

Химическая физика, 2021, T. 40, № 4, стр. 18-26
Многообразие кинетических проявлений цепных разветвленных реакций
А. А. Манташян *
Институт химической физики им. А.Б. Налбандяна Национальной Академии наук
Республики Армения
Ереван, Армения
* E-mail: Adolph@chph.sci.am
Поступила в редакцию 01.10.2020
После доработки 01.10.2020
Принята к публикации 20.10.2020
Аннотация
На основе прямых экспериментальных данных по свободным радикалам проанализированы и обобщены результаты исследований по установлению кинетических особенностей и механизма цепных вырожденно-разветвленых реакций. Рассмотрены результаты исследований природы явлений отрицательного температурного коэффициента скорости реакции, термокинетических осцилляций, холодных и прерывистых пламен.
ВВЕДЕНИЕ
Открытие цепных разветвленных реакций и создание общей теории цепных реакций Н.Н. Семёновым [1–3] обогатили теорию химического превращения и показали, что химические превращения могут протекать также по сложному механизму. Развитие процесса химического превращения при этом характеризуется многообразием конкурирующих элементарных актов, протекающих с участием атомов и свободных радикалов.
По цепному вырожденно-разветвленному механизму осуществляются множество промышленно-важных процессов. По такому механизму протекают, в частности, процессы окислительного превращения природного углеводородного сырья в такие соединения, как альдегиды, спирты, пероксиды, олефины и другие, которые являются исходным сырьем для различных промышленных процессов и используются также в различных технически целях. В силу этих обстоятельств механизм окисления – медленного горения углеводородов был и остается предметом многосторонних исследований на протяжении уже многих десятилетий [4, 5].
Особый интерес как с точки зрения теории химического превращения, так и осуществления этих процессов на практике представляют сопровождающие их явления – отрицательного температурного коэффициента (ОТК) скорости реакции, холодных пламен и осцилляций, о которых упоминает Н.Н. Семёнов в своей известной монографии [2]: “Мы думаем, что разобраться в механизме этих реакций не удастся, пока мы отчетливо не поймем явление отрицательного температурного коэффициента реакции окисления, присущего большинству углеводородов…”.
Необходимо было выяснить, например, что явление ОТК есть результат кинетических особенностей, проявляющихся в процессе развивающегося сложного цепного процесса с вырожденными разветвлениями цепей, или имеет место нарушение фундаментального положения теории химического превращения – закона Аррениуса. Для получения ответа на этот и другие принципиально важные вопросы, связанные с механизмом и особенностями этих сложных химических процессов, протекающих с участием атомов и свободных радикалов, весьма эффективным оказалось применение такого мощного метода исследований, как электронный парамагнитный резонанс (ЭПР) [6], который позволил не только обнаруживать атомы и свободные радикалы, но и изучать кинетическое поведение этих активных центров в сложных радикально-цепных процессах химического превращения. Для изучения цепных газофазных реакций окисления углеводородов был разработан и применен кинетический метод вымораживания радикалов [7, 8], позволивший обнаруживать многоатомные радикалы и изучать их кинетические закономерности в этих сложных процессах. Применение этой методики позволило получить данные об особенностях таких процессов на радикальном уровне.
О МЕТОДИКАХ, ПРИМЕНЕННЫХ В ИССЛЕДОВАНИЯХ ПРОЦЕССОВ ОКИСЛЕНИЯ УГЛЕВОДОРОДОВ
Кинетический метод вымораживания радикалов применялся для изучения процессов как в статических, так и в проточных условиях. Как в том, так и в другом случае в стенку цилиндрического кварцевого реактора была впаяна диафрагма с узкой щелью, через которую по кварцевой трубке в ходе процесса из реактора небольшая доля реагирующих газов при низких давлениях направлялась в узел вымораживания, помещенный в резонатор ЭПР. Отбор и поток отбираемых из реактора газов осуществлялся с помощью вакуумного насоса. Кинетика накопления радикалов в узле вымораживания (при температуре жидкого азота) отражала кинетику накопления их в реакторе непосредственно в зоне реакций.
Для исследования явления “холодных пламен” был сконструирован специальный двухсекционный проточный реактор, состоящий из двух последовательно соединенных через узкую кварцевую трубку цилиндрических сосудов. Секции имели раздельный электрообогрев. Каждая секция была снабжена описанным выше устройством для отбора газов для вымораживания и регистрации свободных радикалов радиоспектрометром ЭПР (рис. 1).
Рис. 1.
Схема установки: I – предпламенная зона; II – зона стабилизированного холодного пламени; 1 и 1' – диафрагма со щелью; 2 и 2' – пальцеобразный отросток для вымораживания и накопления радикалов; 3 и 3' – резонатор спектрометра ЭПР; 4 и 4' – термопара.
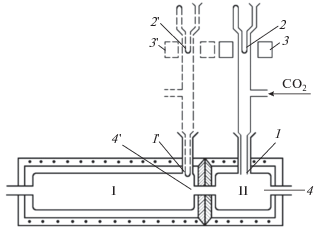
Исследования кинетики процессов разреженных водород-кислородных пламен с добавками диоксида серы осуществлялись также в кварцевых цилиндрических проточных реакторах, снабженных электрообогревом. Для регистрации световых вспышек к реактору прикреплялся светочувствительный приемник. Световые вспышки преобразовывались специальным устройством и передавались для записи на компьютер.
ОСНОВНЫЕ РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЙ, ВЫПОЛНЕННЫХ НА РАДИКАЛЬНОМ УРОВНЕ
Применение разработанной методики вымораживания радикалов позволило впервые в вырожденно-разветвленных цепных реакциях окисления углеводородов обнаружить свободные радикалы и зарегистрировать спектры этих парамагнитных частиц методом ЭПР. Было установлено, что в реакциях как фотохимического, так и термического окисления углеводородов образуются и в наибольших концентрациях накапливаются пероксидные радикалы [8] (рис. 2, 3).
Впервые на основе прямых экспериментальных данных по радикалам в реакции термического окисления метана [9] была определена константа равновесия элементарной реакции
Согласно этим данным пероксидные радикалы, регистрируемые в реакциях окисления углеводородов, действительно, должны быть устойчивы в широком диапазоне температур вплоть до 500–600 °С. Согласно полученным в работах [10, 11] данным, концентрации этих радикалов достигают неожиданно высоких значений: ∼1013 частиц/см3 и более. Исходя из этих результатов, был сделан вывод, что основной реакцией пероксидных радикалов RO2 в этих условиях является их квадратичное взаимодействие с переходом в более активные радикалы RO:
Реакции радикалов RO ответственны за дальнейшее развитие цепей и образование продуктов реакции.
Согласно данным работ [10, 11], экспериментально измеренная общая скорость реакции окисления как метана [10], так и пропана [11] оказывается пропорциональной концентрации радикалов во второй степени:
Константа пропорциональности k по своему значению совпадает с экспериментально определенной в работах [12, 13] величиной константы скорости элементарной реакции квадратичного взаимодействия пероксидных радикалов между собой с образованием алкоксильных радикалов.
Из этих результатов следует [10], что при окислении метана продуктами реакции должны быть метанол и формальдегид, образующиеся в результате следующих элементарных актов:
Эти продукты действительно образуются в цепной реакции окисления метана. Между тем в рассмотренном Н.Н. Семёновым механизме реакции окисления метана как модельной цепной реакции с вырожденными разветвлениями цепей в качестве промежуточного продукта реакции представлен лишь формальдегид [2], а метан в этой модели отсутствует. Образование метанола в процессе окисления метана отмечено в работе [14] Ньюитом еще в 1934 году. Согласно Н.Н. Семёнову, радикалы СН3, образующиеся в реакции окисления метана, быстро переходят в пероксидные радикалы СН3О2, которые затем изомеризуются и быстро распадаются с образованием формальдегида и радикала ОН. В предположении, что изомеризация и распад радикалов СН3О2 протекают быстро, образование формальдегида в модели Н.Н. Семёнова представлено идущим в одну стадию: СН3 + О2 → СН2O + НO2. Эта элементарная реакция как основной канал образования формальдегида фигурирует практически во всех работах по окислению углеводородов на протяжении многих лет. Протеканием реакций изомеризации и распада пероксидных радикалов Штерн в работе [5] объясняет образование продуктов реакций окисления как парафиновых, так и олефиновых углеводородов. Эти взгляды в целом были приняты также и другими исследователями [4].
Исследование процесса окисления этилена – олефинового углеводорода на радикальном уровне показало [15–17], что процесс в этом случае протекает с участием тех же пероксидных радикалов, что и окисление этана [18] – парафинового углеводорода. Следует отметить также, что и стабильные промежуточные продукты при окислении этих углеводородов практически одни и те же. В частности, одним из основных продуктов в том и другом случае является ацетальдегид. При окислении этилена образуется также эпоксид – оксид этилена. Исходя из отмеченных результатов, в работе [19] был сделан вывод, что окисление этилена (олефинового углеводорода) протекает с участием тех же алкильных и алкилпероксидных (С2Н5О2) радикалов, что и окисление этана – парафинового углеводорода. Согласно этим представлениям алкильные радикалы С2Н5 образуются в результате присоединения атомов водорода к этилену. Атомы Н образуются в реакциях развития цепей. Следует отметить, что они действительно должны легче присоединяться к этилену, образуя радикал С2Н5, чем отрывать атом водорода, образуя радикал С2Н3. В результате при окислении этилена должны образоваться алкилпероксидные радикалы С2Н5О2 (RO2), которые в основной элементарной реакции (RO2 + C2H4 → эпоксид C2H4O + RO) перейдут в активные радикалы RO, как и при окислении парафиновых углеводородов. Квадратичные реакции пероксидных радикалов при окислении этилена и олефиновых углеводородов, в общем говоря, также могут иметь место. Механизм окисления этилена проанализировали также численным методом [20], и было получено соответствие с механизмом, предложенным на основе экспериментальных данных [21].
Таким образом, первично образующиеся в вырожденно-разветвленных цепных реакциях окисления углеводородов алкилпероксидные радикалы RO2 переходят в более активные алкоксильные радикалы RO, которые в последующих элементарных актах обеспечивают дальнейшее развитие цепей и образование основных стабильных промежуточных продуктов [17]. Обобщая результаты исследований по механизму окисления углеводородов на основе прямых данных по свободным радикалам, принципиальный механизм в общем виде можно представить так:
На основе определенных представлений о поведении свободных радикалов в сложных цепных вырожденно-разветвленных реакциях и возможностей по кинетическому методу вымораживания радикалов были начаты исследования по изучению природы явлений, наблюдаемых при осуществлении процессов окисления углеводородов. Явление ОТК изучалось на примере окисления пропана [22]. На рис. 4 представлены кинетические кривые накопления радикалов в узле вымораживания радикалов, помещенном в резонатор ЭПР, полученные при температурах Т = 348–480 °С в области ОТК.
Рис. 3.
Температурная зависимость концентрации радикалов в стабилизированных холодных пламенах: 1 – изо-С4Н10 + О2; 2 – н-С4Н10 + О2; 3 – С3Н8 + О2. Символами ◻, ⚪, △ обозначены низкие концентрации радикалов в первой секции реактора, различающиеся более чем на два порядка.
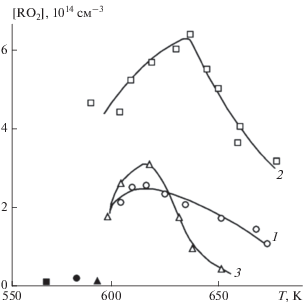
Рис. 4.
Кинетические кривые накопления радикалов в реакции окисления пропана при P = 250 Торр и следующих температурах (в °С): 1 – 348, 2 – 364, 3 – 375, 4 – 392, 5 – 480, 6 – 436. Концентрация радикалов дана в относительных единицах.
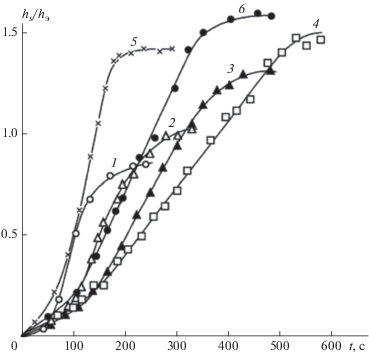
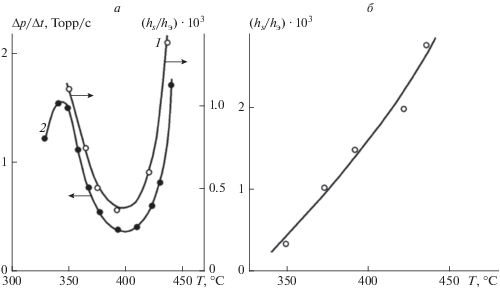
Максимальная скорость реакции окисления достигается при максимальной концентрации радикалов. Кривые накопления радикалов в узле вымораживания радикалов, имеющие S-образный вид, отражают динамику накопления пероксидных радикалов непосредственно в зоне реакции. Точка, соответствующая максимальной концентрации последних на этой кривой, соответствует точке перегиба. Концентрация радикалов в зоне реакции изменяется пропорционально этой величине и, судя по данным рис. 4, изменяется в изученной области температур. Она возрастает с изменением температуры – с повышением до 480 °С, а с дальнейшим ее повышением – уменьшается. Такая температурная зависимость процесса фиксируется и по изменению давления (ΔР) реагирующей смеси по ходу процесса превращения (рис. 5). Однако иной температурной зависимостью характеризуются начальные участки кинетических кривых накопления радикалов (рис. 4). Построенная по этим данным температурная зависимость концентрации радикалов представлена на рис. 5 и характеризует развитие процесса в периоде индукции, т.е. в начале процесса, когда разветвления цепей не играют определяющую роль в развитии процесса. Здесь процесс развивается, следуя закону Аррениуса. Явление ОТК наблюдается в развившемся процессе и очевидным образом должно быть связано с подавлением процесса разветвления цепей в данной области температур. Максимальная скорость цепной реакции достигается при наиболее развитой стадии процесса разветвления цепей.
Рис. 5.
Область отрицательного температурного коэффициента. а – температурные зависимости максимальной концентрации радикалов (1) и максимальной скорости реакции по изменению давления (2); б – температурная зависимость концентрации радикалов, построенная по начальным участкам кривых накопления радикалов.
Согласно теории Н.Н. Семёнова, вырожденное разветвление цепей связано с превращением активного промежуточного продукта цепей. Таким промежуточным продуктом при окислении пропана может быть, например, ацетальдегид – соединение с двумя атомами углерода, который, реагируя с радикалами в процессе окисления пропана, должен образовать ацетильный радикал, который продолжает процесс разветвления цепей:
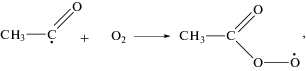
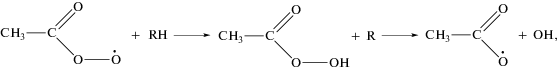
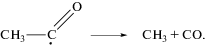
Однако если ацетильный радикал, реагируя с кислородом (реакция (1)) приводит к образованию ацетилпероксидного радикала и дальнейшему развитию процесса разветвления цепей, то его распад (реакция (2)) тормозит разветвление цепей. Радикал СН3 не может продолжить процесс разветвления цепей. Он может привести лишь к образованию малоактивного метилпероксидного радикала СН3О2. С повышением температуры распад ацетильного радикала, ответственного за разветвление цепей, будет усиливаться, процесс разветвления цепей будет ослабевать, и реакция в целом будет затухать. Однако, начиная с некоторой температуры, может начаться другой процесс разветвления цепей, в котором определяющую роль могут играть другие радикалы, например радикал НО2. Таким образом, конкуренция реакций (1) и (2) с повышением температуры может стать причиной явления ОТК.
Как показал численный кинетический анализ процесса окислительного превращения метана с учетом 80 возможных элементарных реакций [23], рассмотренная в [2] модель цепной вырожденно-разветвленной реакции метана упрощенная. Она не только не учитывает факт образования метанола – одного из основных продуктов реакции, но и упрощенно представляет акт разветвления цепей. Как следует из кинетического анализа, выполненного в работе [23], он более сложный. В случае окисления пропана, как мы видим, разветвления цепей могут иметь место по более сложному пути и представлять из себя процесс, в котором основной активный центр, ведущий разветвление цепей, может вступать в две конкурирующие элементарные реакции. В результате такой конкуренции изменение температуры может подавить процесс разветвления цепей и привести, в частности, к явлению ОТК. В уравнении Н.Н. Семёнова
описывающем развитие динамики цепной разветвленной реакции, коэффициент f может иметь более сложную температурную зависимость, которая может включать также влияние температуры на конкуренцию двух элементарных реакций ведущего разветвление цепей радикала. Это обстоятельство приводит к явлению ОТК реакций и тормозит развитие процесса во взрывном режиме, приводя к возникновению другого явления – холодного пламени.
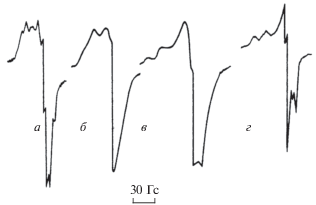
Явление ОТК наблюдается и в режиме холоднопламенного окисления углеводородов, в частности того же пропана. Холодные пламена углеводородов изучались в режиме стабилизированных пламен. Исследования холодных пламен пропана и бутанов проводились в специально разработанном двухсекционном реакторе (рис. 1) с отбором радикалов на вымораживание. Раздельный подогрев секций путем изменения температуры во второй секции, в которой стабилизировалось пламя при определенной постоянно поддерживаемой температуре, позволял иметь развитый на определенной стадии развития холоднопламенный процесс. Изменение температуры во второй секции при постоянной температуре в первой секции позволило получить картину развития и затухания процесса в режиме холодного пламени (рис. 6) [24–27]. Следует отметить, что небольшие изменения параметров, при которых осуществляется процесс (Р, Т, время контакта τ), приводят к переходу последнего в режим осцилляций [26–28]. На рис. 7 представлены различные виды осцилляций и разграничены области протекания окислительного процесса в различных режимах.
Рис. 6.
Различные виды осцилляций в процессе окисления пропана: А – затухающие; Б – гармонические; В – релаксационные; Т = 581–586 °С; С3Н8 : О2 = 1 : 1; Р = 320 Торр. Области существования различных видов осцилляционного и стабильного холоднопламенного окисления пропана в координатах τ – Т: а – медленная реакция; б – стабильные осцилляции; в – затухающие осцилляции; г – стабилизированное холодное пламя.
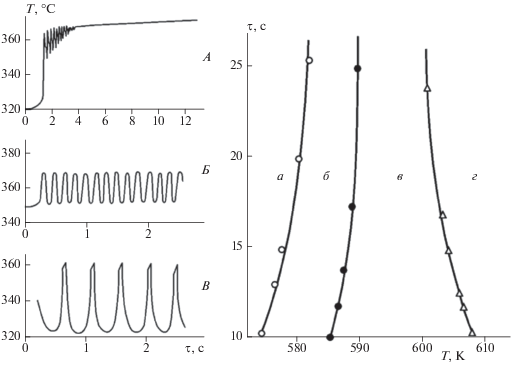
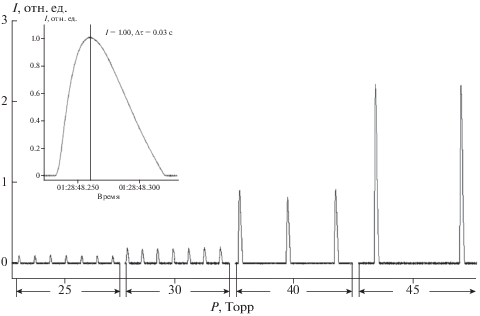
Рис. 7.
Изменение частоты и интенсивности вспышек с повышением давления. На вставке приведен единичный сигнал в развернутом виде; Т = 470 °С; состав смеси: Н2 : О2 : SO2 = 10 : 1 : 1.
Рассмотренные выше результаты по цепным реакциям, а также результаты многих других исследований по механизму цепных разветвленных реакций привели к выводу [29–31], что эти реакции могут служить активной средой, в которой можно осуществлять другие процессы, т.е. осуществлять сопряженные процессы. Такой подход позволил начать исследования по осуществлению процессов химического превращения неорганических соединений под воздействием газофазных реакций. В частности, цепные реакции окисления углеводородов и медленного окисления водорода (над вторым пределом самовоспламенения водород-кислородных смесей) позволили осуществлять окисление SO2 в SO3 пероксидными радикалами [32–35]: SO2 + RO2 (HO2) → SO3 + RO (OH). Такой сопряженный процесс оказывает ускоряющее воздействие на развитие самого цепного процесса, так как переводит пероксидные радикалы в более активные радикалы RО и ОН, продолжающие развитие цепей [1]. Добавки SO2 в реагирующие смеси и других углеводородов также оказывают ускоряющее воздействие на развитие окислительного процесса [36].
Следует подчеркнуть, что диоксид серы в значительных концентрациях присутствует в отходящих газах металлургических производств и тепловых станций. В результате этого возникла большая экологическая проблема – борьба с загрязнениями окружающей среды сернистым газом. В связи с этим ведутся исследования по утилизации и превращению SO2 в другие полезные и используемые соединения и продукты.
Возникает вопрос, какие превращения может претерпевать молекула SO2 под воздействием других цепных реакций, в которых в наибольших концентрациях образуются не пероксидные, а другие свободные радикалы и атомы. Такой является цепная реакция окисления водорода в области самовоспламенения. При осуществлении ее в режиме низкотемпературных разреженных пламен водород-кислородных смесей в наибольших концентрациях накапливаются атомы водорода [6, 37, 38]. Добавки SO2 в реагирующую смесь в таком режиме подвергаются химическому превращению с образованием элементарной серы [39–45] и приводят к возникновению нового явления, называемого “прерывистые пламена” (рис. 7 и 8). Параметрами, определяющими условия возникновения “прерывистых пламен”, являются: скорость прохождения реагирующей смеси через реактор (время контакта), скорость развития процесса самовоспламенения реагирующей водород-кислородной смеси, а также время заполнения реактора критическим количеством поступающей в реактор реагирующей смеси для возникновения самовоспламенения.
Рис. 8.
Динамика развития вспышки во времени при Т = 470 °С; состав смеси: Н2 : О2 : SO2 = 10 : 1 : 1; Р = 30 Торр.

Возможные пути химического превращения SO2 в среде разреженного низкотемпературного пламени с позиции термодинамики и химической кинетики проанализированы в работах [39, 40]. Анализ показывает, что в условиях разреженного пламени водород-кислородной смеси атомы водорода присоединяются к SO2, образуя частицы HSO2, которые затем распадаются по бимолекулярной реакции реагентов: HSO2 + M → SO + OH + M. Затем частицы SO в квадратичной реакции SO + + SO → S + SO2 образуют атомы серы. Атомы серы оказываются не менее активными, чем атомы водорода, и ускоряют общий сопряженный процесс, разветвляя цепи по реакции S + O2 → SO + O наряду с реакцией Н + О → ОН + О. В результате общий цепной сопряженный процесс превращения реализуется с большей скоростью, чем в случае смеси без добавок SO2. Это, вероятно, и является главной причиной в конкуренции отмеченных выше параметров, определяющих динамику развития сопряженного процесса, приводя к явлению “прерывистые пламена”. Совокупность рассмотренных результатов по превращению SO2 приводит к выводу, что сопряженными могут быть реакции, которые протекают с участием одних и тех же активных частиц – атомов и радикалов. Выражаясь в терминологии классического определения, “сопряженные реакции” в формальной химической кинетике – это когда “ведомая реакция” Н2 + SO2 реализуется под воздействием первой “ведущей реакции” Н2 + О2. “Ведомая реакция” самостоятельно, без участия “ведущей реакции” в данном случае не может генерировать начальные активные центры – атомы водорода. Их образование обеспечивается первой “ведущей цепной реакцией” окисления водорода. Этот принцип действовал во всех рассмотренных случаях.
Кинетический анализ сопряженного химического превращения диоксида серы позволил выявить основные элементарные реакции, которые обеспечивают химическое превращение SO2, и на этой основе сделать вывод: превращение SO2 с участием Н2 может протекать как самостоятельная цепная реакция – “цепная реакция окисления водорода диоксидом серы” [45]. При этом цепная реакция окисления водорода в сопряженном процессе является генератором атомов водорода, которые инициируют цепную реакцию окисления водорода кислородом, содержащимся в молекуле SO2. Цепная реакция окисления водорода диоксидом серы может быть представлена следующей последовательностью элементарных реакций:
(0)
${{{\text{Н}}}_{2}} + {\text{S}}{{{\text{O}}}_{2}} \to {\text{H}} + {\text{HS}}{{{\text{O}}}_{2}},$Стадия зарождения цепей по реакции (0) затруднена и может протекать гетерогенно на стенках реактора. Атомы серы не могут осуществлять разветвления, как они это делают в сопряженном процессе, из-за отсутствия свободного кислорода. Численный кинетический анализ рассмотренной модели показал следующее: если допустить, что реакция зарождения цепей (0) протекает гетерогенно с существенно заниженной энергией активации, то представленная модель описывает экспериментально установленные кинетические закономерности цепной реакции окисления водорода диоксидом серы.
Таким образом, исходя из изложенного выше, можно сделать вывод, что многие известные химические реакции превращения различных соединений, считающиеся молекулярными, в действительности протекают с участием атомов и свободных радикалов. Например, принято считать, что восстановление серы из ее диоксида водородом происходит в результате реакции
Между тем, как мы видели, сера из ее диоксида восстанавливается в результате цепной реакции, а данное уравнение является балансовой реакцией, т.е. брутто-реакцией. Сера в действительности восстанавливается в результате цепной реакции “окисления водорода диоксидом серы”.
ЗАКЛЮЧЕНИЕ
В целом можно заключить, что неорганические соединения вступают в такие же реакции с атомами и свободными радикалами, что и органические. Изложенные в данной статье результаты исследований в области цепных реакций являются следствием представлений, внесенных Н.Н. Семёновым в теорию химического превращения. Его революционные идеи и открытия предопределили развитие теории и практики сложных процессов химического превращения.
Список литературы
Семёнов Н.Н. Цепные реакции. Л.: Госхимтехиздат, 1934.
Семёнов Н.Н. О некоторых проблемах химической кинетики и реакционной способности. М.: Изд-во. АН СССР, 1958.
Семёнов Н.Н. Избранные труды. М.: Наука, 2004.
Льюис Б., Эльбе Г. Горение, пламя и взрывы в газах. М.: Мир, 1968.
Штерн В.Я. Механизм окисления углеводородов в газовой фазе. М.: Изд-во АН СССР, 1960.
Панфилов В. Н., Цветков Ю. Д., Воеводский В.В. // Кинетика и катализ. 1960. Т. 1. С. 333.
Манташян А.А., Налбандян А.Б. // ЖФХ. 1972. Т. 46. № 12. С. 3030.
Налбандян А.Б., Манташян А.А. Элементарные процессы в медленных газофазных реакциях. Ереван: Изд-во АН Арм. ССР, 1975.
Khachatryan L.A., Niazyan O.M., Mantashyan A.A., Vedeneev V.I., Teitelboim M.A. // Intern. J. Chem. Kin. 1982. V. 14. № 11. P. 1231.
Манташян А.А., Хачатрян Л.А., Ниазян О.М. // ЖФХ. 1977. Т. 51. № 2. С. 341.
Поладян Е.А., Григорян Г.Л., Хачатрян Л.А., Манташян А.А. // Кинетика и катализ. 1976. Т. 17. № 2. С. 304.
Heiklen J. // Intern. Oxidation Sympos. 1967. V. 15. P. 343.
Parkes D.A. // Proc. 15th Symp. on Combust. Pittsburgh: The Combust. Inst. 1974. P. 150.
Newitt D.M., Thornes L.S. // J. Chem. Soc. 1937. P. 1656.
Арсентьев С.Д., Манташян А.А. // Армян. хим. журн. 1978. Т. 31. № 9. С. 643.
Манташян А.А. // Армян. хим. журн. 1979. Т. 32. № 6. С. 417.
Arsentiev S. D., Mantashyan A.A. // React. Kinet. Catal. Lett. 1980. V. 13. Issue 2. P. 125.
Манташян А.А., Хачатрян Л.А., Ниазян О.М. // Армян. хим. журн. 1978. Т. 31. № 1. С. 49.
Манташян А.А. // Армян. хим. журн. 1979. Т. 32. № 6. С. 417.
Манташян А.А., Аракелян Э.А., Арсентьев С.Д. // Хим. физика. 2003. Т. 22. № 5. С. 43.
Mantashyan A.A., Khachatryan L.A., Niazyan O.M., Arsentiev S.D. // Combust. and Flame. 1981. V. 43. P. 221.
Манташян А.А., Григорян Г.Л., Саакян А.С., Налбандян А.Б. // ДАН СССР. 1972. Т. 204. № 6. С. 1392.
Манташян А.А., Макарян Э.М., Чарчян А.В. // Физика горения и взрыва. 2019. Т. 55. С. 3.
Гукасян П.С., Манташян А.А., Саядян Р.А. // Физика горения и взрыва. 1976. Т. 12. № 5. С. 789.
Манташян А.А., Гукасян П.С. // ДАН СССР. 1977. Т. 234. № 2. С. 379.
Mantashyan A.A., Gookasyan P.S., Sayadyan R.H. // React. Kinet. Catal. Lett. 1979. V. 11. № 3. P. 225.
Mantashyan A.A. // Proc. Combust. Inst. 1994. V. 25. P. 927.
Mantashyan A.A., Bernatosyan S.G., Simonyan T.R. // Oxid. Commun. 1983. V. 5. № 1–2. P. 207.
Манташян А.А // Хим. журн. Армении. 1996. Т. 49. № 4. С. 5.
Mantashyan A.A. // Chem. Phys. Rep. 2001. V. 19. № 11. P. 2163.
Манташян А.А. // Хим. физика. 2007. Т. 26. № 9. С. 43.
Манташян А.А., Макарян Э.М., Аветисян А.М., Элоян А.Э. // Хим. журн. Армении. 2002. Т. 55. № 4. С. 130.
Манташян А.А., Аветисян А.М., Макарян Э.М. // Хим. журн. Армении. 2003. Т. 56. № 3. С. 5.
Манташян А.А., Ванг Х., Аветисян А.М., Мака-рян E.M. // Хим. журн. Армении. 2006. Т. 59. № 4. C. 9.
Манташян А.А., Микаелян А.Ж. // Хим. журн. Армении. 2005. Т. 58. № 1–2. С. 18.
Мкрян Т.Г., Гукасян П.С., Манташян А.А. // Хим. физика. 2002. Т. 21. № 11. С. 33.
Кондратьев В.Н. Спектроскопическое изучение химических газофазных реакций. М.–Л.: АН СССР, 1944.
Панфилов В.Н. // Кинетика и катализ. 1962. Т. 3. С. 643.
Манташян А.А., Макарян Э.М., Арутюнян А.А., Геворгян Г.М. // Физика горения и взрыва. 2016. Т. 52. № 6. С. 26.
Манташян А.А., Макарян Э.М., Арутюнян А.А., Геворгян Г.М. // Физика горения и взрыва. 2016. Т. 52. № 6. С. 35.
Манташян А.А. // Физика горения и взрыва. 2016. Т. 52. № 2. С. 3.
Манташян А.А. // Хим. журн. Армении. 2014. Т. 67. № 2–3. C. 168.
Манташян А.А., Макарян Э.М., Арутюнян А.А. // Хим. журн. Армении. 2016. Т. 69. № 3. С. 226.
Манташян А.А., Макарян Э.М., Эвинян M.A., Акопян А.Г. // Хим. журн. Армении. 2020. Т. 73. № 1. С. 25.
Манташян А.А., Макарян Э.М., Аракелян Л.С. // Физика горения и взрыва. 2019. Т. 55. № 5. С. 3.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика