

Химическая физика, 2020, T. 39, № 8, стр. 40-45
Инициирование воспламенения водород-кислородных смесей поверхностью кварцевого реактора, предварительно активированной пламенем
С. Н. Козлов *
Институт биохимической физики им. Н.М. Эмануэля Российской академии наук
Москва, Россия
* E-mail: kozlovse@yandex.ru
Поступила в редакцию 15.01.2020
После доработки 15.01.2020
Принята к публикации 20.02.2020
Аннотация
Исследовано влияние гетерогенной автокаталитической реакции на кварцевой поверхности реактора, возникающей после проведения воспламенения в среде H2/O2 вблизи третьего предела, на инициирование взрыва водород-кислородной смеси в холодном объеме примыкающих к реактору коммуникаций. При соблюдении определенных условий в большинстве опытов в холодном объеме возникает вторая вспышка. Предложен вероятный химический механизм происходящих процессов.
ВВЕДЕНИЕ
Изучение взаимодействия пламени и взрывчатых газовых смесей с поверхностями актуально в связи с проблемами в области промышленной бе-зопасности на атомных электростанциях, в технологических процессах, при хранении и использовании горючих газов. Сценарий аварийной ситуации с возможностью взрыва может реализоваться при накоплении водорода на атомных электростанциях, а также при неконтролируемой утечке любого горючего газа в атмосферу закрытых помещений. Взрывоопасная концентрация реагентов может быть достигнута и при неравномерном перемешивании горючего с кислородом воздуха. Случайное инициирование может привести к взрыву. Из-за произвольного перемешивания могут сформироваться зоны неполного выгорания с высокой концентрацией активных частиц в газе и следов обработанных пламенем предметов. При поступлении новой порции смеси с места утечки могут произойти повторные взрывы.
Экспериментальные установки и реакторы для лабораторных исследований пламен чаще всего изготавливают из чистого кварцевого стекла, поскольку этот материал считается наиболее нейтральным и инертным. Далее речь будет идти только о реакции окисления водорода в чистых кварцевых реакторах, которой посвящено большое число работ. Мы коснемся некоторых из них. Ранее было установлено [1–4], что при реакции смеси H2/O2 в кварцевом реакторе между вторым и третьим пределами воспламенения происходит медленная реакция окисления водорода, имеющая в ряде случаев автокаталитический характер. Проф. Чирков Н.М. [2] пытался объяснить автокатализ воздействием на реагирующую систему продукта этой реакции, т.е. паров воды, но эта идея не была поддержана. После экспериментов с малым временем контакта реагентов и малыми превращениями в продуктах реакции было обнаружено большое количество перекиси водорода (до 70%) [5]. На основании этого было предположено [5], что первичным продуктом реакции является перекись водорода, которая в дальнейших стадиях преобразуется в конечный продукт – воду. Перекись водорода также зарегистрирована в продуктах реакции водорода с кислородом вблизи первого [6] и третьего предела воспламенения [7]. В этих случаях важная роль в производстве перекиси была отдана газофазной реакции HO2 + Н2 → H2O2 + H. В более поздних работах [8–11] показано, что скорость медленной реакции окисления водорода зависит от очередности запуска реагентов в реактор. Установлено, что скорости газофазной реакции в этих случаях изменяются почти на порядок величины, что говорит о ее гетерогенной природе (на 90%).
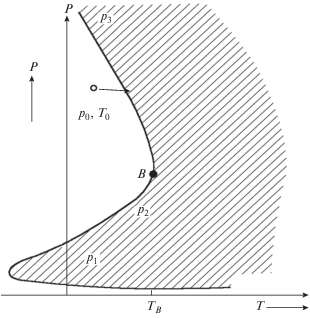
В работах [8–10] были проведены эксперименты с самовоспламенением не полностью перемешанных водород-кислородных смесей вблизи третьего предела (см. рис. 1) и с неполным выгоранием. В этих случаях происходит активация поверхности и возникает автокаталитическая гомогенно-гетерогенная реакция (с характерной S-образной кинетической кривой скорости реакции по изменению давления) образования воды из оставшихся исходных реагентов. При этом в опытах в качестве промежуточных продуктов реакции спектроскопическим методом зарегистрированы перекись водорода и малоактивный радикал HO2.
Рис. 1.
Схема трех пределов воспламенения гремучей смеси: p1, p2, p3 – первый, второй и третий пределы соответственно; p0 – начальное давление; T0 – начальная температура стенки и газа; B – точка перехода второго предела в третий; TB – температура точки перехода.
Цель работы – исследование возможности инициирования воспламенения H2/O2 смесей поверхностью, активированной предварительным самовоспламенением той же смеси, и выяснение способности автокаталитической реакции с участием поверхности реактора производить радикальные частицы в количестве, необходимом для воспламенения водород-кислородной смеси в холодном объеме реактора.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Эксперименты проводили в кварцевом реакторе, получившем название “реактор с рубашкой”, позволяющем изучать реакции на поверхности с малой каталитической активностью (рис. 2). Реактор обладает 30%-ным холодным объемом. Холодные зоны реактора состоят из подводящих стеклянных и кварцевых трубок до двух манометров (образцового и электронного с внутренними объемами ≈10 см3) общей длиной около 1 м и внутренним диаметром 5–6 мм и трубки внутренним диаметром 8 мм и длиной ≈20 см до первого вакуумного крана. Система ввода – капиллярная, объем <1 см3. Объем реактора без холодной части составляет ≈155 см3, с холодным объемом – ≈221 см3, холодный объем – ≈66 см3.
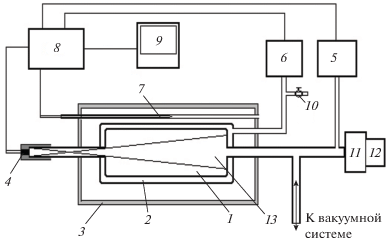
Рис. 2.
Схема эксперимента: 1 – кварцевый реактор; 2 – “рубашка” реактора; 3 – печь; 4 – фотодиод или водородная разрядная лампа ДДС-30; 5, 6 – датчики давления; 7 – термопара для регистрации температуры печи; 8 – многовходовой блок регистрации сигналов; 9 – компьютер; 10 – вакуумный вентиль; 11 – монохроматор BM-25; 12 – фотоэлектронный умножитель; 13 – зона реактора, освещаемая водородной разрядной лампой ДДС-30. Внутренний диаметр реактора – 36 мм, внешний диаметр – 40 мм, внешний диаметр “рубашки” – 50 мм, толщина стенок – 2 мм, длина реактора – 150 мм. Торцевые окна реактора изготовлены из кварцевого стекла марки КУ.
Соотношение компонентов в реакторе и в холодном объеме сразу после ввода практически одинаковое. Концентрация частиц реагентов, приведенная к атмосферному давлению в холодной части реактора, составляет 2.46 ⋅ 1019 частиц/см3 и в горячей части – 0.82 ⋅ 1019 частиц/см3 (при соотношении температур 300 и 900 K соответственно). Время перемешивания в холодном объеме в 5–6 раз больше, чем в горячем реакторе (200 и 35 с соответственно). Подробнее экспериментальная установка описана в нашей работе [8]. Давление в смеси реагентов при вводе регистрировали образцовым вакуумметром и, параллельно, дифференциальным манометром “Сапфир-22Д” с чувствительностью 30 Па. Давление в “рубашке” реактора регистрировали более чувствительным манометром (1 Па) той же марки. Регистрация изменения температуры с точностью ΔT ≈ 0.01 К по схеме газового термометра (что соответствует изменению давления в “рубашке реактора” Δp ≈ 1 Па) [8] позволяет следить за малыми количествами тепла, выделяемыми в реакции. Температуру печи измеряли хромель-алюмелевой термопарой, которая находилась на расстоянии 2–3 мм от внешней поверхности “рубашки” реактора. Свечение в реакторе регистрировали кремниевым фотодиодом типа C30839 (RCA Solid State Division, USA) с чувствительностью на длинах волн в диапазоне 200–1000 нм. Для зондирования зоны реакции в работе использовали водородную разрядную лампу ДДС-30, монохроматор BM-25 и фотоэлектронный умножитель ФЭУ-71.
Процедура проведения опытов
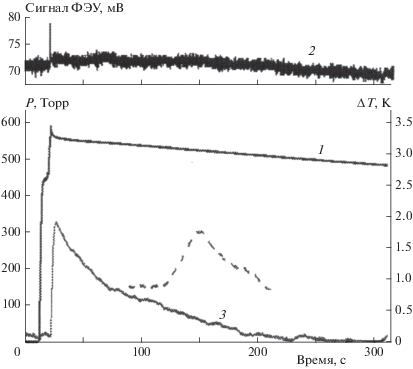
При последовательном вводе реагентов – водорода и кислорода в реактор (см. рис. 3) доводили давление смеси до появления первичной вспышки и перекрывали подачу кислорода. Видимое воспламенение длится доли секунды и вслед за ним, наряду с гетерогенной реакцией окисления водорода, начинается процесс активации поверхности реактора, контактировавшей с пламенем (отрезок BC, кривая 1). Под активацией поверхности мы понимаем образование дополнительных поверхностных активных центров, катализирующих реакцию окисления водорода. Практически одновременно на активированной поверхности развивается автокаталитическая реакция окисления водорода (отрезок CD, кривая 1). Максимум скорости реакции совпадает с максимумом скорости тепловыделения (температурная кривая 3, отрезок EF) и достигается за время ≈ 100 с от начала ввода второго реагента. В этот момент существует наибольшая вероятность для инициирования воспламенения смеси реагентов, диффундирующих из холодного объема реактора. Видимо, в данном опыте этого времени не хватило для достаточного перемешивания смеси в холодном объеме. На рис. 4 приведены результаты другого опыта, где время достижения максимума скорости автокаталитической реакции составило более 200 с. За это время из холодного объема к границе горячей части реактора в результате диффузии поступает уже перемешанная смесь водорода и кислорода. При контакте перемешанной смеси с продуктами автокаталитической гетерогенной реакции в большинстве случаев происходит воспламенение в холодной части реактора. Взрыв происходит в момент максимальной скорости тепловыделения (кривая 2) в гетерогенной автокаталитической реакции при давлении в реакторе гораздо ниже третьего предела воспламенения. Тепловыделение от повторной вспышки не регистрируется, поскольку стенки холодной части реактора не являются одновременно стенками газового термометра. Свет вспышки не всегда фиксируется фотодиодом из-за отдаленного положения источника излучения, т.е. пламени. Разберем случаи, когда и при каких условиях эксперимента не происходит инициирования взрыва в холодном объеме реактора (см. рис. 3 и 5).
Рис. 3.
Изменение давления (кривая 1: AB – ввод реагентов, CD – автокаталитическая реакция) и температуры (кривая 3 – EF) в реакторе в результате автокаталитической реакции на поверхности после проведенной вспышки для активации поверхности; сигнал фотодиода от свечения вспышки – кривая 2.
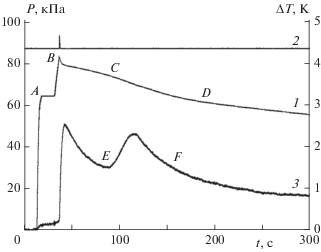
Рис. 4.
Инициирование воспламенения смеси H2/O2 в холодном объеме реактора продуктами автокаталитической гетерогенной реакции. После 205 с процесса происходит резкое падение давления в реакторе (кривая 1). Повторный взрыв происходит в холодном объеме реактора. Кривая 3 – фиксация фотодиодом сигнала от свечения вспышки.
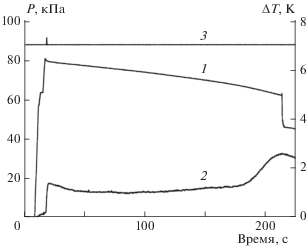
Рис. 5.
Воздействие излучения газоразрядной водородной лампы ДДС-30 со спектром излучения 190–250 нм на реагирующую смесь: кривая 1 – регистрация изменения давления; кривая 2 – регистрация излучения газоразрядной лампы; кривая 3 – изменение температуры (ΔT) стенки реактора в результате вспышки и гетерогенной реакции окисления водорода.
1. После ввода реагентов, даже при наличии активирующего первоначального воспламенения, не возникает автокаталитической реакции, что возможно при хорошем перемешивании смеси и более полном выгорании исходных реагентов в первоначальной вспышке.
2. Давление смеси после ввода реагентов в реактор не превышает предельного значения и не происходит активирующего поверхность реактора воспламенения; в этом случае автокаталитическая реакция возможна.
3. Автокаталитическая реакция после активирующей поверхность реактора вспышки слишком быстро достигает максимума скорости; за это время смесь в холодном объеме не успевает достаточно перемешаться для успешного инициирования взрыва.
4. Воздействие на объем реактора (во время проведения эксперимента) УФ-излучением разрядной водородной лампы (ДДС-30) (190–250 нм, где его интенсивно поглощают промежуточные продукты реакции HO2 и H2O2), приводит к замедлению и практическому исчезновению автокаталитической реакции (рис. 5, где штриховой линией показано вероятное положение кривой 3 без излучения (ср. с рис. 3)). Вследствие этого повторных взрывов не возникает. Вероятно, излучение в основном воздействует на радикал HO2, являющийся основным “сырьем” для производства перекиси водорода на поверхности, и на саму H2O2, разрушая их. При таком воздействии наибольшую вероятность имеют следующие реакции фото-распада:
Фотолиз радикала HO2 в газовой фазе уменьшает вероятность образования H2O2 на поверхности. Фотолиз перекиси с распадом ее на два радикала продолжает разрушать саму перекись до конечного продукта при помощи последующих вторичных реакций:
Суммарный квантовый выход этого процесса 1.7 ± ± 0.4 [12], что не противоречит нашей оценке, что на три разрушенные молекулы перекиси заново образуется только одна. Этими факторами обусловлен распад инициаторов вторичного взрыва в холодном объеме реактора.
5. Тот же эффект, но в меньшей степени, вызывает вымораживание промежуточных продуктов реакции HO2, H2O2 и воды в отростке вблизи реактора при температуре жидкого азота во время проведения эксперимента. Уменьшение концентрации активных промежуточных продуктов существенно замедляет автокаталитическую реакцию и не позволяет развиться повторному взрыву в холодном объеме реактора.
Как было указано выше, в работе [8] в продуктах автокаталитической реакции нами экспериментально зарегистрировано присутствие H2O2 и радикала HO2 в газовой фазе по поглощению света на длине волны 200 нм. Сечение поглощения на этой длине волны у радикала HO2 на порядок выше, чем у перекиси водорода, 4.2 ⋅ 10–18 и 4.7 ⋅ 10–19 см2 соответственно [13]. Мы не можем реально измерить их концентрации раздельно, поэтому относим поглощение света к общему присутствию этих двух компонентов в газовой фазе. Если отнести общее поглощение к одному продукту H2O2, то, по оценке, данной в работе [8], парциальное давление перекиси водорода по оси реактора в разных опытах составляло от 2 до 5 Торр. Если учесть, что характерное время распада перекиси при 850 К составляет 0.25 с, а время диффузии перекиси от стенки, где она, в основном, образуется составляет 4 с, то концентрация ее вблизи стенки реактора, как минимум, на порядок выше, чем на оси. Смешение смеси из холодного объема реактора с промежуточными продуктами автокаталитической реакции заметно облегчает воспламенение в пространстве холодного объема даже при гораздо меньшей температуре газа.
Вероятный химический механизм происходящих процессов
Активные центры (АЦ) гетерогенного катализа на поверхности кварца, необходимые для формирования автокаталитической гетерогенной реакции, надежно идентифицированы и изучены в работах Радцига В.А. с сотр. [14–16]. Одни АЦ существуют на поверхности кварца как разнообразные дефекты кристаллической решетки, построенной из SiO2, и их заселенность зависит от характера поверхности и температуры. Другие поверхностные АЦ образуются при взаимодействии молекулярных компонентов газовой фазы (в нашем случае – взрывчатой смеси) со структурными дефектами решетки и при ликвидации смеси имеют ограниченный срок жизни. Поверхностные образования $ \equiv $Si*, $ \equiv $SiO*, =Si*–OH, =Si*–H, =Si=O, =Si:, представляют собой наиболее устойчивые парамагнитные и диамагнитные активные центры на поверхности кварца при температурах наших опытов (750–950 K). Парамагнитные центры активно реагируют с молекулами газовой фазы, создавая на поверхности центры катализа. Диамагнитные центры, количество которых, как правило, на порядок больше, охотно присоединяют к себе радикальные частицы из газовой фазы [17], образуя при этом на поверхности парамагнитные. Если представить центры катализа в виде RH и ROH, где R – радикальные группы =Si*H, ≡Si*, =Si*(OH), то механизм образования перекиси водорода на стенке, предложенный в работе [8], можно описать следующими реакциями:
(1)
${\text{RH}} + {\text{H}}{{{\text{O}}}_{2}} \to {\text{R}} + {{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{2}},$(3)
${\text{ROH}} + {\text{H}}{{{\text{O}}}_{2}} \to {\text{RO}} + {{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{2}},$Гидроперекисный радикал HO2 в больших количествах образуется в объеме при давлениях смеси между вторым и третьим пределами [18] и на поверхности кварца, вероятно при термическом распаде пероксидных радикалов типа =Si(H)(O–O*) и оксирадикалов =Si(OH)(O*) при взаимодействии с молекулой водорода из газовой фазы [19]. Варианты образования на поверхности радикала HO2 описаны также в работе [20]. Радикал является “сырьем” для образования перекиси водорода при взаимодействии с поверхностью. Этот процесс, по-видимому, происходит по цепному механизму [8], и каждое звено цепи состоит из стадии образования перекиси водорода (реакции (1) и (3)) и стадии восстановления активного центра под действием молекулярного реагента из газовой фазы (реакции (2) и (4)). Роль перекиси водорода в развитии воспламенения сводится к инициированию разветвленно-цепных процессов в газе около поверхности продуктами своего распада. Ее термический распад при температурах вблизи третьего предела приводит к образованию двух активных радикалов OH: H2O2 + M → 2OH + M, которые, через экзотермическую реакцию
продолжают цепи и способствуют инициированию и развитию воспламенения. Также существует вероятность атомарному водороду, образовавшемуся в реакциях (2), (4), (5), присоединиться на поверхности к диамагнитному центру, образуя новые парамагнитные поверхностные радикалы
либо покинуть поверхность, продолжая реакционную цепь в газовой фазе и превратиться в малоактивный радикал HO2.
Возможные причины автокатализа в медленной реакции окисления водорода
На первый взгляд, три фактора могут влиять на ускорение реакции гомогенно-гетерогенного окисления водорода:
1. В результате реакции происходит разогрев пристеночного слоя реактора примерно на два градуса.
2. Увеличивается число активных парамагнитных центров на поверхности путем захвата атомарного водорода диамагнитными АЦ, которые тоже участвуют в этом процессе. Парамагнитные центры в результате реакции с молекулярным водородом превращаются в центры катализа для гетерогенной реакции производства H2O2.
3. Образование паров воды в начальной стадии реакции влияет на скорость тримолекулярной реакции образования радикала HO2. Эффективность паров воды в качестве третьего тела в этой реакции в семь раз больше, чем у водорода, и в 18 раз больше, чем у кислорода [21].
Увеличение концентрации HO2 вызывает рост гетерогенного образования H2O2, что ведет к увеличению скорости ее распада. Продукты распада ‒ радикалы OH в результате экзотермической реакции (5) дополнительно нагревают поверхность, добавляя атомы H в газовую фазу, и способствуют увеличению скорости гетерогенных реакций. Результаты настоящей работы также могут быть использованы в качестве объяснения возможных причин возникновения очагового воспламенения, когда дефлаграционное распространение пламени от очага способствует выгоранию водородно-воздушной смеси гораздо раньше, чем произойдет ее гомогенное самовоспламенение [22, 23].
ВЫВОДЫ
1. Экспериментально зарегистрировано инициирование воспламенения смеси H2/O2 в холодном объеме реактора продуктами гетерогенной автокаталитической реакции, возникающей на активированной пламенем поверхности.
2. Эксперименты показали, что для инициирования воспламенения в холодном объеме реактора самым важным является наличие предварительного активирующего воспламенения и гомогенно-гетерогенной автокаталитической реакции.
3. Предложен вероятный химический механизм происходящих процессов с участием цепных гомогенно-гетерогенных реакций.
4. Выявлены факторы, влияющие на увеличение скорости каталитической реакции при ее наличии.
Список литературы
Hinshelwood C.N., Thompson N.W. // Proc. R. Soc. London. Ser. A. 1928. V. 118. P. 170.
Чирков Н.М. // Аста physicochimica URSS. 1937. Т. 6. С. 915.
Воеводский В.В. // ЖФХ. 1946. Т. 20. № 11. С. 1285.
Полторак В.А., Воеводский В.В. // Там же. 1950. Т. 24. № 3. С. 299.
Pease R. // J. Amer. Chem. Soc. 1930. V. 52. P. 5106.
Кондратьев В.Н. Спектроскопическое изучение химических газовых реакций. М.; Л.: Изд-во АН СССР, 1944.
Gas Phase Combustion V. 17 / Eds. Compton R.G., Bamford C.H., Tippert C.F.H. 1977. P. 21.
Александров Е.Н., Маркевич Е.А., Кузнецов Н.М., Козлов С.Н., Частухин Д.С. // Физика горения и взрыва. 2013. Т. 49. № 1. С. 3.
Александров Е.Н., Кузнецов Н.М., Козлов С.Н. // Физика горения и взрыва. 2010. Т. 46. № 5. С. 48.
Александров Е.Н., Козлов С.Н., Кузнецов Н.М., Частухин Д.С. // ДАН. 2011. Т. 440. № 2. С. 178.
Александров Е.Н., Козлов С.Н., Кузнецов Н.М., Маркевич Е.А. // Хим. физика. 2013. Т. 32. № 11. С. 75.
Окабе Х. Фотохимия малых молекул. Пер. с англ. М.: Мир, 1981.
JPL Publication 02-25. 2003. C. 4–10, 4–11.
Радциг В.А. // Кинетика и катализ. 1979. Т. 20. № 2. С. 456.
Радциг В.А., Баскир Э.Г., Королев В.А. // Там же. 1995. Т. 36. № 1. С. 154.
Бобышев А.А., Радциг В.А. // Хим. физика. 1988. Т. 7. № 7. С. 950.
Радциг В.А., Козлов С.Н. // Кинетика и катализ. 2001. Т. 42. № 1. С. 62.
Льюис Б., Эльбе Г. Горение, пламя и взрывы в газах. Пер. с англ. М.: Мир, 1968. С. 46.
Радциг В.А., Берестецкая И.В., Козлов С.Н. // Хим. физика. 2002. Т. 21. № 3. С. 52.
Козлов С.Н., Маркевич Е.А., Александров Е.Н. // Хим. физика. 2017. Т. 36. № 4. С. 13.
Troe J. // Proc. Combust. Inst. 2000. V. 28. P. 1463.
Medvedev S.P., Agafonov G.L., Khomik S.V., Gelfand B.E. // Combust. and Flame. 2010. V. 157. № 7. P. 1436.
Медведев С.П., Гельфанд Б.Е., Хомик С.В., Агафонов Г.Л. // Инж.-физ. журн. 2010. Т. 83. № 6. С. 1104.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика