | 0.6204 | 0.4684 |
| Керосин | 20 | – | – | 1.5 |
| Дизельное топливо зимнее | 20 | 0.8752 | 8.25 | 7.22 |
| Дизельное топливо летнее | 20 | 0.8787 | 8.94 | 7.858 |
| Масло веретенное АУ (ГОСТ 1642-75) | 20 | 0.912 | 49 | 44.69 |
| Масло моторное SAE 10 | 20 | – | – | 65 |
| Нефть легкая (тулвинское месторождение) | 20 | – | – | 42.8 |
| Нефть тяжелая (москудьинское месторождение) | 20 | – | – | 161.5 |
| Масло моторное SAE 40 | 20 | – | – | 319 |
| Мазут М100 | 18 | 1.015 | 2022 | 2052 |
| Битум | 20 | – | – | 2000–15000 |
Для проведения экспериментальных исследований готовились смеси нефтепродуктов с ГО, концентрация которого изменялась в интервале C ≈ 1–8 вес. %. Величины изученных концентраций C приведены в табл. 2. Там же даны значения параметров η0, ηm, p, tinf 1, tinf 2, h1, h2, рассчитанные в ходе аппроксимации экспериментальных данных теоретической зависимостью (3).
Таблица 2.
Концентрации смесей нефтепродуктов с ГО, а также значения параметров, определенных в ходе аппроксимации экспериментальных данных уравнением (3)
| Нефтепродукт | C, вес.% | η0 | ηm | p | tinf1 | tinf2 | h1 | h2 |
|---|---|---|---|---|---|---|---|---|
| Бензин АИ-93 летний | 0.9781 | 0.4675 | 237.24 | 0.094 | 8.1462 | 63.29 | 0.2547 | 0.1244 |
| 1.9808 | 0.4673 | 480.59 | 0.0963 | 6.58 | 61.782 | 0.2586 | 0.1236 | |
| 2.9729 | 0.4621 | 719.23 | 0.0978 | 5.326 | 60.535 | 0.2422 | 0.1254 | |
| 3.9963 | 0.4637 | 967.06 | 0.0977 | 4.465 | 59.597 | 0.2375 | 0.1248 | |
| 4.9988 | 0.4615 | 1210.3 | 0.0964 | 3.3 | 58.875 | 0.2477 | 0.1245 | |
| 6.0222 | 0.4592 | 1467.9 | 0.101 | 2.585 | 58.105 | 0.2453 | 0.1246 | |
| 6.9928 | 0.4571 | 1702.7 | 0.1029 | 2.351 | 57.469 | 0.2263 | 0.1264 | |
| 8.0267 | 0.4684 | 1957.3 | 0.0996 | 1.802 | 56.805 | 0.2395 | 0.1256 | |
| Керосин | 0.9558 | 1.501 | 239.48 | 0.0919 | 11.5927 | 66.679 | 0.274 | 0.12458 |
| 1.9692 | 1.508 | 493.67 | 0.0947 | 9.5561 | 64.879 | 0.2416 | 0.1248 | |
| 2.9825 | 1.503 | 744.27 | 0.0966 | 7.5979 | 63.009 | 0.2413 | 0.12342 | |
| 4.0168 | 1.517 | 1002.6 | 0.0993 | 6.1097 | 61.431 | 0.2479 | 0.12524 | |
| 5.0087 | 1.517 | 1253.4 | 0.0941 | 5.1697 | 60.216 | 0.256 | 0.12408 | |
| 5.9901 | 1.507 | 1499.3 | 0.0995 | 3.9164 | 59.211 | 0.2327 | 0.1261 | |
| 7.0453 | 1.512 | 1690.7 | 0.0958 | 3.0548 | 57.466 | 0.2375 | 0.1264 | |
| 8.0264 | 1.503 | 1936.8 | 0.0893 | 2.82 | 56.8 | 0.2598 | 0.12554 | |
| Дизельное топливо зимнее | 0.9723 | 7.21 | 246.15 | 0.0636 | 24.752 | 79.915 | 0.5791 | 0.1213 |
| 1.9548 | 7.19 | 493.59 | 0.0835 | 20.287 | 75.6 | 0.3444 | 0.1228 | |
| 2.9687 | 7.21 | 743.96 | 0.0869 | 16.762 | 71.978 | 0.3284 | 0.1239 | |
| 4.0035 | 7.25 | 1006.6 | 0.0897 | 13.943 | 68.946 | 0.2835 | 0.1234 | |
| 5.0171 | 7.2 | 1264.3 | 0.0957 | 11.201 | 66.632 | 0.2489 | 0.1236 | |
| 6.0093 | 7.23 | 1513.8 | 0.1035 | 9.4 | 64.71 | 0.2371 | 0.1249 | |
| 7.0331 | 7.2 | 1781.7 | 0.1014 | 7.833 | 63.047 | 0.2404 | 0.1241 | |
| 8.0252 | 7.22 | 2023.8 | 0.1004 | 6.188 | 61.188 | 0.2416 | 0.1265 | |
| Дизельное топливо летнее | 0.9712 | 7.843 | 251.86 | 0.0609 | 27.729 | 83.177 | 0.7843 | 0.1188 |
| 1.9433 | 7.844 | 497.38 | 0.0786 | 23.107 | 78.517 | 0.355 | 0.1234 | |
| 2.989 | 7.861 | 765.82 | 0.0916 | 19.269 | 74.648 | 0.2903 | 0.1236 | |
| 3.9711 | 7.84 | 1017.9 | 0.0944 | 16.136 | 71.518 | 0.2518 | 0.1231 | |
| 4.9847 | 7.849 | 1266.4 | 0.0939 | 13.473 | 68.699 | 0.2502 | 0.1258 | |
| 6.0194 | 7.851 | 1531.1 | 0.0923 | 10.809 | 66.11 | 0.2957 | 0.1254 | |
| 7.0115 | 7.845 | 1786.2 | 0.0941 | 9.0078 | 64.135 | 0.2437 | 0.126 | |
| 8.0248 | 7.858 | 2054.8 | 0.1 | 7.4413 | 62.836 | 0.2227 | 0.1266 |
В качестве ГО была выбрана композиция тетраалкоксибората лития, содержащая эквимолярные количества трет-бутилата лития Li–O–C(CH3)3 и триалкоксибората B(OC8H17)3, а также 0.1%-ный раствор фенолфталеина в этиловом спирте, являющегося индикатором процесса гелеобразования. Образцы смесей нефтепродуктов с ГО во время испытаний хранились в герметично закрытых емкостях без контакта с атмосферой, так как взаимодействие гелей с воздушной средой сопровождается разрушением их структуры и выделением жидкой фазы на поверхности геля. Стабильность структурированных продуктов во времени при их контакте с воздухом – один из факторов выбора рациональной концентрации ГО.
Динамические вязкости η(t) образцов смесей определялись в ходе экспериментов на протяжении 4 сут через каждые ~2−3 ч с помощью вискозиметра камертонного типа FVM Micro Motion, а также выборочно для контроля у образцов с низкой вязкостью с помощью калиброванного стеклянного вискозиметра в соответствии с процедурой, описанной в ГОСТ 33-2000 [44].
Зависимости динамической вязкости исследованных нефтепродуктов от времени при значениях концентраций ГО C(%), приведенных в табл. 2, представлены на рис. 4а–г. На этих рисунках точками показаны экспериментальные данные зависимостей η(t). Сплошные линии рассчитаны с помощью уравнения (3) с параметрами, указанными в табл. 2. Как видно, наблюдается высокое качество аппроксимации экспериментальных кривых теоретическими: коэффициенты детерминации во всех расчетах составляли R2 > 0.999.
Рис. 4.
Зависимости динамической вязкости нефтепродуктов от времени в процессе образования в них геля. Точки – экспериментальные данные. Сплошные линии рассчитаны с помощью уравнения (3) с параметрами, представленными в табл. 2; а – бензин АИ-93 летний, концентрации ГО (вес.%): 1 – 0.9781, 2 – 1.9808, 3 – 2.9729, 4 – 3.9963, 5 – 4.9988, 6 – 6.0222, 7 – 6.9928, 8 – 8.0267; б – керосин, концентрации ГО (вес.%): 1 – 0.9558, 2 – 1.9692, 3 – 2.9825, 4 – 4.0168, 5 – 5.0087, 6 – 5.9901, 7 – 7.0453, 8 – 8.0264; в – дизельное топливо зимнее, концентрации ГО (вес.%): 1 – 0.9723, 2 – 1.9548, 3 – 2.9687, 4 – 4.0035, 5 – 5.0171, 6 – 6.0093, 7 – 7.0331, 8 – 8.0252; г – дизельное топливо летнее, концентрации ГО (вес.%): 1 – 0.9712, 2 – 1.9433, 3 – 2.989, 4 – 3.9711, 5 – 4.9847, 6 – 6.0194, 7 – 7.0115, 8 – 8.0248.
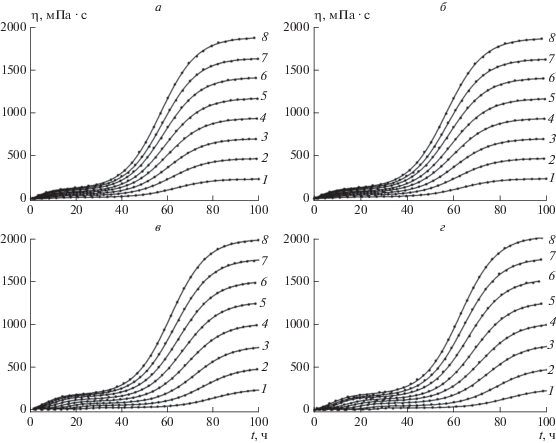
Отметим очень важное сходство между формами кривых для всех изученных нефтепродуктов: все они бимодальные. Выше мы уже отмечали, что бимодальность зависимости вязкости от времени, возможно, связана с изменением механизма реакции при переходе от первой стадии процесса, протекающего в низковязкостной жидкости, ко второй стадии, когда процесс протекает в условиях среды с высокой вязкостью.
В рассматриваемом случае гелеобразования в нефтепродуктах процесс протекает сначала через первую стадию образования супрамолекулярных гелей, получающихся из низкомолекулярных гелеобразующих агентов – трет-бутилата лития Li–O–C(CH3)3 и триалкоксибората B(OC8H17)3, а затем – через вторую стадию – реакция образования сольватов тетраалкоксиборатов лития с углеводородами нефтепродуктов.
Основываясь на результатах исследований процессов комплексообразования в системах, содержащих алкоксиды бора и лития [25, 26, 45–49], можно предположить, что структура углеводородных гелей образуется по приведенной на рис. 5 схеме. На этой схеме условно показана структура комплексного соединения тетраалкоксибората лития с представителем алканов – н-деканом, хотя, конечно, гели в углеводородах ННП образуются в результате реакций комплексообразования со всеми типами их углеводородных компонентов.
На приведенном на рис. 5 графике бимодальной зависимости вязкости от времени отмечено три временны́х интервала.
Рис. 5.
Схема процесса гелеобразования в смесях углеводородов с гелеобразующими агентами – трет-бутилатом лития Li–O–C(CH3)3 и триалкоксиборатом B(OC8H17)3. Линии-указки: 1 – Li–O–C(CH3)3, 2 – B(OC8H17)3, 3 – н-декан, 4 – тетраалкоксиборат лития, 5 – гель. Пояснения см. в тексте.
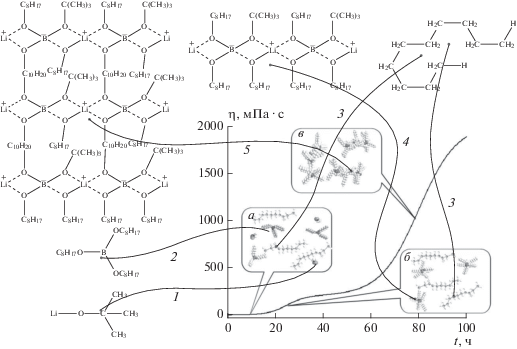
Интервал ~1–18 ч – начальное смешивание компонентов трет-бутилата лития Li–O–C(CH3)3 (1), триалкоксибората B(OC8H17)3 (2) и н-декана (3). Все три компонента изображены на вставке а в виде молекулярных моделей. Указатель на вставке а показывает на 10 ч. Для каждой из этих моделей с помощью пронумерованных указок 1–3 показаны их химические формулы. На этой стадии процесса между компонентами осуществляется только ван-дер-ваальсово взаимодействие. Вязкость такой смеси трех компонентов минимальная и мало отличается от вязкости н-декана.
Интервал ~21–25 ч, как мы уже отмечали выше, соответствует началу процесса гелеобразования (наличие точки перегиба tinf 1 на кривой зависимости вязкости от времени). На этой стадии процесса образуется комплексное соединение – тетраалкоксиборат лития, отмеченный на схеме рис. 5 указкой 4. В реакционной смеси в этом временнóм интервале находятся главным образом два компонента: тетраалкоксиборат лития и н-декан, молекулярные модели которых показаны на вставке б, где указатель показывает на точку перегиба tinf 1 ≈ 10 ч. Вязкость такой смеси относительно исходной незначительно возрастает за счет образования комплексного соединения 4, концентрация которого мала (1–8 вес.%).
Интервал 70–90 ч, в котором наблюдается точка перегиба tinf 2, согласно сказанному выше, соответствует времени образования стабильного геля. В реакционной смеси в этот момент начинает преобладать комплексное соединение тетраалкоксибората лития с н-деканом, молекулярные модели которого изображены на вставке в, а соответствующая химическая формула показана указкой 5. Указатель на вставке в показывает на точку перегиба tinf 2 ≈ 80 ч. Во временнóм интервале > 70 ч резко возрастает вязкость среды и, как уже сказано, образуется стабильный гель. Повторим, что хотя на схеме рис. 5 дана иллюстрация процесса гелеобразования в н-декане, в общем случае аналогичные процессы протекают с участием любых углеводородных компонентов ННП.
Предложенная нами математическая обработка бимодальной экспериментальной зависимости вязкости от времени путем ее аппроксимации уравнением (3) позволяет относительно просто и с минимальными ошибками определить значения некоторых параметров из (3), представленных в табл. 2, и тем самым провести предварительный анализ кинетических особенностей процесса. Проиллюстрируем это на нескольких примерах.
Зависимости времени начала процесса гелеобразования tinf 1 и времени образования стабильного геля tinf 2 от концентрации C гелеобразующего агента
Зависимости tinf 1 и tinf 2 от C, полученные по данным табл. 2, приведены на рис. 6 и 7. Из анализа хода кривых зависимостей tinf 1 и tinf 2 от C, представленных на этих рисунках, следует, что
Рис. 6.
Зависимости времени начала процесса гелеобразования tinf 1, от концентрации C ГО для бензина АИ-93 летнего (1), керосина (2), дизельного топлива зимнего (3), дизельного топлива летнего (4).
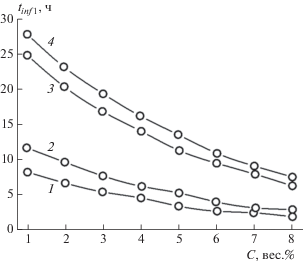
Рис. 7.
Зависимости времени образования стабильного геля tinf 2 от концентрации C ГО для бензина АИ-93 летнего (1), керосина (2), дизельного топлива зимнего (3), дизельного топлива летнего (4).
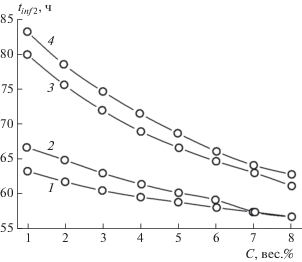
1) общий вид и порядок расположения вдоль оси ординат кривых зависимостей в интервалах времени начала процесса гелеобразования (0–30 ч) и времени образования стабильного геля (55−85 ч) подобны для всех изученных нефтепродуктов;
2) анализируя процесс гелеобразования на начальной стадии, можно предсказать время образования стабильного геля;
3) начало процесса гелеобразования и образование стабильного геля в низковязкостных продуктах нефтепереработки происходят при гораздо меньших концентрациях ГО, чем в нефтепродуктах с более высокой вязкостью. Например, время начала гелеобразования в смеси керосина с ГО с концентрацией, равной ~1 вес.%, равно tinf 1 = 66.679 ч и приблизительно совпадает с аналогичным временем tinf 1 = 66.11 ч для смеси дизельного топлива летнего с ГО с концентрацией ~6 вес.%, или, например, время образования стабильного геля из смеси бензина АИ-93 летнего с ГО с концентрацией, равной ~1 вес.%, tinf 2 = 63.29 ч, что близко к таковому tinf 2 = 63.047 ч для смеси дизельного топлива зимнего с ГО при его концентрации ~7 вес.%. Другими словами, для образования стабильного геля в бензине при схожих временны́х условиях требуется в 7 раз меньшее количество ГО, чем в дизельном топливе.
Таким образом, анализ временны́х параметров tinf 1 и tinf 2 из уравнения (3) позволяет выбрать оптимальные условия для проведения процесса гелеобразования.
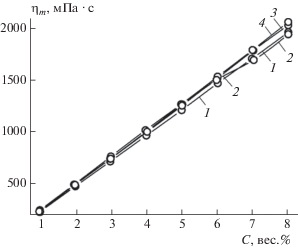
Зависимости максимальной вязкости ηm образующихся гелей от концентрации С гелеобразующего агента
Зависимости максимальной вязкости ηm гелей, образующихся в смесях с гелеобразующим агентом, от его концентрации C в ряду изученных по данным табл. 2 ННП приведены на рис. 8. Характерной особенностью зависимостей, изображенных на рис. 8, является их линейность. Исключение составляет зависимость, полученная для керосина (линия 2), которая при концентрации >6 вес.% отклоняется от линейной. Углы наклона зависимостей для всех изученных ННП близки между собой, что указывает на схожесть реологических свойств и, возможно, строения финишных гелей, образующихся из различных ННП. Зависимости, показанные на рис. 8, позволяют подобрать оптимальную (минимальную) концентрацию ГО для того, чтобы в результате загущения образовался гель, подходящий для сбора разливов ННП механическими средствами или вручную.
Вклад реакций первой и второй стадий в бимодальный процесс гелеобразования
Как уже отмечалось выше, величина параметра p в уравнении (3) характеризует долю реакции первой стадии в бимодальном процессе гелеобразования. Это – реакция образования комплексного соединения тетраалкоксибората лития (4) в результате взаимодействия трет-бутилата лития Li–O–C(CH3)3 (1) и триалкоксибората B(OC8H17)3 (2) в эквимолярном соотношении (нумерация дана по схеме рис. 5). Именно тетраалкоксиборат лития, “степень полимеризации” которого, а следовательно, и его молекулярная масса, могут достигать достаточно высоких значений, является главным гелеобразующим веществом на этой стадии.
По данным, представленным в табл. 2, построены графики зависимостей p от C для всех изученных нефтепродуктов, показанные на рис. 9. Отметим, что при концентрации C ≥ 3 вес.% значения p лежат в интервале 0.1 ≥ p ≥ 0.087 для всех нефтепродуктов. Это означает, что реакция первой стадии занимает по времени лишь 9–10% от всего процесса загущения. Однако при уменьшении концентрации ГО ниже 3 вес.% величина p уменьшается. Особенно это заметно для дизельных топлив, как зимнего, так и летнего, когда при C ≈ 1 вес.% p ≈ 0.06. Такое уменьшение вклада первой стадии бимодального процесса гелеобразования при C < 3 объясняется недостатком количества гелеобразующего агента в смеси.
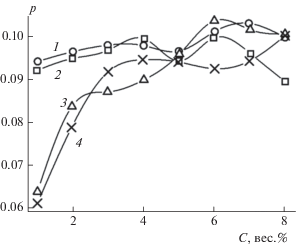
Зависимости параметров h1 и h2 из уравнения (3), характеризующих скорости реакций гелеобразования на 1-й и 2-й стадиях процесса
В пояснениях к уравнению (3) мы уже отмечали, что параметры h1 и h2 характеризуют скорости реакций комплексообразования в двухстадийном процессе. На рис. 10 и 11 представлены зависимости этих параметров от концентрации ГО, построенные по данным табл. 2. К сожалению, в настоящее время еще не проведены фундаментальные исследования кинетики и механизма реакций в системах, подобных рассматриваемым в настоящей статье. Поэтому в качестве первого приближения мы полагаем, что параметр h1 можно условно ассоциировать с константой скорости образования комплексного соединения – тетраалкоксибората лития (4) в результате реакции эквимолярных количеств трет-бутилата лития Li–O–C(CH3)3 (1) с триалкоксиборатом B(OC8H17)3 (2) – см. рис. 5. Аналогично параметр h2 мы ассоциируем с константой скорости образования геля (5) в результате реакции тетраалкоксибората лития с углеводородами ННП.
Рис. 10.
Зависимости параметра h1 из уравнения (3) от концентрации ГО. Обозначения те же, что и на рис. 9.
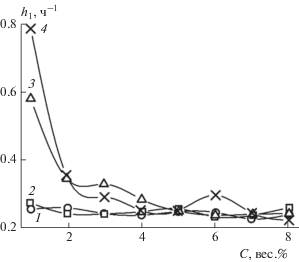
Рис. 11.
Зависимости параметра h2 из уравнения (3) от концентрации ГО. Обозначения те же, что и на рис. 9.
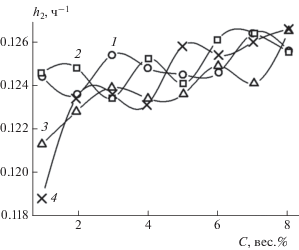
Из рис. 10 и 11 видно, что при концентрации ГО C ≥ 3 вес.% значения констант для всех ННП находятся в интервалах 0.25 ≤ h1 ≤ 0.35 ч–1 и 0.123 ≤ ≤ h2 ≤ 0.127 ч–1. То есть значения констант реакций в последовательных стадиях различаются примерно в два раза, хотя их разброс внутри интервалов невелик. Более того, константы скорости h1 для бензина и керосина имеют еще меньший разброс: 0.225 ≤ h1 ≤ 0.275 ч–1, причем во всем интервале изученных концентраций. Значительные отклонения величин h1 и h2 от их значений в указанных интервалах наблюдаются только для дизельных топлив – как зимнего, так и летнего. Например, при концентрации ГО ~1 вес.% величины констант равны h1 = 0.784 ч–1, h2 = 0.119 ч–1, что находится далеко за пределами указанных интервалов.
Наблюдаемое отклонение поведения величин p, h1, h2 при концентрациях ГО C < 3 вес.% от соответствующего поведения при других изученных концентрациях ГО 3 < C < 8 вес.% возможно удастся объяснить в дальнейшем при более глубоком теоретическом исследовании механизма бимодального процесса гелеобразования.
ЗАКЛЮЧЕНИЕ
В заключение отметим, что на практике структурирование нефти или продуктов ее переработки с помощью низкомолекулярных гелеобразователей, загущение и, если необходимо, отверждение, значительно облегчают процедуру их механического и/или ручного сбора в случае чрезвычайной ситуации разлива на грунте или водной поверхности. Для изучения процесса гелеобразования в ННП в большинстве опубликованных в настоящее время работ с целью регистрации экспериментального факта гелеобразования использовался так называемый “тест на переворачивание” сосуда, к сожалению малоинформативный.
В настоящей работе для исследования процесса гелеобразования в вязкотекучих жидкостях, в частности в нефти и нефтепродуктах, под воздействием гелеобразующих агентов нами был использован метод вискозиметрии. Определены экспериментальные зависимости вязкости бензина, керосина, дизельного топлива зимнего и летнего от времени. Полученные экспериментальные кривые зависимостей вязкости среды от времени имеют типичную бимодальную форму, именуемую в научной литературе формой “доза–эффект”, содержащей две точки перегиба, каждая из которых расположена между ее отрезками с выпуклостями, направленными вниз и вверх.
В случае гелеобразования в нефтепродуктах для описания наблюдаемой бимодальной кинетической кривой было предположено, что процесс протекает через две стадии: 1) реакция эквимолярных количеств гелеобразующих агентов – трет-бутилата лития Li–O–C(CH3)3 с триалкоксиборатом B(OC8H17)3 с образованием комплексного соединения – тетраалкоксибората лития; 2) реакция образования геля в результате взаимодействия тетраалкоксибората лития с углеводородами ННП.
Предложена формула для описания кинетики реакции в виде зависимости вязкости от времени – η(t). В эту формулу входят параметры подгонки η0, ηm, p, tinf 1, tinf 2, h1, h2, которые определялись в процессе аппроксимации экспериментальных данных предложенной формулой. Проведен анализ зависимости измеренных величин ηm, p, tinf 1, tinf 2, h1, h2 от концентрации ГО. Получены выводы об особенностях кинетики бимодального процесса гелеобразования, протекающего в нефтепродуктах под воздействием гелеобразующих агентов.
Работа выполнена в рамках Программы фундаментальных исследований ИХФ РАН № 46.15 (номер темы ФАНО: 0082-2014-0005; номер государственной регистрации ЦИТИС: АААА-А17-117091220076-4).
Список литературы
Lee K.Y., Mooney D.J. // Chem. Rev. 2001. V. 101. № 7. P. 1869.
Rudra J.S., Sun T., Bird K.C. et al. // ACS Nano. 2012. V. 6. P. 1557.
Gasiorowski J.Z., Collier J.H. // Biomacromolecules. 2011. V. 12. Issue 10. P. 3549.
Wu E.C., Zhang S., Hauser C.A.E. // Adv. Funct. Mater. 2012. V. 22. Issue 3. P. 456.
Li X., Wang Y., Yang C. et al. // Nanoscale. 2014. V. 6. P. 14488.
Skilling K.J., Citossi F., Bradshaw T.D. et al. // Soft Matter. 2014. V. 10. P. 237.
Rodrigues M., Calpena A.C., Amabilino D.B. et al. // J. Mater. Chem. B. 2014. V. 2. P. 5419.
Tiller J.C. // Angew. Chem. Intern. Ed. 2003. V. 42. P. 3072.
Xing B., Yu C.-W., Chow K.-H. et al. // J. Amer. Chem. Soc. 2002. V. 124. P. 14846.
Uhrich K.E. // Chem. Rev. 1999. V. 99. P. 3181.
Chen J., Wu W., McNeil A.J. // Chem. Comm. 2012. V. 48. P. 7310.
Yoshimura I., Miyahara Y., Kasagi N. et al. // J. Amer. Chem. Soc. 2004. V. 126. P. 12204.
Wadhavane P.D., Izquierdo M.A., Galindo F. et al. // Soft. Matter. 2012. V. 8. P. 4373.
Oishi K., Maehata Y. // Chemosphere. 2013. V. 91. P. 302.
Shirsath S.R., Patil A.P., Patil R. et al. // Ultrason. Sonochem. 2013. V. 20. P. 914.
Chen H.-Y., Lin L., Yu X.-Y. et al. // Electrochim. Acta. 2013. V. 92. P. 117.
Rong Y., Li X., Liu G. et al. // J. Power Sources. 2013. V. 235. P. 243.
Choudhury N.A., Ma J., Sahai Y. // Ibid. 2012. V. 210. P. 358.
An L., Zhao T.S. Zeng L. // Appl. Energy. 2013. V. 109. P. 67.
Bhattacharya S., Krishnan-Ghosh Y. // Chem. Commun. 2001. P. 185.
Basak S., Nanda J., Banerjee A. // J. Mater. Chem. 2012. V. 22. P. 11658.
Konda M., Maity I., Rasale D.B., Das A.K. // ChemPlusChem. 2014. V. 79. № 10. P. 1482.
Raju Ch.S.K., Pramanik B., Kar T. et al. // RSC Adv. 2016. V. 6. Issue 58. P. 53415.
Wang D., Niu J., Wang Z., Jin J. // Langmuir. 2015. V. 31 № 5. P. 1670.
Кумпаненко И.В., Рощин А.В., Усин В.В. и др. // Хим. физика. 2014. Т. 33. № 10. С. 66; https://doi.org/10.7868/S0207401X14100069
Кумпаненко И.В., Рощин А.В., Берлин А.А., Мясоедов Б.Ф. // Хим. безопасность. 2017. Т. 1. № 1. С. 30; https://doi.org/10.25514/CHS.2017.1.11430
Roy S., Dasgupta A., Das P.K. // Langmuir 2007. V. 23. P. 11769; https://doi.org/10.1021/la701558m
Mitra R.N., Das D., Roy S., Das P.K. // J. Phys. Chem. B. 2007. V. 111. P. 14107; https://doi.org/10.1021/jp076495x
Das D., Dasgupta A., Roy S. et al. // Chem. Eur. J. 2006. V. 12. P. 5068; https://doi.org/10.1002/chem.200501638
Menger F.M., Caran K.L. // J. Amer. Chem. Soc. 2000. V. 122. P. 11679; https://doi.org/10.1021/ja0016811
García-Maté M., Londono-Zuluaga D., De la Torre A.G. et al. // Cem. Concrete Comp. 2016. V. 72. P. 39; https://doi.org/10.1016/j.cemconcomp.2016.05.021
Perazzo A., Nunes J.K., Guido S., Stone H.A. // Proc. Natl. Acad. Sci. 2017. V. 114. № 41. P. E8557; https://doi.org/10.1073/pnas.1710927114
Mourant D., Riedl B., Rodrigue D. et al. // J. Appl. Polym. Sci. 2007. V. 106. P. 1087.
Jiao Y., Gyawali D., Stark J.M. et al. // Soft Matter. 2012. V. 8. P. 1499.
Chever F., Duboscq K., Receveur J., Audegong C., Guyomarch J. // Proc. Thirty-ninth AMOP Technical Seminar, Environment and Climate Change. Canada, Ottawa, ON, 2016. P. 916.
Wang G.-X., Wang Z.-H., Chen Z.-Q. // Chinese J. Chem. 1995. V. 13. № 2. P. 97.
Jia H., Ren Q., Li Y.-M., Ma X.P. // Petroleum. 2016. V. 2. P. 90.
Bai Y., Xiong C., Wei F. et al. // Energy Fuels. 2015. V. 29. № 2. P. 447.
Deene Y.De, Venning A., Hurley C., Healy B.J., Baldock C. // Phys. Med. Biol. 2002. V. 47. P. 2459; https://doi.org/10.1088/0031-9155/47/14/307
Wang C., Wang Y., Li Y. et al. // Nat. Commun. 2015. V. 6. № 1. P. 1; https://doi.org/10.1038/ncomms
Tiwari D.K., Arai Y., Yamanaka M. et al. // Nat. Methods. 2015. V. 12. P. 515.
Fingas M. A Review of Literature Related to Oil Spill Solidifiers 1990–2008 / Ed. I.C.C.o.O.P.R. (ICCOPR). Edmonton, Alberta, Canada: Spill Science, 2008.
Fingas M. Review of Solidifiers. An Update 2013; http://www.pwsrcac.org/wp-content/uploads/filebase/ programs/environmental_monitoring/non-dispersing_ response_technologies/Review%20of%20Solidifiers% 20An%
ГОСТ 33-2000 (ИСО 3104-94). Нефтепродукты. Прозрачные и непрозрачные жидкости. Определение кинематической вязкости и расчет динамической вязкости. М.: Стандартинформ, 2008.
Дмитриева З.Т., Хатьков Н.Д. // Инж.-физ. журн. 2007. Т. 80. № 4. С. 13.
Дмитриева З.Т. // ЖПХ. 2007. Т. 80. № 1. С. 111.
Дмитриева З.Т., Бондалетов В.Г. // Изв. Томск. политехн. ун-та. 2009. Т. 315. № 3. С. 69.
Дмитриева 3.Т. // Там же. 2011. Т. 319. № 3. С. 139.
Dmitrieva Z.T. // Mol. Cryst. Liq. Cryst. Inc. Nonlin. Opt. 1988. V. 161. P. 529.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика