

Химическая физика, 2020, T. 39, № 8, стр. 58-63
Кинетика конверсии продуктов газификации полипропилена и автомобильных покрышек
А. М. Тереза 1, *, Г. Л. Агафонов 1, Э. К. Андержанов 1, С. П. Медведев 1, С. В. Хомик 1, С. К. Петров 2, М. В. Чернышов 2
1 Федеральный исследовательский центр химической физики им. Н.Н. Семёнова
Российской академии наук
Москва, Россия
2 Балтийский государственный технический университет “ВОЕНМЕХ”
им. Д.Ф. Устинова
Санкт-Петербург, Россия
* E-mail: tereza@chph.ras.ru
Поступила в редакцию 15.01.2020
После доработки 10.02.2020
Принята к публикации 20.02.2020
Аннотация
Проведено численное моделирование кинетики конверсии продуктов газификации полипропилена и изношенных автомобильных шин при атмосферном давлении в диапазоне температуры от 800 до 1300 К. За начальные условия были приняты экспериментальные данные, представленные в литературе. Установлено, что в результате реакций взаимодействия газофазных продуктов пиролиза полипропилена температура смеси понижается, в то время как для изношенных шин – повышается. Показано, что повышение начальной температуры смеси уменьшает конверсию его газофазных продуктов как для полипропилена, так и для изношенных шин. Установлено, что пути протекания конверсии алкенов и алканов С1–С4 сильно зависят от начальной температуры и их исходного состава.
ВВЕДЕНИЕ
Под пиролизом бытовых отходов обычно подразумевают термохимическое разложение органических веществ и материалов при высокой температуре в отсутствие кислорода или в присутствии малого количества воздуха, находящегося непосредственно в исходном материале [1]. Этот процесс всегда приводит к частичной газификации. В зависимости от географических и экономических особенностей местности в различных странах на сегодняшний день преобладают те или иные методики утилизации технологического мусора и отходов жизнедеятельности человека. В странах с развитыми технологиями в основном преобладает пиролиз продуктов нефтехимии, в то время как в других – это утилизация отходов растительного происхождения. В то же время развитие технологий и экономическая география также определяются экономикой стран и их потребностями в сырьевых источниках, которыми могут быть продукты пиролиза отходов. В работах [1, 2] дан анализ влияния экономической географии и технологического развития ведущих стран мира на методики пиролитического разложения отходов с целью получения максимально полезных для этих стран продуктов. Для стран с развитой нефтехимической отраслью промышленности наиболее актуальным является пиролиз полимерных изделий и каучуксодержащих материалов, в частности изношенных автомобильных шин.
Пиролиз отходов производства полиэтилена, полипропилена, резины и пластмассы, а также бытового мусора, содержащего эти ингредиенты, активно изучается уже несколько последних десятилетий [1–5]. Однако ввиду различных конструктивных особенностей пиролитических установок и многообразия бытовых отходов продукции нефтехимического синтеза на сегодняшний день затруднительно сформировать единый механизм утилизации [1, 3, 6]. Пиролиз пластмассовых отходов, отработанных шин и биополимеров осуществляется в плавильных емкостях, доменных печах, автоклавах, проточных реакторах, вращающихся и шнековых печах, камерах коксования и реакторах с псевдоожижженным слоем [3, 6]. В представленных в литературе обзорах показано, что для пластиковых отходов при температурах в диапазоне 750–1000 К выход газовой фазы составляет от нескольких процентов весовой концентрации у лигнина и полистирола до 90% у политетрафторэтилена [3]. При этом остаток в виде шлака может составлять от долей процента до 50%, как в случае с каучуком и лигнином. Экологически вредным продуктом пиролиза является стирол, содержание которого может достигать 65 мас.% [3]. Часто к этим продуктам относят и другие ароматические углеводороды, объединенные английской аббревиатурой BTX – по первым буквам латинских названий бензол–толуол–ксилол [1, 7, 8]. Кроме того, при пиролизе некоторых видов пластика образуются хлор- и цианидсодержащие компоненты [3, 9]. Эти продукты относятся к максимально токсичным соединениям и требуют полной утилизации. Одним из путей при их утилизации является повышение температуры и давления процессов. Однако такой подход связан с огромными энергетическими затратами и сложностью технологических процессов при высоких температурах (выше 1000 К), когда линейное тепловое расширение и механические нагрузки требуют создания конструкций из специальных дорогостоящих тугоплавких материалов с высокими прочностными характеристиками. Это обуславливает выбор величин давления в пределах от одной до нескольких атмосфер. В подобных условиях пиролиз полиэтилена и каучуксодержащих материалов наиболее приемлем при температуре до 1200 К, когда получается устойчивая газовая фаза, содержащая в основном простые компоненты [1].
Наиболее оптимальным для получения газообразной фазы продуктов пиролиза бытовых отходов является метод неподвижного слоя, который характеризуется тем, что в этом случае при температуре от 700 до 1000 К выход газовой фазы составляет до 65 мас.%. По данным работ [5, 6, 10–12] установлено, что основными компонентами газовой фазы продуктов пиролиза полиэтилена являются водород и предельные углеводороды группы алканов и алкенов: метан (СН4), этан (С2Н6), этилен (С2Н4), пропен (С3Н6), пропан (С3Н8), 1-бутен (С4Н8), н-бутан (С4Н10), 1.3-бутадиен (С4Н6) и изобутилен (С4Н8). В случае неподвижного слоя на стенках пиролитической установки с ростом температуры начинает образовываться и ощутимая доля ароматических углеводородов (ВТХ) вместе со стиролом и этилбензолом [13]. В работах [14, 15] появление этой группы углеводородов объясняется каталитическими реакциями. С другой стороны, каталитический пиролиз требует применения цеолитов H-beta [16], что усложняет и делает экономически неоправданным увеличение доли газовой фазы за счет роста экологически вредной ароматической группы ВТХ. В случае каучуксодержащих материалов, в частности изношенных автомобильных шин, доля группы ВТХ в продуктах газовой фазы активно увеличивается [8]. В работе [17] показано, что с ростом температуры алкены начинают переходить в ароматические соединения, тем самым увеличивая выход ВТХ. Если же в исходном материале присутствует воздух, то образуются в достаточном количестве СО и СО2, которые понижают выход более тяжелых углеводородов [11]. В этом случае посредством фильтрационного горения можно понижать выход не только пиролизных смол, но и серосодержащих химических соединений, образующихся при газификации автомобильных покрышек [18]. Тяжелые углеводороды в основном представлены следующими алканами и алкенами: деценом-1 (С10Н20), деканом (С10Н22), ундеценом-1 (С11Н20), ундеканом (С11Н22), додеканом (С12Н26), тридеценом-1 (С13Н26) и т.д. [19, 20]. С учетом того, что при температурах ниже 600 К эти углеводороды быстро конденсируются, рассмотрение их участия в газовой фазе неактуально. Таким образом, газовая фаза будет определяться в основном водородом, алканами, алкенами от С1 до С4 и ароматической группой ВТХ. В работе [21] было показано, что для отделения газовой фазы углеводородов С1–С4 необходимо сразу проводить ее отбор, чтобы не началась обратная полимеризация. Учитывая, что выход газообразных продуктов пиролиза сильно зависит от условий, в которых он проводится, и исходного состава отходов [1, 3, 6], возникает проблема кинетической конверсии этих продуктов во времени. Данный процесс важен с точки зрения дальнейшего использования этих продуктов, которое может пойти по двум путям. Первый путь – это сепарация продуктов пиролиза с целью получения ценных с точки зрения экономики веществ, используемых в промышленности. Второй – дальнейшая утилизация, которая связана с повышением температуры и давления. Одним из перспективных направлений в этом случае является использование детонационного режима для утилизации продуктов пиролиза. В этом случае при высоких температуре и давлении происходит быстрая фрагментация больших и средних молекул до малоатомных молекул и радикалов [10], кинетика взаимодействия которых намного лучше описывается с помощью детальных кинетических механизмов (ДКМ), чем кинетика взаимодействия молекул и радикалов С3 и выше. Использование ДКМ для углеводородов С1 и С2 позволяет разрабатывать метод абсорбирования фрагментарных продуктов детонации в малотоксичные соединения. Однако для выяснения детонационной способности газообразных продуктов пиролиза бытовых и технологических отходов необходимо знать их устойчивый состав смеси, которую предполагается напускать в детонационную камеру.
В данной работе проведено численное моделирование кинетики конверсии прямых продуктов пиролиза полипропилена и автомобильных шин с целью получения дополнительной информации об устойчивом составе смеси, которую целесообразно утилизировать в детонационном режиме.
ЧИСЛЕННОЕ МОДЕЛИРОВАНИЕ
Для моделирования пиролиза углеводородов С1–С4 в настоящее время в литературе представлено достаточно много ДКМ [22–29]. Основной сложностью при этом является неопределенность химического состава исходного материала, подвергающегося термическому разложению. Однако, поскольку основными продуктами, определяющими газовую фазу, являются алкены и алканы, то целесообразно рассматривать ДКМ, формально содержащие блоки реакций с участием углеводородов С1–С4. Такой подход вполне обоснован и широко применяется при численном моделировании различных физико-химических процессов, включающих в себя пиролиз твердых и жидких материалов. Так, например, при численном моделировании быстрого пиролиза полиэтилена и полибутадиена [30, 31] рассматривались сразу прямые продукты пиролиза, а именно, С2Н4 (этилен) и С4Н6 (бутин).
Все расчеты в представленной работе проводились с помощью программного модуля CHEMKIN III [32] при условии V = const. Расчетный интервал времени был определен равным 1 ч. Данные по исходному составу газообразных продуктов пиролиза полипропилена были взяты из работы [10] и соответствовали температуре ≈950 К в газогенераторе и ≈850 К на выходе в зоне отбора проб. Для расчетов сначала массовые доли были переведены в мольные концентрации. Затем были отброшены за малостью вклада компоненты изо-C4H8, 2-Me-Buten-1 и остаток тяжелых фракций. Их атомные веса были распределены пропорционально между следующими составляющими рабочей смеси (смесь 1):
Для численного моделирования кинетики конверсии продуктов пиролиза автомобильных шин в газовой фазе были использованы экспериментальные данные из работы [6] для Т ≈ 900 К, полученные в реакторе с кипящим слоем. Проведя процедуру, аналогичную описанной выше полипропилена, определим состав смеси 2:
Сравнение смесей 1 и 2 показывает приблизительно равные начальные значения содержания метана и пропана. В то же время в смеси 2 присутствует намного больше молекулярного водорода и в разы меньше С3Н6 и С2Н6. Следует отметить, что из составляющих смеси 1 и 2 исходных компонент все углеводороды, кроме метана и молекулярного водорода, в стехиометрических смесях с кислородом и воздухом имеют приблизительно одинаковую задержку воспламенения с точностью до фактора неопределенности 3 при температурах выше 1200 К [33]. Поскольку задержка воспламенения наряду с энергией активации смеси является важным параметром, определяющим переход дефлаграционного горения газовой смеси в режим детонации [34], то можно предположить, что соотношение газообразных компонентов смесей при дальнейшей их утилизации будет влиять и на пределы детонации [35].
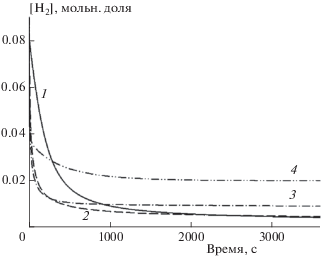
В качестве параметров процесса химического взаимодействия продуктов пиролиза были выбраны температура и концентрация молекулярного водорода. Температура, будучи глобальным параметром, характеризует процесс термодинамического и химического равновесия в целом. Молекулярный водород, являясь наиболее активным компонентом реагирующей смеси, отображает развитие кинетики химических реакций. Численное моделирование в рамках ДКМ [22] показало, что при Т < 800 К ни температура, ни концентрация водорода практически не изменяются в течение часа. С повышением начальной температуры смеси 1 в реакторе начинаются химические процессы, вызывающие изменение температуры. На рис. 1 представлена временнáя зависимость изменения концентрации молекулярного водорода с ростом температуры для смеси 1 при давлении 0.1 МПа. Видно, что концентрация водорода падает и выходит на квазиравновесное значение. Значение квазиравновесной концентрации при T0 < 1100 K приблизительно в 10 раз меньше начальной, но с увеличением T0 она начинает расти, что объясняется увеличением роли реакций распада углеводородных молекул.
Рис. 1.
Временнáя зависимость изменения концентрации молекулярного водорода с ростом температуры для смеси 1 при Р = 0.1 МПа. Линии соответствуют расчетам по ДКМ из работы [22] при следующих значениях температуры T0 (К): 1 – 900, 2 – 1000, 3 – 1100, 4 – 1300.
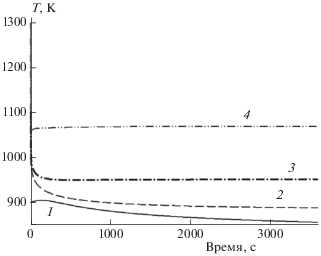
На рис. 2 представлена зависимость изменения температуры со временем для смеси 1. Видно, что вначале температура повышается, а затем наблюдаются ее снижение от начального значения и выход на стационарный уровень. Если при Т0 = 900 К это снижение малозаметно, то в дальнейшем падение температуры составляет уже 20% от начального значения. В то же время, чем выше начальная температура, тем быстрее устанавливается температурное квазиравновесное состояние. Падение температуры объясняется реакциями распада, которые всегда являются эндотермичными.
Рис. 2.
Изменение температуры со временем для смеси 1 при Р = 0.1 МПа и различных начальных температурах Т0 (К): 1 – 900, 2 – 1000, 3 – 1100, 4 – 1300. Линии соответствуют расчетам по ДКМ [22].
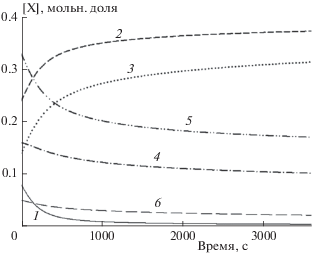
Из рис. 1 и 2 можно сделать вывод, что, хотя с повышением температуры квазистационарный уровень энергетически выгодного молекулярного водорода увеличивается и появляются более мелкие фрагменты распада исходных молекул, тем не менее общие затраты энергии на пиролиз могут оказаться неэффективными. Таким образом, пиролиз целесообразно проводить в диапазоне температуры от 800 до 1300 К. В ходе расчетов не было обнаружено влияния давления вплоть до его значения, равного 1 МПа, на температурные зависимости. Следовательно, диапазон давления от одной до нескольких атмосфер вполне приемлем, что значительно упрощает создание конструкции для исследования пиролиза полипропилена. На рис. 3 представлена временнáя зависимость изменения концентраций исходных компонентов смеси при Т0 = 900 К. Видно, что концентрации метана и этилена плавно приближаются к максимальному значению, в то время как концентрации других компонент падают до стационарного уровня. В целом рост метана и этилена незначителен, а это означает, что отбор пробы продуктов пиролиза полипропилена в работе [10] был произведен в условиях, близких к квазистационарному равновесию, соответствующему указанной в данной работе температуре.
Рис. 3.
Временны́е зависимости изменения концентрации исходных продуктов пиролиза полипропилена при Т0 = 900 К и Р = 0.1 МПа для смеси 1. Линии соответствуют расчетам по ДКМ [22]; 1 – [Н2], 2 – [СН4], 3 – [С2Н4], 4 – [С2Н6], 5 – [С3Н6], 6 – [С3Н8].
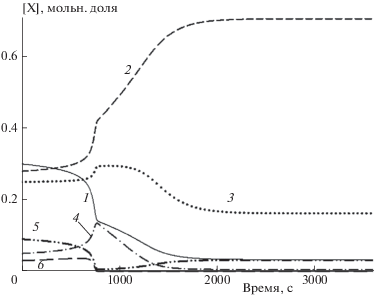
Совершенно иная зависимость температуры от времени наблюдается для кинетики конверсии первичных продуктов пиролиза автомобильных шин (рис. 4). Видно, что при Т0 < 1000 К температура растет с ускорением, затем резко меняет темп роста и уже в более плавном режиме выходит на стационарное значение, значительно превышающее начальное. В случае Т0 > 1000 К температура резко падает, а затем начинает расти и быстро выходит на квазиравновесное значение, которое несколько больше начальной температуры. Во всем диапазоне рассчитанных температур, от 800 до 1200 К, температура конверсии смеси продуктов пиролиза автомобильных покрышек в конечной фазе превышает начальную. При этом с ростом начальной температуры величина этого превышения уменьшается. Почему же изменение температуры имеет такой характер в газовой фазе продуктов пиролиза автомобильных шин? Это можно понять из рис. 5, на котором представлены временны́е зависимости изменения концентраций продуктов пиролиза. Рисунок 5 показывает, что резкое изменение концентраций водорода, метана, этана и этилена соответствует ускоряющемуся расходованию пропена, которого в смеси 2 приблизительно в 4 раза меньше, чем в смеси 1. С окончанием расходования С3Н6 то количество пропана, составляющее несколько процентов в исходной смеси, фактически снижается до нулевого значения. К 14-той минуте прекращается и незначительный рост содержания этилена и этана. Из всех концентраций компонентов только концентрация метана продолжает расти и достигает 70% от общей концентрации, что превышает [СН4]0 в ~2.5 раза. Таким образом, численный расчет показывает, что в смеси 2 посредством ее саморазогрева происходит конверсия исходных компонентов в метан. При этом разогрев характеризуется индукционной задержкой. С ростом температуры эта задержка “вырождается” (рис. 4). Помимо расчетов по ДКМ из работы [22], были проведены тестовые расчеты для смеси 2 при условиях, показанных на рис. 5, по ДКМ из работ [28, 29]. В целом ДКМ из [28] и [29] показали ту же положительную температурную динамику при конверсии смеси 2, что и ДКМ из [22], но приблизительно в 3 раза с более быстрым разогревом. При этом в случае ДКМ из [28] температура смеси 2 повысилась до ~1230 К, а при расчетах с ДКМ из [29] – до ~1070 К.
Рис. 4.
Изменение температуры со временем для смеси 2 при Р = 0.1 МПа и различных начальных температурах Т0 (К): 1 – 800, 2 – 900, 3 – 1000, 4 – 1100, 5 – 1200. Линии соответствуют расчетам по ДКМ [22].
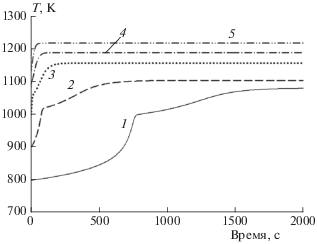
Рис. 5.
Временны́е зависимости изменения концентрации исходных продуктов пиролиза автомобильных шин при Т0 = 800 К и Р = 0.1 МПа для смеси 2. Линии соответствуют расчетам по ДКМ [22]; 1 – [Н2], 2 – [СН4], 3 – [С2Н4], 4 – [С2Н6], 5 – [С3Н6], 6 – [С3Н8].
Следует учитывать, что данные расчеты имеют только оценочный качественный характер. К тому же газификация полипропилена и автомобильных шин проводилась на принципиально разных установках [6, 10], что может сказываться на значениях концентраций продуктов газификации. В работе [6] показано, что технические особенности процессов газификации изношенных автомобильных шин не влияют кардинально на соотношения концентраций продуктов выхода. Поэтому в целом на основе проведенного численного моделирования можно утверждать, что характер конверсии продуктов газификации полипропилена и автомобильных шин имеют принципиально разный. Представленные в настоящее время в литературе ДКМ [23–29] способны количественно и качественно описывать пиролиз и конверсию алкенов и алканов при повышенных температурах. Однако для выбора более приемлемого ДКМ с включением ароматических углеводородов необходимо проведение проверки имеющихся ДКМ [23–26, 28, 29, 32] на основе надежных экспериментальных данных по пиролизу ароматических соединений типа BTX [36].
ВЫВОДЫ
Подводя итог, можно сказать, что продукты, получаемые при пиролизе пропилена и каучуксодержащих отходов, в условиях стандартных пиролитических процессов сильно зависят от вида исходного материала. Тем не менее, в большинстве своем, это линейные алканы и алкены С1–С4 в совокупности с ароматическими углеводородами от С6 и выше (ВТХ). Алканы и алкены С1–С4 могут взаимно трансформироваться с ростом начальной температуры. При этом температура смеси может падать, как в случае продуктов газификации полипропилена, и расти, что характерно для продуктов газификации автомобильных шин. Характер конверсии углеводородов С1–С4 сильно зависит от начальной температуры и их исходного состава.
Работа выполнена при финансовой поддержке Министерством науки и высшего образования Российской Федерации в рамках Программы “Исследования и разработки по приоритетным направлениям развития Российского научно-технического комплекса на период 2014–2020” (грант № 075-15-2019-1843, идентификатор проекта RFMEFI60719X0307).
Список литературы
Czajczyn’ska D., Anguilano L., Ghazal H. et al. // Therm. Sci. Engineer. Prog. 2017. V. 3. P. 171.
Zhang G.-H., Zhua J.-F., Okuwaki A. // Resources, Conservation and Recycling. 2007. V. 50. № 3. P. 231.
Kaminsky W. // J. Phys. IV. 1993. V. 3. P. 1543.
Anene A.F., Fredriksen S.B., Satre K.A., Tokheim L.-A. // Sustainability. 2018. V. 10. P. 3979.
Ademiluyi T., Adebayo T.A. // J. Appl. Sci. Environ. Manage. 2007. V. 11. № 2. P. 21.
Williams P.T. // Waste Manage. 2013. V. 33. P. 1714.
Cho M.-H., Jung S.-H., Kim J.-S. // Energy Fuels. 2010. V. 24. P. 1389.
Ramirez-Canon A., Munoz-Camelo Y.F., Singh P. // Environments. 2018. V. 5. P. 72.
Vijayakuma A., Sebastian J. // IOP Conf. Series: Mater. Sci. and Eng. 2018. V. 396. P. 012062.
Фролов С.М., Звегинцев В.И., Аксёнов В.С. и др. // Горение и взрыв. 2018. Т. 11. № 4. С. 44.
Buah W.K., Cunliffe A.M., Williams P.T. // Process Saf. and Environ. Prot. 2007. V. 85. № B5. P. 450.
Zattini G., Leonardi Ch., Mazzocchetti L. et al. // Proc. Intern. Conf. Sustainable Design Manufacturing. Cham. Germany: Springer, 2017. P. 480; https://doi.org/10.1007/978-3-319-57078-5
Kiran N., Ekinci E., Snape C.E. // Resources, Conservation and Recycling. 2000. V. 29. P. 273.
Nishino J., Itoh M., Fujiyoshi H., Uemichi Y. // Fuel. 2008. V. 87. P. 3681.
Gaurh P., Pramanik H. // Waste Manage. 2018. V. 77. P. 114.
Zeaiter J. // Fuel. 2014. V. 133. P. 276.
Onwudili J. A., Insura N., Williams P.T. // J. Anal. Appl. Pyrolysis. 2009. V. 86. P. 293.
Кислов В.М., Цветкова Ю.Ю., Глазов С.В. и др. // Хим. физика. 2020. Т. 39. № 8.
Kumar S., Singh R.K. // J. Petroleum Eng. 2013. P. 987568; https://doi.org/10.1155/2013/987568
Ahmad I., Khan M. I., Khan H. et al. // Intern. J. Green Energy. 2015. V. 12. P. 663.
Sezgi N.A., Cha W.S., Smith J.M., McCoy B.J. // Ind. Eng. Chem. Res. 1998. V. 37. P. 2582.
Tereza A.M., Medvedev S.P., Smirnov V.N. // Acta Astronaut. 2019. V. 163. P. 18; https://doi.org/10.1016/j.actaastro.2019.03.001
Westbrook C.K., Pitz W.J., Herbinet O., Curran H.J., Silke E.J. // Combust. and Flame. 2008. V. 156. P. 181.
Wang H., Dempsey A.B., Yao M., Jia M., Reitz R.D. // Energy and Fuels. 2014. V. 28. № 8. P. 5480; https://www.erc.wisc.edu/chemicalreaction.php
Wang H., Warner S.J., OehlschlaegerM. A. et al. // Combust. and Flame. 2010. V. 157. P. 1976.
Zhou C-W., Li Y., Burke U. et al. // Ibid. 2018. V. 197. P. 423; http://www.nuigalway.ie/combustionchemistrycentre/ mechanismdownloads/
Healy D., Kalitan D.M., Aul C.J., Petersen E.L., Bourque G., Curran H.J. // Energy and Fuels. 2010. V. 24. № 3. P. 1521.
Battin-Leclerc F. // Prog. Energy Comb. Sci. 2008. V. 34. № 4. P. 440.
Ranzi E., Frassoldati A., Grana R. et al. // Ibid. 2012. V. 38. № 4. P. 468; http://creckmodeling.chem.polimi.it/
Arisawa H., Brill T.B. // Combust. and Flame. 1996. V. 106. № 1–2. P. 131.
Morinigo J.A., Hermida-Quesada J. // Aerospace Sci. Technol. 2016. V. 58. P. 358.
Kee R.J., Rupley F.M., Meeks E., Miller J.A. CHEMKIN III. Livermore, CA: Sandia National Laboratories, Tech. Report № SAND96-8216.
Тереза А.М., Андержанов Э.К. // Хим. физика. 2019. Т. 38. № 8. С. 58.
Хомик С.В., Медведев С.П., Вейссьер Б. и др. // Изв. АН. Сер. хим. 2014. № 8. С.1666.
Медведев С.П., Поленов А.Н., Хомик С.В., Гельфанд Б.Е. // Хим. физика. 2010. Т. 29. № 1. С. 58.
Tereza A., Agafonov G., Anderzhanov E. et al. // High Temp. Mater. Processes. 2019. V. 29. Issue 4. P. 291; https://doi.org/10.1615/HighTempMatProc.2019033024
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика