

Химическая физика, 2020, T. 39, № 4, стр. 11-18
Ab initio расчет низших $^{1}\Sigma _{g}^{ + }$-состояний димера Na2
С. О. Адамсон 1, *, Д. Д. Харлампиди 2, Г. В. Голубков 1, 3, Ю. А. Дьяков 1, 4, Ш. Ш. Набиев 3, М. Г. Голубков 1
1 Федеральный исследовательский центр химической физики им. Н.Н. Семёнова
Российской академии наук
Москва, Россия
2 Московский педагогический государственный университет
Москва, Россия
3 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
4 Академия Синика
Тайбэй, Тайвань
* E-mail: sergey.o.adamson@gmail.com
Поступила в редакцию 16.04.2019
После доработки 26.06.2019
Принята к публикации 22.07.2019
Аннотация
Проведены расчеты основного и десяти низших возбужденных адиабатических электронных $^{1}\Sigma _{g}^{ + }$‑состояний димера Na2 с использованием метода псевдопотенциала. Применение базиса [7s6p5d4f] атомных орбиталей позволило расширить интервал межъядерных расстояний (1.7–50 Å). Показано, что теоретические значения молекулярных постоянных Te и De хорошо согласуются с экспериментальными данными. Проведены пробные расчеты матричных элементов неадиабатического связывания и показана возможность перехода к базису квазидиабатических состояний. Установлено, что для электронных состояний, коррелирующих с пределом 3s + 5p и вышележащими диссоциационными пределами, модифицированный радиус ле Роя смещает границу асимптотической области в сторону меньших межъядерных расстояний.
1. ВВЕДЕНИЕ
В настоящее время сформировалось новое научное направление — радиохимическая физика атмосферы, в задачу которого входит исследование особенностей распространения радиоволн на высотах 60–110 км от поверхности Земли [1–3]. В сферу этого направления попадает ряд научно-технических приложений [4], включая проблему ошибок позиционирования глобальных навигационных спутниковых систем [5]. Важнейшая роль в радиохимической физике атмосферы отводится процессам l-перемешивания, диссоциативной рекомбинации, ассоциативной ионизации и обмена, протекающим через образование промежуточных высоковозбужденных (ридберговских) состояний двухатомных молекул [6]. Именно эти процессы в основном определяют параметры неравновесной атмосферной плазмы. При расчете сечений указанных выше процессов возникает естественный вопрос о необходимости учета вкладов от ридберговских состояний, локализованных в области от 5Re до 15Re, где Re – равновесное межъядерное расстояние молекулы. Поэтому для разработки корректных теоретических подходов целесообразно рассмотреть хорошо исследованную систему, например димер Na2 [7].
Димер натрия (Na2) является одной из немногих двухатомных молекул, спектры поглощения и испускания которой многократно исследовались экспериментально и теоретически на протяжении более чем 100 лет [7–10]. Теоретические исследования молекулы Na2 начались во второй половине 1960-х годов с расчетов равновесного межъядерного расстояния и энергии диссоциации основного электронного состояния этой молекулы [11–13]. С ростом вычислительных возможностей были сделаны расчеты возбужденных электронных состояний, коррелирующих с диссоциационными пределами I–III [14–17], дипольных моментов переходов X→A и X→B и времен жизни возбужденных колебательно-вращательных состояний [18–21].
С кратким обзором экспериментальных и теоретических исследований, опубликованных ранее, можно ознакомиться в работе [8]. Из поздних работ следует указать на расчеты поверхности потенциальной энергии (ППЭ) возбужденных состояний [22–30], дальнодействующих потенциалов [31–36], а также дипольных моментов перехода и времен жизни возбужденных колебательно-вращательных состояний [10, 37, 38].
Причиной интереса к возбужденным электронным состояниям Na2 являются значительный разброс оптических свойств и нарушения монотонности колебательных и вращательных составляющих энергий возбужденных колебательно-вращательных состояний, относимых к одному и тому же наблюдаемому возбужденному электронному состоянию [10, 37, 38]. Данная особенность системы объясняется резонансной (нестационарной) природой возбужденных колебательно-вращательных состояний, возникающей вследствие их делокализации по близколежащим электронным состояниям. Упрощая физическую картину, нарушения монотонности свойств колебательно-вращательных состояний ассоциируют со сложным поведением адиабатических потенциальных кривых (наличие нескольких минимумов, потенциальные барьеры, псевдопересечения и т.д.), объединяющих фрагменты диабатических состояний ковалентного и ионного (M+ + M–) характера [37, 38].
В настоящее время для молекулы Na2 с высокой точностью рассчитаны 83 адиабатических электронных состояния в интервале ~5–40 а.е. (2.6–21.2 Å), включая (1–11)$^{1}\Sigma _{g}^{ + }$-состояния, коррелирующие с диссоциационными пределами, лежащими ниже предела Na+(1S) + Na–(1S) [28, 29, 37]. Несмотря на значительную величину интервала, его левая граница не позволяет отталкивательным ветвям адиабатических потенциальных кривых достичь областей энергии, превышающих соответствующие энергии диссоциации. Правая граница захватывает лишь псевдопересечение (5–6)$^{1}\Sigma _{g}^{ + }$-состояний. К сожалению, данного интервала межъядерных расстояний недостаточно для расчета характеристик возбужденных колебательно-вращательных состояний с энергиями, близкими или превышающими энергию пересечения адиабатических потенциальных кривых (5–6)$^{1}\Sigma _{g}^{ + }.$
Цель нашего исследования – построение адиабатических потенциальных кривых (1–11)$^{1}\Sigma _{g}^{ + }$-состояний в более широком, чем в работе [28], интервале межъядерных расстояний. Метод расчета изложен в разд. 2 статьи, в разд. 3 проведен подробный анализ полученных результатов. В Заключении указаны пути и основные трудности дальнейшего развития теории.
2. МЕТОД РАСЧЕТА
Неэмпирическое исследование возбужденных адиабатических электронных состояний молекулы Na2 проводилось ранее методами многоконфигурационного самосогласованного поля [16, 20], ограниченного конфигурационного взаимодействия [15, 17, 22, 23] и полного конфигурационного взаимодействия (ПКВ) [24, 28, 37, 38]. Во всех методиках расчета молекула Na2 рассматривалась как двухэлектронная система: атомные остовы [Na+] “замораживались” в полноэлектронных базисах атомных орбиталей (АО) [16, 20] или заменялись эффективными остовными потенциалами [15, 17, 22–24, 28, 37, 38].
Среди предложенных вариантов параметризации остовного потенциала наиболее удачным оказался неэмпирический псевдопотенциал Дьюрана–Бартела [39]. Его применение в комбинации с базисом (7s6p5d2f)/[6s5p4d2f] безузловых функций гауссова типа и поляризационным остовным потенциалом [40] позволило достичь лучшего согласия с экспериментальными результатами [28] по сравнению с расчетами [23, 24]. Позднее для правильного воспроизведения атомного состояния 2F(4f) набор диффузных функций f-типа был расширен по схеме (3f) /[2f ] [41, 42]. В такой форме набор валентных АО при сохранении параметризации остовного и поляризационного потенциалов применялся для расчетов возбужденных состояний молекул NaH [43], LiNa [44, 45], NaCs [46], NaAr [41] и иона LiNa+ [42, 47]. На основе базиса валентных АО [28] были построены еще два набора АО, использованных в расчетах электрических дипольных моментов гетероядерных димеров щелочных металлов [48] и при моделировании взаимодействия Na2 с кластерами аргона [30].
Для молекулы NaH были предприняты расчеты возбужденных 1.3Σ+-состояний с применением для атома Na комбинации неэмпирического псевдопотенциала [49, 50] с поляризационным потенциалом [51, 52] и базисом валентных АО [7s6p3d1f] [53]. Данный подход обеспечил воспроизведение энергий возбуждения 3s–5s-состояний атома натрия с отличием от экспериментальных значений на ~10 см–1, но был менее точен в описании основного состояния NaH по сравнению с псевдопотенциалом [41, 42].
Анализируя результаты расчетов адиабатических электронных состояний молекул Na2 и NaH, несложно видеть, что лучшее согласие с экспериментальными данными обеспечивается при использовании комбинации остовного псевдопотенциала и базиса АО [42]. Однако для молекулы Na2 при межъядерных расстояниях меньше 2.4 Å (~4.5 а.е.) минимальное собственное значение матрицы интегралов перекрывания в данном базисе АО оказывается меньше 10–6, что указывает на появление вычислительной линейной зависимости. При исключении из базиса АО наиболее диффузной функции p-типа с показателем экспоненты αp = 0.0023 минимальное собственное значение матрицы интегралов перекрывания возрастает почти в три раза, но снижается точность расчета атомных состояний 2P(3p–5p). Эта проблема может быть решена в результате оптимизации нескольких диффузных функций p-типа, но их показатели достаточно близки (~2.49–2.52), и попытки оптимизации с высокой степенью вероятности должны приводить к вычислительной линейной зависимости. В этой ситуации наиболее разумным представлялось провести полную оптимизацию набора базисных АО.
Расчет атома Na проведен в настоящей работе для псевдопотенциала [39], дополненного базисом [(7s6p5d4f)]/[7s6p5d4f] валентных АО (табл. 1). Остовно-валентная корреляция учитывалась включением в схему расчета остовного поляризационного потенциала с αs = 0.9947, ρs = 0.9605 а.е. [51, 52]. При построении набора АО сначала минимизировалась полная энергия атома Na в базисе четырех функций s-типа, а затем оптимизировались показатели экспонент остальных функций с целью лучшего воспроизведения возбужденных атомных 3p–4f-состояний. Максимальное отклонение от экспериментальных данных составляет около 11 см–1 для состояния 2D(4d) (табл. 2). Как и в предыдущих работах [24, 28, 37, 38], двухэлектронная волновая функция молекулы Na2 строилась в приближении ПКВ. Все расчеты проводились с использованием пакета программ MOLPRO [54].
Таблица 1.
Базис безузловых гауссовых функций для атома натрия
| Орбиталь | Показатели экспоненты |
|---|---|
| s | 2.662241, 0.853282, 0.273488, 0.087656, 0.028095, 0.009492, 0.002746 |
| p | 1.048871, 0.325736, 0.106338, 0.031416, 0.006500, 0.002500 |
| d | 0.314937, 0.065639, 0.022201, 0.009123, 0.003435 |
| f | 0.053883, 0.016108, 0.005857, 0.002130 |
Таблица 2.
Энергии возбуждения электронных состояний атома натрия (см–1)
| Состояние | Eexc | ΔEexc | ||||
|---|---|---|---|---|---|---|
| [28]* | [53] | [41] | [43, 47] | расчет** | ||
| 2S(3s) | 0 | 0 | 0 | 0 | 0 | 0 |
| 2P(3p) | 16 968 | +1 | +11 | +1 | +1 | +7 |
| 2S(4s) | 25 740 | –19 | +9 | –2 | –2 | –9 |
| 2D(3d) | 29 173 | +1 | –536 | 0 | 0 | +1 |
| 2P(4p) | 30 271 | +3 | +8 | –1 | –1 | –1 |
| 2S(5s) | 33 201 | –10 | +8 | –6 | –5 | –8 |
| 2D(4d) | 34 549 | –7 | – | –13 | –13 | –11 |
| 2F(4f) | 34 587 | –16 | – | – | –25 | –7 |
| 2P(5p) | 35 042 | –13 | – | –14 | – | –40 |
| 1S0(2p6) | 41 450 | +1 | 0 | 0 | 0 | –1 |
* Расчет методом псевдопотенциала (метод А). ** Расчет, выполненный в данной работе. Примечание: Eexc – экспериментальная энергия возбуждения [55], ΔEexc – разность экспериментальной и теоретической энергий возбуждения.
Для оценки параметров псевдопересечений адиабатических состояний была построена потенциальная кривая диабатического ионного состояния, коррелирующего с диссоциационным пределом Na++Na– [29]. Для параметризации этой кривой был использован потенциал ионизации натрия 41 449.45 см–1 [55], энергия сродства к электрону 4419.32 см–1 [56] и поляризуемости α(Na+) = 0.998033$a_{0}^{3}$ [57], α(Na–) = 1090.2$a_{0}^{3}$ [58], хорошо согласующиеся с теоретическими и экспериментальными оценками [59, 60].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Комбинирование остовного псевдопотенциала с поляризационным потенциалом и базисом [7s6p5d4f] АО натрия дало возможность построить адиабатические потенциальные кривые (1–11)$^{1}\Sigma _{g}^{ + }$-состояний в интервале межъядерных расстояний 1.7–50 Å. (рис. 1). Смещение левой границы интервала на 0.9 Å в область меньших межъядерных расстояний по отношению к данным работы [28] позволило довести отталкивательные ветви потенциальных кривых до энергий, превышающих энергию диссоциационного предела 3s + 5p. В области больших межъядерных расстояний ограничения в 50 Å оказалось достаточно, чтобы обнаружить псевдопересечение (10–11)$^{1}\Sigma _{g}^{ + }$-состояний (рис. 1). Показано, что найденные значения электронных термов (Te) и энергий диссоциации (De) (1–6)$^{1}\Sigma _{g}^{ + }$-состояний лучше согласуются с существующими экспериментальными результатами, чем данные из работы [28]. При этом расчетные значения равновесных межъядерных расстояний (Re) оказываются меньше экспериментальных на ~0.01 Å. Значительная ошибка в расчетных значениях Re является, скорее всего, следствием применения адиабатического приближения, не учитывающего неадиабатических и релятивистских эффектов. Для лежащих выше $^{1}\Sigma _{g}^{ + }$-состояний известны только экспериментальные значения Te = 34 587.58, 34 976.59 см–1 [61], удовлетворительно согласующиеся с термами (7–8)$^{1}\Sigma _{g}^{ + }$-состояний. Для состояний (9–11)$^{1}\Sigma _{g}^{ + }$ экспериментальные значения Te и De отсутствуют, а теоретические – расходятся на 600–2000 см–1 (табл. 3).
Рис. 1.
Адиабатические электронные (4–11)$^{1}\Sigma _{g}^{ + }$-состояния Na2 в области 1.7–20.5 Å (а) и 13–50 Å (б). Штриховые линии соответствуют основному состоянию иона ${\text{Na}}_{{\text{2}}}^{{\text{ + }}}$ (◻) и эмпирической ионной диабатической кривой (⚫), сплошные линии – $^{1}\Sigma _{g}^{ + }$-состояния Na2. Нумерация кривых $^{1}\Sigma _{g}^{ + }$-состояний соответствует номенклатурным обозначениям. За точку отсчета шкалы энергии принято положение минимума энергии основного электронного состояния (см–1).
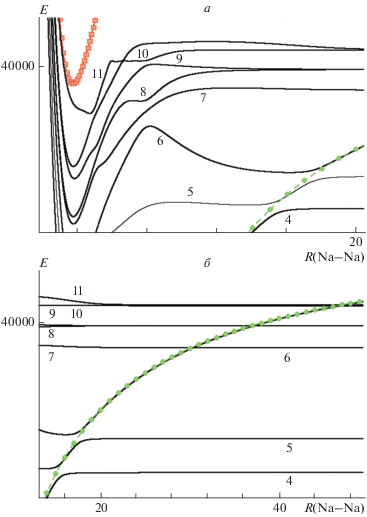
Таблица 3.
Молекулярные постоянные низших электронных $^{{\mathbf{1}}}\Sigma _{g}^{ + }$-состояний (равновесные межъядерные расстояния Re приведены в Å, электронные термы Te и энергии диссоциации De – в см–1)
| Состояние | Предел | Эксперимент | Теория | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Re | Te | De | ссылка | Re | Te | De | ссылка | ||
| 1$^{1}\Sigma _{g}^{ + }$ | 3s + 3s | 3.080 | 0 | 6022 | [62] | 3.085 | 0 | 5892 | [28]* |
| 3.069 | 0 | 5987 | наст. работа | ||||||
| 2$^{1}\Sigma _{g}^{ + }$ | 3s + 3p | 4.450 | 19 338 | 3658 | [63] | 4.445 | 19 349 | 3509 | [28] |
| 4.446 | 19 325 | 3623 | наст. работа | ||||||
| 3$^{1}\Sigma _{g}^{ + }$ | 3s + 4s | 3.563 | 25 692 | 6071 | [63] | 3.556 | 25 615 | 6035 | [28] |
| 3.569 | 25 692 | 6070 | [64, 65] | 3.551 | 25 662 | 6075 | наст. работа | ||
| 4$^{1}\Sigma _{g}^{ + }$ | 3s + 3d | 4.073 | [65] | 4.069 | 28 240 | 6825 | [28] | ||
| 4.074 | 28 327 | 6868** | [66] | 4.061 | 28 277 | 6883 | наст. работа | ||
| 5$^{1}\Sigma _{g}^{ + }$ | 3s + 4p | 3.638 | 31 770 | 4525 | [67] | 3.625 | 31 700 | 4464 | [28] |
| 3.626 | 31 737 | 4522 | наст. работа | ||||||
| 6$^{1}\Sigma _{g}^{ + }$ | 3s + 5s | 3.735 | 32 563 | 6660 | [36, 68] | 3.710 | 32 460 | 6643 | [28] |
| 3.698 | 32 506 | 6691 | наст. работа | ||||||
| 7$^{1}\Sigma _{g}^{ + }$ | 3p + 3p | 34 587 | [61] | 3.625 | 34 572 | 5253 | [28] | ||
| 3.614 | 34 631 | 5271 | наст. работа | ||||||
| 8$^{1}\Sigma _{g}^{ + }$ | 3p + 3p | 34 977 | [61] | 3.625 | 35 025 | 4801 | [28] | ||
| 3.604 | 34 876 | 5026 | наст. работа | ||||||
| 9$^{1}\Sigma _{g}^{ + }$ | 3s + 4d | 3.598 | 34 724 | 5539 | [28] | ||||
| 3.677 | 36 176 | 4372 | наст. работа | ||||||
| 10$^{1}\Sigma _{g}^{ + }$ | 3p + 4f | 3.577 | 35 900 | 4416 | [28] | ||||
| 3.612 | 36 590 | 3992 | наст. работа | ||||||
| 11$^{1}\Sigma _{g}^{ + }$ | 3p + 5p | 3.577 | 36 063 | 4693 | [28] | ||||
| 4.556 | 38 398 | 2671*** | наст. работа | ||||||
* Для состояний (1–8)$^{1}\Sigma _{g}^{ + }$ представлены результаты расчетов методом псевдопотенциала (метод А), для прочих состояний – методом модельного потенциала (метод Б) [28]. ** Энергия дисссоциационного предела 3s + 3d [65]. *** Энергия диссоциационного предела оценена для межъядерного расстояния в 95 Å.
По аналогии с работой [65], в дополнение к молекулярным постоянным были рассчитаны параметры псевдопересечений адиабатических потенциальных кривых. Для (1–6)$^{1}\Sigma _{g}^{ + }$-состояний точки пересечения (Rc) оказались на 0.01–0.02 Å сдвинуты в область бóльших расстояний по сравнению с точками пересечения потенциалов Ридберга–Кляйна–Риса (РКР). Для тех же электронных состояний энергии (Ec(Rc)) и ширины пересечений (ΔE(Rc)) отличаются от экспериментальных значений незначительно – на 30–60 см–1 (табл. 4).
Таблица 4.
Параметры псевдопересечений адиабатических электронных $^{{\mathbf{1}}}\Sigma _{g}^{ + }$-состояний
| n | Эксперимент* | Расчет** | $R_{{LR{\text{ - }}m}}^{{***}}$ | Rd | ||||
|---|---|---|---|---|---|---|---|---|
| Rc | ΔE(Rc) | Ec(Rc) | Rc | ΔE(Rc) | Ec(Rc) | |||
| 1 | 5.289 | 14 989.2 | 12 441.2 | 5.282 | 15 007.5 | 12 412.6 | 9.2 | – |
| 2 | 7.557 | 5001.8 | 25 459.1 | 7.529 | 5018.1 | 25 400.7 | 13.6 | 7.1 |
| 3 | 11.259 | 1383.3 | 32 038.4 | 11.240 | 1323.1 | 31 977.1 | 16.5 | 10.9 |
| 4 | 15.457 | 414.8 | 35 253.9 | 15.434 | 419.8 | 35 208.5 | 19.4 | 15.1 |
| 5 | 17.633 | 425.5 | 36 369.0 | 17.640 | 361.2 | 36 305.1 | 25.5 | 17.5 |
| 6 | – | – | – | 30.429 | 9.223 | 39 197.9 | 27.0 | 30.4 |
| 7 | – | – | – | 37.255 | 0.399 | 39 901.9 | 18.0 | 37.6 |
| 8 | – | – | – | 37.256 | 0.223 | 39 902.3 | 18.0 | 37.6 |
| 9 | – | – | – | 46.931 | 0.070 | 40 547.7 | 34.2 | 46.9 |
| 10 | – | – | – | 47.581 | 0.046 | 40 581.6 | 29.4 | 47.6 |
Примечание: n – номер низшего по энергии адиабатического состояния (n$^{1}\Sigma _{g}^{ + }$); Rc – межъядерное расстояние, отвечающее минимальной разности энергий состояний (Å); ΔE(Rc) – минимальная разность энергий состояний (см–1); Ec(Rc) – положение центра пересечения состояний (см–1), RLR-m – модифицированный радиус ле Роя (Å); Rd – оценка левой границы асимптотической области по энергии ионного диабатического состояния (Å). За точку отсчета шкалы энергии принято положение минимума энергии основного электронного состояния (см–1). * Найдены для РКР-потенциалов, литературные источники указаны в табл. 3. ** Расчет, выполненный в данной работе. *** Использованы значения квантового дефекта [71].
Таблица 5.
Матричные элементы радиального неадиабатического связывания (R(Na–Na) = 11.25 Å)
| δR | Метод А | Метод Б | ||||
|---|---|---|---|---|---|---|
| A21 | A32 | A43 | A21 | A32 | A43 | |
| 0.004 | –0.134020 | –0.050170 | –0.207886 | –0.134054 | –0.049966 | –0.207273 |
| 0.002 | –0.134124 | –0.049733 | –0.206240 | –0.134127 | –0.049736 | –0.206247 |
| 0.001 | –0.134126 | –0.049735 | –0.206244 | –0.134127 | –0.049736 | –0.206247 |
| 0.0005 | –0.134127 | –0.049736 | –0.206245 | –0.134127 | –0.049736 | –0.206247 |
| 0.00025 | –0.134127 | –0.049736 | –0.206246 | –0.134127 | –0.049736 | –0.206247 |
Примечание: метод А – оценка справа, метод Б – оценка центральной разностью; обозначения Anm соответствуют матричным элементам вида ${{A}_{{nm}}} = \left\langle {{{\psi }_{n}}\left| {{d \mathord{\left/ {\vphantom {d {dR}}} \right. \kern-0em} {dR}}} \right|{{\psi }_{m}}} \right\rangle ,$ где ψn – электронная волновая функция состояния ${{n}^{1}}\Sigma _{g}^{ + }$ и R – межъядерное расстояние; все величины представлены в атомных единицах.
Псевдопересечения в системе (6–11)$^{1}\Sigma _{g}^{ + }$-состояний локализованы в небольших интервалах межъядерных расстояний (~0.1–0.5 Å) и имеют малые ширины (~0.05–9 см–1). Эти особенности указывают на необходимость учета неадиабатического связывания при расчете свойств возбужденных колебательно-вращательных состояний. В частности, эта проблема может быть решена посредством перехода к базису квазидиабатических состояний с последующим решением системы связанных уравнений [69]. Учитывая, что области псевдопересечений пространственно локализованы, необходимо оценить устойчивость процедуры вычисления матричных элементов радиального неадиабатического связывания (МЭНС) (интегралов перекрывания адиабатических волновых функций, если использовать для построения квазидиабатических состояний вариационную теорию эффективных гамильтонианов [69]) к выбору величины приращения межъядерного расстояния. Проверочные расчеты МЭНС были выполнены с использованием двух вариантов конечно-разностной аппроксимации первой производной электронной волновой функции для R(Na–Na) = 11.25 Å. В первом варианте предполагалась замена производной выражением для ее оценки справа по двум точкам (метод А), а во втором – двухточечной центральной разностью (метод Б). Найдено, что приращения в 0.0001–0.000025 а.е. достаточно, чтобы вычислить значения МЭНС с точностью до пяти значащих цифр (табл. 5). Эти результаты позволяют сделать вывод о том, что (6–11)$^{1}\Sigma _{g}^{ + }$-состояния, после перехода к квазидиабатическому базису, могут быть включены в систему связанных уравнений, описывающих движение ядер молекулы.
На больших межъядерных расстояниях ab initio или РКР-потенциалы могут быть продолжены дальнодействующими (асимптотическими) потенциалами, вид которых зависит от природы диссоциирующей системы. На примере (3–5)$^{1}\Sigma _{g}^{ + }$-состояний Na2 было показано, что положение левой границы асимптотической области может быть оценено с помощью модифицированного радиуса ле Роя (Le Roy) – RLR-m [7, 70]. Сравнивая величины RLR-m и Rc, несложно увидеть, что, начиная с псевдопересечения (6–7)$^{1}\Sigma _{g}^{ + }$-состояний, выполняется условие Rc > RLR-m, т.е. радиус RLR-m смещает левую границу асимптотической области в область меньших межъядерных расстояний (табл. 4). Более разумную оценку можно получить, если границу асимптотической области рассматривать как точку кривой ионного диабатического состояния с энергией, равной энергии диссоциации электронного состояния (табл. 4).
ЗАКЛЮЧЕНИЕ
Проведенные расчеты с применением нового базиса валентных атомных орбиталей позволили уточнить характеристики адиабатических электронных (1–11)$^{1}\Sigma _{g}^{ + }$-состояний в широком диапазоне межъядерных расстояний, включая и области псевдопересечений. Найдено, что для 6$^{1}\Sigma _{g}^{ + }$-(предел 3s + 5p) и вышележащих электронных состояний оценка границы асимптотической области с помощью модифицированного радиуса ле Роя приводит к ошибочным результатам. Показано также, что матричные элементы неадиабатического связывания могут быть вычислены с высокой точностью, что в перспективе дает возможность перехода к базису квазидиабатических электронных состояний.
Полученные результаты могут быть использованы для расчета сечения процесса тушения при столкновении ридберговских атомов натрия. В дальнейшем аналогичные расчеты необходимо провести для систем, содержащих атомы азота и кислорода. При этом необходимо учитывать, что сечения и константы скорости основных процессов, определяющих параметры неравновесной атмосферной плазмы, существенно зависят от плотности нейтральной среды [72–75].
Работа выполнена в рамках государственного задания Министерства науки и высшего образования Российской Федерации (тема 0082-2019-0017, регистрационный номер АААА-А19-119010990034-5).
Список литературы
Голубков Г.В., Манжелий М.И., Карпов И.В. // Хим. физика. 2011. Т. 30. № 5. С. 55.
Голубков Г.В., Манжелий М.И., Лушников А.А. // Хим. физика. 2014. Т. 33. № 7. С. 101.
Голубков Г.В., Манжелий М.И., Берлин А.А. и др. // Хим. физика. 2018. Т. 37. № 7. С. 33.
Голубков Г.В., Манжелий М.И., Берлин А.А. и др. // Хим. физика. 2016. Т. 35. № 1. С. 5.
Голубков Г.В., Голубков М.Г., Манжелий М.И. // Хим. физика. 2014. Т. 33. № 2. С. 64.
Kuverova V.V., Adamson S.O., Berlin A.A. et al. // Adv. Space Res. 2019. V. 64. № 10. P. 1876.
Stwalley W.C. // J. Mol. Spectrosc. 2016. V. 330. P. 14.
Verma K.K., Bahns J.T., Rajaei-Rizi A.R., Stwalley W.C., Zemke W.T. // J. Chem. Phys. 1983. V. 78. № 6. P. 3599.
Jones K.M., Tiesinga E., Lett P.D., P.S. Julienne P.S. // Rev. Mod. Phys. 2006. V. 78. № 2. P. 483.
Jayasundara N., Anunciado R.B., Burgess E., Ashman S., Hüwel L. // J. Chem. Phys. 2019. V. 150. № 6. P. 064301.
Szasz L., McGinn G. // Ibid. 1966. V. 45. № 8. P. 2898.
Szasz L., McGinn G. // Ibid. 1968. V. 48. № 7. P. 2997.
Goodfriend L. // J. Mol. Spectrosc. 1969. V. 30. № 1–3. P. 111.
Roach A.C. // Ibid. 1972. V. 43. № 1. P. 27.
Bardsley J.N., Junker B.R., Norcross D.W. // Chem. Phys. Lett. 1976. V. 37. № 3. P. 502.
Konowalow D.D., Rosenkrantz M.E., Olson M.L. // J. Chem. Phys. 1980. V. 72. № 4. P. 2612.
Valance A., Nguyen Tuan Q. // Phys. Lett. A. 1981. V. 82. № 3. P. 116.
Tango W.J., Zare R.N. // J. Chem. Phys. 1970. V. 53. № 8. P. 3094.
Lam L.K., Gallagher A., Hessel M.M. // Ibid. 1977. V. 66. № 8. P. 3550.
Stevens W.J., Hessel M.M., Bertoncini P.J., Wahl A.C. // Ibid. № 4. P. 1477.
Konowalow D.D., Julienne P.S. // Ibid. 1980. V. 72. № 11. P. 5815.
Jeung G.H., Malrieu J.P., Daudey J.P. // Ibid. 1982. V. 77. № 7. P. 3571.
Jeung G. // J. Phys. B: At., Mol. Phys. 1983. V. 16. № 23. P. 4289.
Jeung G.H. // Phys. Rev. A. 1987. V. 35. № 1. P. 26.
Henriet A., Masnou-Seeuws F. // J. Phys. B: At., Mol. Phys. 1987. V. 20. № 4. P. 671.
Henriet A., Masnou-Seeuws F. // Ibid. 1990. V. 23. № 2. P. 219.
Henriet A., Masnou-Seeuws F., Dulieu O. // Z. Phys. D. 1991. V. 18. № 3. P. 287.
Magnier S., Millié Ph., Dulieu O., Masnou-Seeuws F. // J. Chem. Phys. 1993. V. 98. № 9. P. 7113.
Magnier S., Aubert-Frkon M., Bouty O. et al. // J. Phys. B: At., Mol. Opt. Phys. 1994. V. 27. № 9. P. 1723.
Gross M., Spiegelmann F. // J. Chem. Phys. 1998. V. 108. № 10. P. 4148.
Barrow R.F., Verges J., Effantin C., Hussein K., d’Incan J. // Chem. Phys. Lett. 1984. V. 104. № 2–3. P. 179.
Vigne-Maeder F. // Chem. Phys. 1984. V. 85. № 1. P. 139.
Bussery B., Aubert-Frecon M. // J. Mol. Spectr. 1986. V. 115. № 1. P. 169.
Marinescu M., Dalgarno A. // Phys. Rev. A. 1995. V. 52. № 1. P. 311.
Marinescu M. // Ibid. 1997. V. 56. № 6. P. 4764.
Laue T., Pellegrini P., Dulieu O. et al. // Eur. Phys. J. D. 2003. V. 26. № 2. P. 173.
Sanli A., Beser B., Edwardson J.R. et al. // J. Chem. Phys. 2015. V. 143. № 10. P. 104304.
Sanli A., Pan X., Magnier S. et al. // J. Chem. Phys. 2017. V. 147. № 20. P. 204301.
Maynau D., Daudey J.P. // Chem. Phys. Lett. 1981. V. 81. № 2. P. 273.
Foucrault M., Millie Ph., Daudey J.P. // J. Chem. Phys. 1992. V. 96. № 2. P. 1257.
El Hadj Rhouma M.B., Berriche H., Lakhdar Z.B., Spiegelman F. // Ibid. 2002. V. 116. № 5. P. 1839.
Berriche H. // J. Mol. Struct. (Theochem). 2003. V. 663. № 1–3. P. 101.
Khelifi N. // J. Russ. Laser Res. 2008. V. 29. № 3. P. 274.
Mabrouk N., Berriche H. // J. Phys. B: At., Mol. Opt. Phys. 2008. V. 41. № 15. P. 155101.
https://doi.org/10.1080/00268976.2019.1605098
Mabrouk N., Berriche H. // Russ. J. Phys. Chem. A. 2017. V. 91. № 8. P. 1475.
Khelifi N., Dardouri R., Al-Dossary O.M., Oujia B. // J. Russ. Laser Res. 2009. V. 30. № 2. P. 172.
Aymar M., Dulieu O. // J. Chem. Phys. 2005. V. 122. № 20. P. 204302
Fuentealba P., Preuss H., Stoll H., von Szentpaly L. // Chem. Phys. Lett. 1982. V .89. № 5. P. 418.
Leininger T., Nicklass A., Kuchle W. et al. // Ibid. 1996. V. 255. № 4–6. P. 274.
Müller W., Flesch J., Meyer W. // J. Chem. Phys. 1984. V. 80. № 7. P. 3297.
Müller W., Meyer W. // Ibid. P. 3311.
Lee H.S., Lee Y.S., Jeung G.H. // Chem. Phys. Lett. 2000. V. 325. № 1–3. P. 46.
Werner H.-J., Knowles P.J., Knizia G., Manby F.R., Schütz M. et al. // MOLPRO. Ver. 2010.1; a package of ab initio programs; http://www.molpro.net
Sansonetti J.E. // J. Phys. Chem. Ref. Data. 2008. V. 37. № 4. P. 1659.
Hotop H., Lineberger W.C. // Ibid. 1985. V. 14. № 3. P. 731.
Gray L.G., Sun X., MacAdam K.B. // Phys. Rev. A. 1988. V. 38. № 10. P. 4985.
Pyper N.C., Pike C.G., Edwards P.P. // J. Amer. Chem. Soc. 1993. V. 115. № 4. P. 1468.
Masili M., Carlos S., De Groote J.J. // Phys. Rev. A. 2004. V. 70. № 5. P. 054501.
Mitroy J., Safronova M.S., Clark C.W. // J. Phys. B: At., Mol. Opt. Phys. 2010. V. 43. № 20. P. 202001.
Taylor A.J., Jones K.M., Schawlow A.L. // J. Opt. Soc. Amer. 1983. V. 73. № 8. P. 994.
Zemke W.T., Stwalley W.C. // J. Chem. Phys. 1994. V. 100. № 4. P. 2661.
Effantin C., d’Incan J., Ross A.J., Barrow R.F., Verges J. // J. Phys. B: At., Mol. Phys. 1984. V. 17. № 8. P. 1515.
Tsai Chin-Chun, Whang T.-J., Bahns J.T., Stwalley W.C. // J. Chem. Phys. 1993. V. 99. № 11. P. 8480.
Tsai Chin-Chun, Bahns J.T., Wang H., Whang T.-J., Stwalley W.C. // Ibid. 1994. V. 101. № 1. P. 25.
Wang H., Whang T.-J., Lyyra A., Li L., Stwalley W.C. // Ibid. 1991. V. 94. № 7. P. 4756.
Tsai Chin-Chun, Bahns J.T., Stwalley W.C. // Ibid. 1994. V. 100. № 2. P. 768.
Tsai Chin-Chun, Bahns J.T., Stwalley W.C. // J. Mol. Spectr. 1994. V. 167. № 2. P. 429.
Jellali S., Habli H., Mejrissi L., Oujia B., Gadea F.X. // J. Phys. Chem. A. 2019. V. 123. № 2. P. 544.
Ji Bing, Tsai Chin-Chun, Stwalley W.C. // Chem. Phys. Lett. 1995. V. 236. № 3. P. 242.
Martin W.C. // J. Opt. Soc. Amer. 1980. V. 70. № 7. P. 784.
Голубков Г.В., Иванов Г.К., Балашов Е.М. и др. // ЖЭТФ. 1998. Т. 114. Вып. 1. С. 100.
Голубков Г.В., Иванов Г.К. // Хим. физика. 2003. Т. 22. № 10. С. 25.
Голубков Г.В., Иванов Г.К., Голубков М.Г. // Хим. физика. 2005. Т. 24. № 6. С. 3.
Голубков Г.В., Иванов Г.К., Голубков М.Г. // Хим. физика. 2005. Т. 24. № 9. С. 3.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика