

Химическая физика, 2021, T. 40, № 5, стр. 55-67
Вопрос о механизме захвата O3 на покрытии из метановой сажи
В. В. Зеленов 1, *, Е. В. Апарина 1
1 Федеральный исследовательский центр химической физики им. Н.Н. Семёнова
Российской академии наук
Москва, Россия
* E-mail: zelenov@binep.ac.ru
Поступила в редакцию 30.10.2020
После доработки 10.11.2020
Принята к публикации 20.11.2020
Аннотация
Исследован захват O3 на свежем покрытии из метановой сажи при T = 256 и 295 К в диапазоне концентраций O3 1.1 ⋅ 1012–4.1 ⋅ 1013 cм–3 с использованием проточного реактора с подвижной вставкой и масс-спектрометрической регистрацией. Установлено, что захват носит многостадийный характер. Обе стадии захвата, быстрая и медленная, происходят по механизму реакции на поверхности с адсорбированной частицей. На базе лэнгмюровской концепции адсорбции зависимость коэффициента захвата γ озона на сажевом покрытии представлена в виде γ = γ0/(1 + KL[O3]) c параметрами для первой стадии захвата γ0 (295 К) = (6.6 ± 0.4) ⋅ 10–4 и γ0 (256 К) = (8.1 ± 2.6) ⋅ 10–4, KL (295 К) = = (7 ± 1) ⋅ 10–14 и KL (256 К) = (22 ± 9) ⋅ 10–14 см3. Параметры для второй стадии захвата: γ0 (295 К) = = (1.3 ± 0.2) ⋅ 10–4 и γ0 (256 К) = (1.2 ± 0.5) ⋅ 10–4, KL (295 К) = (11 ± 2) ⋅ 10–14 и KL (256 К) = (28 ± 13) ⋅ ⋅ 10–14 см3. Из зависимости γ от времени экспозиции сажевого покрытия к потоку О3 оценена константа скорости мономолекулярного распада поверхностного комплекса с адсорбированной молекулой озона: kr (295 К) = (0.39 ± 0.01) с–1, kr (256 К) = (0.15 ± 0.01) с–1 для быстрой стадии и kr (295 К) = (2.8 ± 0.1) ⋅ 10–2 с–1, kr (256 К) = (1 ± 0.04) ⋅ 10–2 с–1 для медленной стадии. В пределе малых концентраций γ слабо зависит от [O3] и T вследствие соотношения γ0 ∼ kr/kd и близости по величине энергии активации константы скорости kr реакции и теплоты адсорбции константы скорости kd десорбции.
ВВЕДЕНИЕ
Основными источниками озона в тропосфере являются фотохимические реакции с участием углеводородов, перекисных радикалов и свободных радикалов NOx. Дополнительный вклад вносят грозы и поступление озона из стратосферы, где его концентрация в 90 раз выше. Последний источник носит спорадический характер из-за обратного температурного профиля в атмосфере. Тем не менее благодаря этому источнику локальные концентрации озона могут превышать на 40 ppb его базовый уровень в 70 ppb [1].
Озон является одним из парниковых газов с вкладом в парниковый эффект, составляющим 20% от вклада углекислого газа [2]. Типичные приземные концентрации озона составляют 10–70 ppb, но вблизи индустриальных регионов могут достигать 100–500 ppb [3–7].
Доля органического аэрозоля в атмосфере с глобальной эмиссией 12.6–24 Тг/год [8, 9] составляет 30–50% [10, 11]. Большую часть этого аэрозоля составляет сажа. Сажевый аэрозоль образуется при неполном сгорании топлив и биомасс, в частности при сгорании метана как основного компонента горючих газообразных ископаемых, а также побочного продукта добычи нефти. Сажевый аэрозоль состоит из нерастворимого углеродного остова и растворимой органической фракции, адсорбированной на поверхности твердой частицы [12, 13]. Сажа влияет на глобальный излучательный баланс планеты посредством эффективного поглощения коротковолнового солнечного спектра [14, 15]. Кроме того, он является мощным погодообразующим фактором. Свежие сажевые частицы гидрофобны по своей природе [16], но при окислении активными компонентами атмосферы (NOx, HNO3, O3) становятся гидрофильными [17–19]. На такой модифицированной поверхности может конденсироваться атмосферная влага, тем самым эти компоненты служат центрами нуклеации облаков [20–22]. Продукты окисления, которые в виде осадков возвращаются на землю, зачастую представляют угрозу здоровью человека [23, 24].
Работы по исследованию захвата озона на органическом аэрозоле достаточно многочисленны. Условно их можно разделить на две группы. Первая из них включает работы по анализу состава твердых продуктов этой гетерогенной реакции и их токсикологические характеристики [17, 18, 23–30]. Анализ этих работ находится вне темы данной работы. Вторая группа работ направлена на исследование механизма захвата. Исследовался захват на покрытиях из элементного углерода и саж различного происхождения: молотый древесный уголь [31], элементный углерод [32–35], сажи из метана [36], н-гексана [3, 37, 38], толуола и керосина [39]. Исследовался также захват на индивидуальных органических веществах, нанесенных в виде пленочного покрытия на различные подложки [40–47]. Эти вещества являются либо установленными компонентами различных саж, либо их представителями. Во всех этих работах наблюдалась зависимость коэффициента захвата от времени экспозиции, связанная с ограниченным количеством центров адсорбции [48]. Во многих работах отмечается сложный многостадийный характер захвата. В ряде работ такого характера не обнаружено, но связано это, вероятно, с использованием непроточного реактора и малым временны́м разрешением [3, 32, 34, 35, 37]. В большинстве работ выявлена обратно пропорциональная зависимость начального коэффициента захвата от концентрации озона. В отдельных работах такой зависимости не обнаружено [39, 40, 42]. Исходя из температурной зависимости коэффициента захвата, в ряде работ на базе представления захвата по механизму Лэнгмюра–Хиншельвуда оценены коэффициент Лэнгмюра, теплота адсорбции и энергия активации элементарных реакций [34, 35, 37, 41].
Что касается механизма захвата, то в англоязычной литературе в качестве такого рассматривается механизм Лэнгмюра–Хиншельвуда и даже делается вывод о схожести этого механизма с процессами захвата озона на покрытиях различного состава [38]. Как отмечается авторами работы [49], название обсуждаемого механизма ошибочно. Исходя из кинетических схем, которые приводятся в работах, и вытекающей из них зависимости, подразумевается механизм захвата через “реакцию на поверхности с адсорбированной частицей”. Этот тип захвата предполагает обратимую адсорбцию газа-реагента на химически активных центрах и последующую гетерогенную реакцию адсорбированной молекулы с этими нелетучими компонентами поверхности [49]. Классический механизм Лэнгмюра–Хиншельвуда предполагает гетерогенную реакцию между двумя поверхностными комплексами, содержащими адсорбированные молекулы [50].
В современных моделях захвата газов-реагентов на поверхности жидкостей и твердых тел с учетом конкурентной адсорбции необходимо знание входных параметров, таких как коэффициент Лэнгмюра, константа скорости гетерогенной реакции, включая теплоту адсорбции и энергию активации [49, 51–54]. Достоверность оценки этих параметров из экспериментальных данных определяется правильностью выбора механизма, описывающего всю совокупность исходных данных. Цель данной работы заключалась в получении количественной информации об адсорбционно/десорбционных свойствах системы озон/метановая сажа, а также о ее начальной реакционной способности. В представленной работе получены экспериментальные данные по зависимости коэффициента захвата от времени экспозиции, концентрации газа-реагента и температуры. Проведено модельное описание всей совокупности экспериментальных данных по двум альтернативным механизмам захвата: механизму мономолекулярного распада и классическому механизму Лэнгмюра–Хиншельвуда. На основании полученных результатов, а также с привлечением литературных данных сделан вывод в пользу механизма мономолекулярного распада.
ЭКСПЕРИМЕНТАЛЬНАЯ УСТАНОВКА
Экспериментальные данные получены с использованием проточного реактора с подвижной вставкой и нанесенным не нее пленочным сажевым покрытием из диффузионного пламени горения метана. Реактор сопряжен с масс-спектрометром высокого разрешения с ионизацией низковольтными электронами. Схема реактора приведена и подробно описана ранее [55], поэтому отметим кратко основные особенности. Поток смеси O3/He (гелий – 99.99%) протекает через цилиндрический стеклянный реактор и регулируемый вентиль откачки. Все внутренние поверхности реактора покрыты тефлоновой пленкой (Teflon FEP). Тонкий центральный стержень из нержавеющей стали с сажевым покрытием можно перемещать вдоль оси реактора через тонкую стеклянную трубку из компенсирующего объема в зону контакта сажевого покрытия с газом-реагентом. Перемещение стержня с покрытием осуществляется с помощью внешнего магнита. Исходная смесь O3/He с заданной концентрацией приготавливается заранее в бачке из нержавеющей стали. Бачок и все трубки подачи смеси в реактор были пропассивированы заранее молекулярным фтором. Поток смеси O3/He, ${{Q}_{{{{{{{\text{O}}}_{{\text{3}}}}} \mathord{\left/ {\vphantom {{{{{\text{O}}}_{{\text{3}}}}} {{\text{He}}}}} \right. \kern-0em} {{\text{He}}}}}}},$ измеряется расходомером, встроенным в магистраль подачи перед входом в реактор, с точностью 0.1 см3 ⋅ Торр ⋅ с–1. Через компенсирующий объем подается дополнительный поток гелия, QHe, чтобы избежать неконтролируемого диффузного потока O3 из зоны контакта в этот объем. Изменяя поток гелия через этот ввод и поток смеси O3/He так, чтобы их сумма оставалась постоянной, можно было менять концентрацию озона в реакторе при неизменном времени контакта газа-реагента с сажевым покрытием. Отбор пробы в масс-спектрометр осуществляется путем формирования молекулярного пучка через отверстие диаметром 0.35 мм в вершине напускного конуса, расположенного соосно с внешней трубкой реактора.
Для проведения экспериментов при пониженной температуре реактора были изготовлены две металлические кюветы, заполняемые смесью NaCl/H2O при температуре ее таяния и охватывающие область трубки реактора и компенсирующего объема.
Основные параметры реактора и магистрали подачи озона: внутренний диаметр трубки реактора dR = 1.3 см; внешний диаметр стержня из нержавеющей стали dr = 0.2 см; максимальная длина стержня L = 50 см; давление p = 1–2 Торр; скорость потока газа-носителя u = 100–400 см ⋅ с–1; внутренний диаметр трубки подачи смеси O3/He в реактор – 2 мм; скорость потока смеси O3/He по трубке подачи 0.5–5 см ⋅ с–1.
Приготовление и состав реакционной смеси. Озон приготавливали из кислорода марки ОСЧ, перемораживали в пропассивированный фтором бачок из нержавеющей стали и добавляли гелий до давления в 1 атм. За время транспортировки смеси O3/He в реактор происходит частичное разложение озона, особенно ощутимое при малых подачах. Типичные соотношения концентраций озона и кислорода в реакторе в зависимости от величины потока исходной смеси O3/He приведены в табл. 1. Абсолютная концентрация озона определялась из материального баланса при полном переводе O3 в кислород путем нагрева сегмента трубки подачи. Калибровка масс-спектрометра по кислороду проводилась в отдельном опыте при известном давлении в реакторе и заданном потоке кислорода. Измерения интенсивностей ионных токов O3 (m/z = 48) и O2 (m/z = 32) проводили при энергии ионизирующих электронов Ee = 50 эВ для О3 и 12.5 эВ для О2. В последнем случае исключается вклад осколочного иона ${\text{О}}_{2}^{ + }$ из масс-спектра озона.
Таблица 1.
Соотношение концентраций O3 и О2 в реакторе при изменении массовой скорости подачи смеси ${{Q}_{{{{{{{\mathbf{О}}}_{{\mathbf{3}}}}} \mathord{\left/ {\vphantom {{{{{\mathbf{О}}}_{{\mathbf{3}}}}} {{\mathbf{He}}}}} \right. \kern-0em} {{\mathbf{He}}}}}}}$ [O3] : [He] = 1 : 685
| ${{Q}_{{{{{{О}_{3}}} \mathord{\left/ {\vphantom {{{{О}_{3}}} {He}}} \right. \kern-0em} {He}}}}},$ 1017 с–1 | [О3]*, 1012 cм–3 | [О2]*, 1012 cм–3 |
|---|---|---|
| 3.5 | 0.43 | 1.4 |
| 4.3 | 0.73 | 1.5 |
| 6.7 | 1.4 | 1.9 |
| 10.2 | 2.55 | 2.2 |
| 14 | 3.46 | 3.0 |
| 18.4 | 6.1 | 1.7 |
| 26 | 10.2 | 1.0 |
| 30 | 12 | 0 |
Нанесение сажи. Сажу наносили на металлический стержень в пламени метана с использованием лабораторной горелки, присоединенной к газовой магистрали, без дополнительной подачи сжатого воздуха. При диффузионном горении метана металлический стержень располагали на расстоянии 15–17 см от основания пламени при постоянном вращении стержня вручную. Через 15 мин масса нанесенной на стержень сажи составляла 2–4 мг и определялась путем взвешивания на весах с точностью 0.1 мг. После взвешивания стержень с нанесенной сажей сразу вставляли в реактор и оставляли под вакуумом до начала эксперимента. Поверхностная плотность покрытия сажей составляла ρm = 70–120 мкг ⋅ см–2 и во всех случаях не превышала величины, при которой необходимо учитывать пористость покрытия [39, 56].
Процедура измерения и обработка данных. Экспериментальные зависимости коэффициента захвата озона на свежем сажевом покрытии от времени его экспозиции к потоку озона определяли по относительному изменению концентрации озона на масс-спектральной линии с интенсивностью I48 (m/z = 48) при введении и выведении стержня с покрытием в зону его контакта с озоном. Измерение интенсивности ионного тока проводили в режиме механической модуляции молекулярного пучка с частотой 25 Гц и синхронного счета ионов с минимальным временем накопления 5 с на одну экспериментальную точку, с задаваемым количеством точек в серии. В отсутствие стержня с покрытием при заданном давлении в реакторе и газовых потоках измеряли исходную концентрацию озона. Вводили в зону контакта сегмент стержня (обычно длиной 5 см), одновременно включая режим многократного измерения в течение нескольких сот секунд. Убирали стержень и повторно измеряли уровень концентрации озона. Вновь вводили стержень и повторяли многократный режим измерения до полного прекращения захвата озона. Изменяли исходную концентрацию озона и повторяли всю процедуру измерения при введении следующего сегмента стержня с покрытием. Ошибка измерений ионного тока рассчитывается в автоматическом режиме с использованием программного обеспечения. Дополнительный вклад в ошибку измерений вносят неточности в определении давлений, температуры и потоков; суммарная оценка этого вклада составляет около 5%. Статистические ошибки, приведенные далее в тексте, соответствуют 95%-ному доверительному интервалу.
При наших скоростях потока и давлении в реакторе кинетика расхода озона при его захвате на сажевом покрытии после его ввода в зону контакта описывается уравнением первого порядка:
(1)
${{--d\left[ {{{{\text{O}}}_{3}}\left( {{{t}_{c}},t} \right)} \right]} \mathord{\left/ {\vphantom {{--d\left[ {{{{\text{O}}}_{3}}\left( {{{t}_{c}},t} \right)} \right]} {d{{t}_{c}}}}} \right. \kern-0em} {d{{t}_{c}}}} = {{k}_{w}}\left( t \right)\left[ {{{{\text{O}}}_{3}}\left( {{{t}_{c}},t} \right)} \right],$где [O3] – концентрация озона; tc = [0, ΔL/u] – время контакта озона с покрытием на стержне, введенном в реактор в зону контакта на длину ΔL, u – средняя скорость потока газа-носителя гелия; t – время экспозиции покрытия к потоку газа-реагента. Константа скорости гетерогенной реакции, kw, выражается через кинетический, $k_{w}^{k},$ и диффузионный, $k_{w}^{d},$ пределы:
(2)
$\begin{gathered} {{\text{1}} \mathord{\left/ {\vphantom {{\text{1}} {{{k}_{w}}\left( t \right)}}} \right. \kern-0em} {{{k}_{w}}\left( t \right)}} = {{\text{1}} \mathord{\left/ {\vphantom {{\text{1}} {k_{w}^{k}\left( t \right)}}} \right. \kern-0em} {k_{w}^{k}\left( t \right)}}{\text{ + }}{{\text{1}} \mathord{\left/ {\vphantom {{\text{1}} {k_{w}^{d}}}} \right. \kern-0em} {k_{w}^{d}}}, \\ k_{w}^{k}\left( t \right) = ({{\gamma \left( t \right){{c}_{{{{{\text{O}}}_{{\text{3}}}}}}}} \mathord{\left/ {\vphantom {{\gamma \left( t \right){{c}_{{{{{\text{O}}}_{{\text{3}}}}}}}} 4}} \right. \kern-0em} 4})({{{{S}_{{ef}}}} \mathord{\left/ {\vphantom {{{{S}_{{ef}}}} {{{V}_{R}}}}} \right. \kern-0em} {{{V}_{R}}}}),\,\,\,\,k_{w}^{d} = {{{\text{4}}K(\rho ){{D}_{{{{{\text{О}}}_{{\text{3}}}}}}}} \mathord{\left/ {\vphantom {{{\text{4}}K(\rho ){{D}_{{{{{\text{О}}}_{{\text{3}}}}}}}} {d_{R}^{2}}}} \right. \kern-0em} {d_{R}^{2}}}. \\ \end{gathered} $Здесь ρ = dr/dR; K(ρ = 0.15) = 1.7 [57] – безразмерная константа; ${{c}_{{{{{\text{O}}}_{{\text{3}}}}}}}$ = 3.61(Т/295)1/2 ⋅ 104 см ⋅ с–1 – средняя молекулярная скорость O3 при температуре Т; $p{{D}_{{{{{\text{О}}}_{{\text{3}}}}}}}$ = 394 Торр ⋅ см2 ⋅ с–1 [42] – коэффициент диффузии молекул О3 в гелии; Sef – эффективная площадь покрытия в зоне реакции при выдвижении стержня на длину ΔL; VR – объем реактора, соответствующий этой же длине.
При (dr/dR) $ \ll $ 1 для сажевого покрытия, поверхность которого определяется эффективной площадью Sef, выраженной через массу навески на единичную поверхность ρm и удельную поверхность Sspec, величина $k_{w}^{k}$(t) определяется уравнением
(3)
$k_{w}^{k}\left( t \right) \approx [{{\gamma \left( t \right){{c}_{{{{{\text{О}}}_{{\text{3}}}}}}}{{S}_{{spec}}}{{\rho }_{m}}} \mathord{\left/ {\vphantom {{\gamma \left( t \right){{c}_{{{{{\text{О}}}_{{\text{3}}}}}}}{{S}_{{spec}}}{{\rho }_{m}}} {{{d}_{R}}}}} \right. \kern-0em} {{{d}_{R}}}}]\left( {{{{{d}_{r}}} \mathord{\left/ {\vphantom {{{{d}_{r}}} {{{d}_{R}}}}} \right. \kern-0em} {{{d}_{R}}}}} \right),$где Sspec = 40 м2 ⋅ г–1 – удельная поверхность сажи [58]. В нашем случае даже в начальной стадии быстрого захвата выполнялось условие $k_{w}^{d} \gg k_{w}^{k},$ и можно было считать, что ${{k}_{w}} = k_{w}^{k}.$
Характерное время контакта tc составляет не более 0.1 с, что существенно меньше минимального времени (t = 5 с) интегрирования интенсивности сигнала масс-спектрального пика. На этом основании возможно интегрирование уравнения (1) по tc, и с учетом (3) зависимость коэффициента захвата γ в уравнении (1) от времени экспозиции t при захвате на сажевом покрытии с учетом ВЕТ-поверхности будет иметь вид
(4)
$\gamma {\text{(}}t{\text{)}} = \frac{{{\text{ln}}\left( {{{I_{{{\text{48}}}}^{{\text{0}}}} \mathord{\left/ {\vphantom {{I_{{{\text{48}}}}^{{\text{0}}}} {{{I}_{{48}}}{\text{(}}t{\text{)}}}}} \right. \kern-0em} {{{I}_{{48}}}{\text{(}}t{\text{)}}}}} \right)}}{{{{t}_{c}}}}\frac{{d_{R}^{2}}}{{{{c}_{{{{{\text{O}}}_{{\text{3}}}}}}}{{S}_{{spec}}}{{\rho }_{m}}{{d}_{r}}}},$где $I_{{48}}^{0}(t)$ и I48(t) – интенсивности ионных токов озона без введения стержня с покрытием и с введенным стержнем соответственно.
Времязависимый захват O3на сажевом покрытии. На всех свежих покрытиях из сажи во всех экспериментах наблюдается быстрый начальный захват O3 и последующее его уменьшение практически до нуля. На рис. 1 приведен пример временнóй зависимости коэффициента захвата при нескольких исходных концентрациях озона и комнатной температуре. Каждая из этих зависимостей получена на свежем покрытии. Зависимости получены с использованием формулы (4).
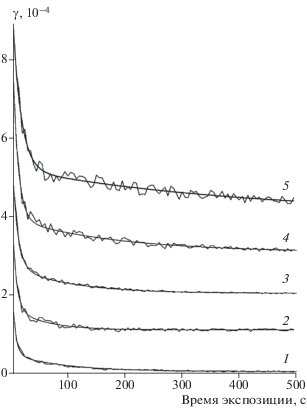
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Экспериментально полученные значения коэффициентов захвата γ(t), приведенные на рис. 1, имеют сложную временнýю зависимость. Такой характер этой зависимости отмечался и в ряде работ, что объяснялось сложным механизмом захвата, при котором последний может происходить через несколько последовательных стадий. Характер зависимости γ от концентрации газа-реагента соответствует двум альтернативным механизмам захвата – обратимой адсорбции с образованием поверхностного комплекса и его мономолекулярным распадом либо гетерогенной бимолекулярной реакции этих комплексов по классическому механизму Лэнгмюра–Хиншельвуда. Сравним результаты описания экспериментальных данных, используя эти два различных механизма.
Кинетическая модель захвата O3на сажевом покрытии, осуществляемого через мономолекулярный распад. Аналитическая зависимость коэффициента захвата от времени экспозиции сажевого покрытия к газу-реагенту и его концентрации можно получить из модифицированной общепринятой лэнгмюровской модели захвата:
(R1)
$\begin{gathered} {{{\text{O}}}_{{\text{3}}}}\left( {\text{г}} \right) + {{{\text{z}}}_{r}}\left( {{\text{тв}}} \right)\underset{{{{k}_{d}}}}{\overset{{{{k}_{a}}}}{\longleftrightarrow}}{{{\text{O}}}_{{\text{3}}}} \ldots {{{\text{z}}}_{r}}, \\ {{{\text{O}}}_{{\text{3}}}} \ldots {{{\text{z}}}_{r}}~\xrightarrow{{{{k}_{{r1}}}}}{{{\text{z}}}_{s}}\left( {{\text{тв}}} \right) + {{{\text{О}}}_{{\text{2}}}}\left( {\text{г}} \right), \\ \end{gathered} $(R2)
$\begin{gathered} {{{\text{O}}}_{{\text{3}}}}\left( {\text{г}} \right) + {{{\text{z}}}_{s}}\left( {{\text{тв}}} \right)\underset{{{{k}_{d}}}}{\overset{{{{k}_{a}}}}{\longleftrightarrow}}{{{\text{O}}}_{{\text{3}}}} \ldots {{{\text{z}}}_{s}}, \\ {{{\text{O}}}_{{\text{3}}}} \ldots {{{\text{z}}}_{s}}\xrightarrow{{{{k}_{{r2}}}}}{{{\text{z}}}_{{fin}}}\left( {{\text{тв}}} \right) + {{{\text{О}}}_{{\text{2}}}}\left( {\text{г}} \right). \\ \end{gathered} $Дополнительно в модель включены две последние стадии. Физически они отражают повторный процесс захвата О3 на менее реакционноспособных центрах zs, которые образуются в результате первичного, быстрого захвата на первичных центрах zr. Повторный, медленный захват также происходит через обратимую адсорбцию О3 на центрах zs с последующим образованием комплекса O3…zs, поверхностная плотность которого составляет θ[zs] [59]. Здесь [zs] – поверхностная плотность центров zs, θ – их доля, занятая адсорбированными молекулами О3. В результате мономолекулярного распада этого комплекса образуется твердый продукт zfin, химически инертный по отношению к озону, но способный к адсорбции.
По этой схеме расход озона определяется согласно уравнению
(5)
$ - {{V}_{R}}\frac{d}{{dt}}{\text{[}}{{{\text{O}}}_{{\text{3}}}}{\text{]}} = \left[ {{{J}_{{rapid}}}(t) + {{J}_{{slow}}}{\text{(}}t{\text{)}}} \right]{{S}_{{ef}}},$где VR и Sef определены в (2). Реакционные потоки
(6)
${{J}_{{rapid}}}\left( t \right) = {{k}_{{r1}}}\theta \left[ {{{{\text{z}}}_{r}}\left( t \right)} \right],\,\,\,\,{{J}_{{slow}}}\left( t \right) = {{k}_{{r2}}}\theta \left[ {{{{\text{z}}}_{s}}\left( t \right)} \right]$зависят от поверхностной плотности центров адсорбции, которая, в свою очередь, определяется системой уравнений
(7)
$ - \frac{d}{{dt}}[{{{\text{z}}}_{r}}] = {{k}_{{r1}}}\theta [{{{\text{z}}}_{r}}],\,\,\,\, - \frac{d}{{dt}}[{{{\text{z}}}_{s}}] = {{k}_{{r2}}}\theta [{{{\text{z}}}_{s}}] - {{k}_{{r1}}}\theta [{{{\text{z}}}_{r}}]$с начальным условием [zr(t = 0)] = [z0] со связью [zr] + [zs] = [z0]. Здесь [z0] – максимальная поверхностная плотность активных центров для данного типа поверхности. Из решения (7) следует явный вид реакционных потоков:
(8)
${{J}_{{slow}}}\left( t \right) = {{k}_{{r2}}}\theta \left[ {{{{\text{z}}}_{0}}} \right]\exp (--{{k}_{{r2}}}\theta t)(1 - \exp (--{{k}_{{r1}}}\theta t).$Из сравнения формального уравнения расхода озона, выраженного через коэффициент захвата,
(9)
$ - {{V}_{R}}\frac{d}{{dt}}[{{{\text{O}}}_{{\text{3}}}}] = \frac{{\gamma (t){{c}_{{{{{\text{O}}}_{{\text{3}}}}}}}}}{{\text{4}}}[{{{\text{O}}}_{{\text{3}}}}]{{S}_{{ef}}},$с уравнением этого же расхода, выраженного через реакционные потоки (5), следует аналитический вид коэффициента захвата:
(10)
$\gamma \left( t \right) = {{\gamma }_{r}}\exp \left( { - {{a}_{r}}t} \right) + {{\gamma }_{s}}\exp \left( { - {{a}_{s}}t} \right)\left[ {1 - \exp \left( { - {{a}_{r}}t} \right)} \right].$Параметры, определяющие эту зависимость, представляют собой комбинацию элементарных констант, описывающих процесс захвата:
(11)
${{\gamma }_{r}} = {{\gamma _{r}^{0}} \mathord{\left/ {\vphantom {{\gamma _{r}^{0}} {\left( {{\text{1}} + {{K}_{L}}\left[ {{{{\text{O}}}_{{\text{3}}}}} \right]} \right)}}} \right. \kern-0em} {\left( {{\text{1}} + {{K}_{L}}\left[ {{{{\text{O}}}_{{\text{3}}}}} \right]} \right)}},\,\,\,\,{{\gamma }_{s}} = {{\gamma _{s}^{0}} \mathord{\left/ {\vphantom {{\gamma _{s}^{0}} {\left( {{\text{1}} + {{K}_{L}}\left[ {{{{\text{O}}}_{{\text{3}}}}} \right]} \right)}}} \right. \kern-0em} {\left( {{\text{1}} + {{K}_{L}}\left[ {{{{\text{O}}}_{{\text{3}}}}} \right]} \right)}},$(12)
$\begin{gathered} \gamma _{r}^{0} = {{{{\alpha }_{s}}{{k}_{{r1}}}} \mathord{\left/ {\vphantom {{{{\alpha }_{s}}{{k}_{{r1}}}} {{{k}_{d}}}}} \right. \kern-0em} {{{k}_{d}}}},\,\,\,\,\gamma _{s}^{0} = {{{{\alpha }_{s}}{{k}_{{r{\text{2}}}}}} \mathord{\left/ {\vphantom {{{{\alpha }_{s}}{{k}_{{r{\text{2}}}}}} {{{k}_{d}}}}} \right. \kern-0em} {{{k}_{d}}}}, \\ {{a}_{r}} = {{k}_{{r{\text{1}}}}}\theta ,\,\,\,\,{{a}_{s}} = {{k}_{{r{\text{2}}}}}\theta , \\ \end{gathered} $(13)
$\theta = {{{{K}_{L}}\left[ {{{{\text{O}}}_{{\text{3}}}}} \right]} \mathord{\left/ {\vphantom {{{{K}_{L}}\left[ {{{{\text{O}}}_{{\text{3}}}}} \right]} {\left( {1 + {{K}_{L}}\left[ {{{{\text{O}}}_{{\text{3}}}}} \right]} \right)}}} \right. \kern-0em} {\left( {1 + {{K}_{L}}\left[ {{{{\text{O}}}_{{\text{3}}}}} \right]} \right)}},\,\,\,\,{{K}_{L}} = {{{{k}_{a}}} \mathord{\left/ {\vphantom {{{{k}_{a}}} {\left( {{{k}_{d}}\left[ {{{{\text{z}}}_{0}}} \right]} \right)}}} \right. \kern-0em} {\left( {{{k}_{d}}\left[ {{{{\text{z}}}_{0}}} \right]} \right)}},$где kr1 и kr2 – константы скорости мономолекулярного распада комплексов О3…zr и O3…zs соответственно; ka = ${{{{\alpha }_{s}}{{c}_{{{{{\text{O}}}_{{\text{3}}}}}}}} \mathord{\left/ {\vphantom {{{{\alpha }_{s}}{{c}_{{{{{\text{O}}}_{{\text{3}}}}}}}} 4}} \right. \kern-0em} 4}$ – константа скорости адсорбции; 0 < αs < 1 – коэффициент поверхностной аккомодации [49, 52, 53, 60, 61]; kd = = νdexp(–Qad/RT) – константа скорости десорбции; KL – константа Лэнгмюра, определяющая долю поверхности, занятую адсорбированными молекулами. Отметим, что при выводе формулы (10) неявно предполагалось, что теплота адсорбции Qad одинакова для центров zr и zs.
Сводные результаты аппроксимации экспериментальных зависимостей γ(t) по формуле (10) при двух температурах реактора и при вариации концентрации озона приведены в табл. 2 и 3. На рис. 2 приведен пример такой аппроксимации для одной из зависимостей γ(t) с соответствующими параметрами из табл. 3. Все полученные зависимости параметров аппроксимации от концентрации озона представлены на рис. 3–6.
Таблица 2.
Параметры аппроксимации по формуле (10) коэффициента захвата γ(t) озона на покрытии из метановой сажи при Т = 295 К
| [O3], 1012 см–3 | γr, 10–4 | ar, с–1 | γs, 10–5 | as, 10–2 с–1 |
|---|---|---|---|---|
| 1.1 | 6.2 ± 2 | 0.07 ± 0.03 | 11.7 ± 2.5 | 0.25 ± 0.15 |
| 1.36 | 6 ± 1 | 0.04 ± 0.02 | 9.3 ± 3 | 0.69 ± 0.24 |
| 1.4 | 6.6 ± 2 | 0.03 ± 0.02 | 9 ± 2 | 0.9 ± 0.3 |
| 1.57 | 6 ± 2.5 | 0.04 ± 0.02 | 10.2 ± 4.2 | 0.42 ± 0.22 |
| 2.17 | 6 ± 2 | 0.06 ± 0.05 | 12.8 ± 8 | 0.52 ± 0.3 |
| 2.55 | 6.9 ± 2 | 0.11 ± 0.01 | 8.7 ± 2.5 | 0.55 ± 0.15 |
| 3.46 | 6.1 ± 2 | 0.10 ± 0.04 | 10.8 ± 2.2 | 0.93 ± 0.47 |
| 5.4 | 4.8 ± 2.6 | 0.12 ± 0.03 | 9.4 ± 4.9 | 1.2 ± 0.4 |
| 6.1 | 4.3 ± 1 | 0.12 ± 0.02 | 6.9 ± 1 | 1 ± 0.3 |
| 6.5 | 3.9 ± 1 | 0.12 ± 0.02 | 8 ± 1 | 0.69 ± 0.3 |
| 7.1 | 4.9 ± 2.2 | 0.15 ± 0.03 | 8.5 ± 2.8 | 1.66 ± 0.5 |
| 9 | 4 ± 2.4 | 0.15 ± 0.02 | 6 ± 1.6 | 1.8 ± 0.5 |
| 10 | 4.4 ± 1.8 | 0.18 ± 0.04 | 7.7 ± 1.8 | 1.4 ± 0.3 |
| 11.2 | 4.2 ± 1 | 0.15 ± 0.02 | 5.8 ± 1.2 | 1.8 ± 0.2 |
| 11.3 | 3.9 ± 1.2 | 0.19 ± 0.04 | 7.3 ± 1 | 1.5 ± 0.2 |
| 12 | 3 ± 1 | 0.17 ± 0.04 | 9.6 ± 3 | 1.7 ± 0.3 |
| 13.3 | 2.8 ± 1 | 0.18 ± 0.05 | 6.8 ± 1 | 1.8 ± 0.5 |
| 15.6 | 3.3 ± 0.8 | 0.21 ± 0.03 | 5.8 ± 1.5 | 1.8 ± 0.4 |
| 15.7 | 2.6 ± 0.7 | 0.19 ± 0.02 | 5 ± 2 | 1.2 ± 0.3 |
| 15.8 | 3 ± 0.3 | 0.17 ± 0.05 | 4.5 ± 0.5 | 1.1 ± 0.2 |
| 20 | 2.9 ± 0.5 | 0.21 ± 0.05 | 4.8 ± 0.6 | 1.76 ± 0.2 |
| 24 | 2.8 ± 0.6 | 0.24 ± 0.02 | 3.7 ± 1 | 2.2 ± 0.4 |
| 26 | 2.3 ± 0.9 | 0.22 ± 0.02 | 2.8 ± 1 | 1.8 ± 0.3 |
| 27 | 2.5 ± 0.4 | 0.24 ± 0.03 | 3.5 ± 0.8 | 1.54 ± 0.3 |
Таблица 3.
Параметры аппроксимации по формуле (10) коэффициента захвата γ(t) озона на саже при Т = 256 К
| [O3], 1012 см–3 | γr, 10–4 | ar, с–1 | γs, 10–5 | as, 10–2 с–1 |
|---|---|---|---|---|
| 1.7 | 6.2 ± 2.3 | 0.05 ± 0.02 | 10.3 ± 2.5 | 0.43 ± 0.11 |
| 3.9 | 4.6 ± 2 | 0.08 ± 0.02 | 7.4 ± 1.3 | 0.69 ± 0.09 |
| 5.5 | 4 ± 1 | 0.1 ± 0.03 | 5.6 ± 0.8 | 0.64 ± 0.15 |
| 6.2 | 3.2 ± 1.3 | 0.11 ± 0.02 | 3.8 ± 0.5 | 0.74 ± 0.12 |
| 8.3 | 3.7 ± 1.3 | 0.1 ± 0.02 | 3 ± 0.5 | 0.62 ± 0.13 |
| 10 | 2.1 ± 0.8 | 0.12 ± 0.01 | 3 ± 0.7 | 0.95 ± 0.18 |
| 12.3 | 2.5 ± 1.2 | 0.10 ± 0.02 | 2.4 ± 0.5 | 0.7 ± 0.17 |
| 14.5 | 1.6 ± 0.6 | 0.13 ± 0.02 | 3.2 ±0.5 | 0.69 ± 0.2 |
| 16.2 | 2.4 ± 0.9 | 0.11 ± 0.02 | 2.5 ± 0.4 | 0.8 ± 0.15 |
| 20 | 1.3 ± 0.4 | 0.11 ± 0.03 | 1.4 ± 0.4 | 0.85 ± 0.13 |
| 23.6 | 1.1 ± 0.3 | 0.12 ± 0.01 | 2 ± 0.4 | 0.85 ± 0.2 |
| 25 | 1.1 ± 0.3 | 0.11 ± 0.02 | 1.6 ± 0.8 | 0.87 ± 0.15 |
| 31 | 1.2 ± 0.4 | 0.14 ± 0.02 | 1.3 ± 0.2 | 0.77 ± 0.17 |
| 35.5 | 0.9 ± 0.3 | 0.12 ± 0.01 | 1.1 ± 0.3 | 0.96 ± 0.2 |
| 41 | 0.8 ± 0.3 | 0.12 ± 0.01 | 1 ± 0.2 | 0.8 ± 0.3 |
Рис. 2.
Вклад быстрой и медленной стадий в суммарный времязависимый захват O3 на свежем покрытии из метановой сажи при T = 256 K и [O3] = 3.9 ⋅ 1012 см–3: кривая с “шумом” – исходные данные, штриховые кривые – аппроксимация по формуле (10) с параметрами из табл. 4.
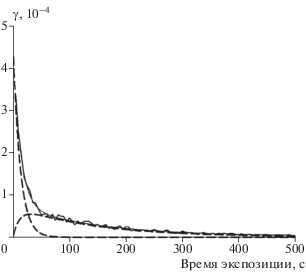
Рис. 3.
Зависимость параметра γr быстрой стадии времязависимого захвата O3 на свежей метановой саже от [O3]: темные символы – данные из табл. 2 при Т = 295 К, светлые символы – данные из табл. 3 при Т = 256 К; сплошные прямые линии – аппроксимация по формуле (11) по механизму мономолекулярного распада с параметрами из табл. 4, штриховая кривая – аппроксимация по формуле (14) захвата по механизму Лэнгмюра–Хиншельвуда с аналитическим продолжением (точечная кривая) при малых концентрациях.
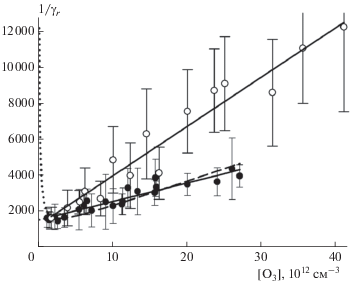
Рис. 4.
Зависимость параметра ar быстрой стадии времязависимого захвата O3 на свежей метановой саже от [O3]: темные символы – данные из табл. 2 при Т = 295 К, светлые символы – данные из табл. 3 при Т = 256 К; сплошные кривые – аппроксимация по формуле (12) по механизму мономолекулярного распада с параметрами из табл. 4.
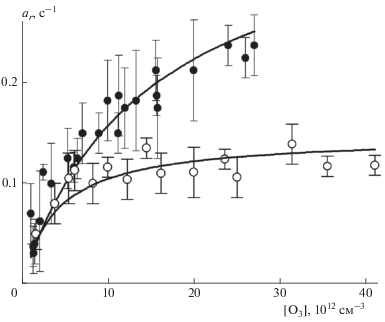
Рис. 5.
Зависимость параметра γs медленной стадии времязависимого захвата O3 на покрытиях из свежей метановой сажи от [O3]: темные символы – данные из табл. 2 при Т = 295 К, светлые символы – данные из табл. 3 при Т = 256 К; сплошные прямые линии – аппроксимация по формуле (11) по механизму мономолекулярного распада с параметрами из табл. 4.
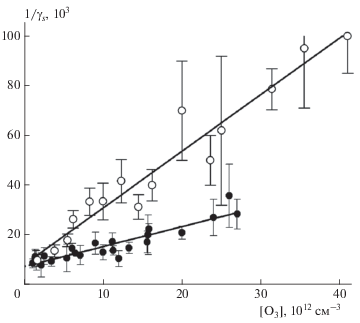
Рис. 6.
Зависимость параметра as медленной стадии времязависимого захвата O3 на свежей метановой саже от [O3]: темные символы – данные из табл. 2 при Т = 295 К, светлые символы – данные из табл. 3 при Т = 256 К; сплошные кривые – аппроксимация по формуле (12) по механизму мономолекулярного распада с параметрами из табл. 4.
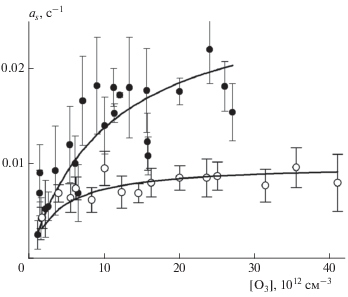
Исходную плотность [z0] активных центров можно оценить, предполагая гибель одного активного центра при необратимом захвате одной молекулы О3:
Усредненное значение поверхностной плотности было найдено путем интегрирования аналитического выражения (10) по всей совокупности данных из табл. 2 и 3. Оно оказывается одинаковым при температурах реактора 256 и 295 К и составляет (2 ± 0.2) ⋅ 1014 см–2 для первой, быстрой стадии и (3.7 ± 0.5) ⋅ 1014 см–2 для повторного захвата. Значение суммы этих плотностей, равное 5.7 ⋅ 1014 см–2, согласуется с аналогичными данными по захвату озона на ряде сажевых покрытий: электроискровая сажа – 6.5 ⋅ 1014 см–2 [34], электроискровая сажа с покрытием бензо[a]пиреном – (5.7 ± 1.7) ⋅ ⋅ 1014 см–2 [41], сажа из толуола – 7 ⋅ 1014 см–2 [39], сажа из н-гексана – (4 ± 2) ⋅ 1014 см–2 [38], керосиновая сажа – (5.8 ± 0.7) ⋅ 1014 см–2 [40]. С другой стороны, поверхностные плотности на аналогичных покрытиях, вычисленные по захвату других газов-реагентов, оказываются существенно ниже и согласуются с нашими данными по быстрой стадии захвата озона: при захвате NO2 на метановой саже [z0] = (2 ± 0.4) ⋅ 1014 см–2 [58], при адсорбции HNO3 на электроискровой саже [z0] = 2.2 ⋅ 1014 см–2 [22], а на н-гексановой саже [z0] = 2 ⋅ 1014 см–2 [62]. Противоречивость данных можно устранить, если допустить, что вторая, медленная стадия захвата озона происходит дважды на одних и тех же активных центрах с поверхностной плотностью 2 ⋅ 1014 см–2. Физически это означает, что в результате мономолекулярного распада комплекса O3…zs по реакции (R2) и образования твердого продукта zfin последний остается химически активным, и на нем повторно происходит захват молекулы О3, образование комплекса и его последующий распад.
Исходя из вида зависимости коэффициента захвата от концентрации газа-реагента, представленного формулой (11), из линейной регрессии обратных величин γr и γs (рис. 3 и рис. 5) находим константу Лэнгмюра KL, а также по точке пересечения аналитической прямой с осью ординат − параметры $\gamma _{r}^{0}$ и $\gamma _{s}^{0}.$ В терминах формулы (12) эти параметры представляют собой отношение αskr/kd для обеих стадий захвата. Из значения KL при [z0] = 2 ⋅ 1014 см–2 по формуле (13) находим величину kd/αs. Конструируем функцию θ([O3]) при известной константе KL и аппроксимируем экспериментальные данные ar и as (рис. 4 и 6) аналитической зависимостью (12) с единственным подгоночным параметром kr. Сводные результаты расчета приведены в табл. 4. В последнем столбце этой таблицы приведено отношение αskr/kd, которое должно быть идентично параметрам $\gamma _{r}^{0}$ и $\gamma _{s}^{0}$ исходя из их определения выражением (12). Из данных табл. 4 следует, что для быстрой стадии захвата это выполняется. Для медленной стадии параметр $\gamma _{s}^{0}$ в 2 раза больше отношения αskr2/kd. Это превышение согласуется с нашим представлением о повторном захвате О3 дважды на одних и тех же центрах. В предположении одинаковых значений скоростей реакции (R2) и последующей за ней это удвоение можно выразить аналитически в виде дополнительного члена в выражении (10):
Таблица 4.
Механизм мономолекулярного распада. Параметры аппроксимации по формулам (11), (12) коэффициента времязависимого захвата О3 на свежем сажевом покрытии
| Первая, быстрая стадия | |||||
|---|---|---|---|---|---|
| T, K | KL, 10–12 см3 | $\gamma _{r}^{0},$ 10–4 | kd/αs, c–1 | kr1, c–1 | αskr1/kd, 10–4 |
| 295 | 0.07 ± 0.01 | 6.6 ± 0.4 | 654 ± 115 | 0.39 ± 0.01 | 6 ± 1 |
| 256 | 0.22 ± 0.09 | 8.1 ± 2.6 | 188 ± 80 | 0.15 ± 0.01 | 8.0 ± 3.4 |
| Вторая, медленная стадия | |||||
| T, K | KL, 10–12 см3 | $\gamma _{s}^{0},$ 10–4 | kd/αs, c–1 | kr2, 10–2 c–1 | αskr2/kd, 10–4 |
| 295 | 0.11 ± 0.02 | 1.3 ± 0.2 | 410 ± 85 | 2.8 ± 0.14 | 0.68 ± 0.14 |
| 256 | 0.28 ± 0.13 | 1.2 ± 0.5 | 150 ± 70 | 0.99 ± 0.04 | 0.67 ± 0.31 |
Временнáя зависимость этого вклада показана на рис. 7. Очевидно, что количественные данные о медленной стадии, приведенные в табл. 4, носят оценочный характер, поскольку реальная временнáя зависимость оказывается более сложной.
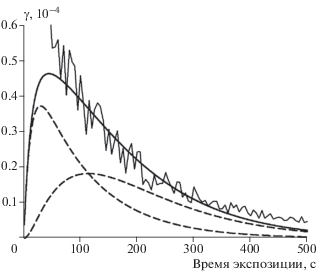
Рис. 7.
Аппроксимация по формуле (10) медленной стадии захвата из рис. 2 с учетом повторного вторичного захвата: кривая с “шумом” – исходные данные, штриховые кривые – вклады вторичного и третичного захватов, сплошная кривая – их сумма.
В предположении аррениусовской зависимости констант скорости десорбции kd = νdexp(–Qad/RT) и мономолекулярного распада kr = Arexp(–Ea/RT) из отношения kr(295 K)/kr(256 K) и kd(295 K)/ kd(256 K) следует оценка теплоты адсорбции Qad и энергии активации Ea обеих стадий захвата. Сводные данные приведены в табл. 5. Из этих данных можно сделать вывод, что теплота адсорбции слабо зависит от химического состава поверхности. Энергия активации обеих стадий захвата практически одинакова, поэтому отличие в 15 раз констант скорости первичного и вторичного захватов может быть обусловлено только различием в предэкспонентах этих констант.
Таблица 5.
Механизм мономолекулярного распада. Теплоты адсорбции О3 на сажевом покрытии и энергии активации константы скорости
| Стадия | Qad, кДж/моль | Ea, кДж/моль |
|---|---|---|
| Быстрая | 20 ± 7 | 15.6 ± 0.6 |
| Медленная | 16 ± 8 | 16.7 ± 0.6 |
В рамках обсуждаемого механизма захвата при малых концентрациях озона, т.е. при KL[O3] $ \ll $ 1, коэффициент захвата определяется отношением констант αskr/kd. Зависимость от температуры этих констант практически одинакова, поэтому в этом диапазоне концентраций озона коэффициент захвата должен слабо зависеть от концентрации и температуры. Этот вывод наглядно демонстрируют рис. 3 и 5, а также величины отношения параметров $\gamma _{r}^{0}$(295 K)/$\gamma _{r}^{0}$(256 K) и $\gamma _{s}^{0}$(295 K)/$\gamma _{s}^{0}$ (256 K) из табл. 4.
Захват по механизму Лэнгмюра–Хиншельвуда. В классическом представлении этого механизма, как и в рассмотренном выше, в результате обратимой адсорбции образуются комплексы O3…zr с поверхностной плотностью θ[zr(t)]. В результате бимолекулярной гетерогенной реакции между этими комплексами образуются конечный твердый продукт zs (тв) и свободный кислород. При этом из одной израсходованной молекулы О3 высвобождается одна молекула О2 [31]:
(R3)
$\begin{gathered} {{{\text{O}}}_{{\text{3}}}}\left( {\text{г}} \right) + {{{\text{z}}}_{r}}\left( {{\text{тв}}} \right)\,\,\underset{{{{k}_{d}}}}{\overset{{{{k}_{a}}}}{\longleftrightarrow}}{\text{ }}{{{\text{O}}}_{{\text{3}}}} \ldots {{{\text{z}}}_{r}}, \\ {{{\text{O}}}_{{\text{3}}}} \ldots {{{\text{z}}}_{{\text{r}}}}{\text{ }} + {\text{ }}{{{\text{O}}}_{{\text{3}}}} \ldots {{{\text{z}}}_{{\text{r}}}}{\text{ }}\xrightarrow{{{{k}_{r}}}}~{{{\text{z}}}_{s}}\left( {{\text{тв}}} \right) + {\text{2}}{{{\text{О}}}_{{\text{2}}}}\left( {\text{г}} \right). \\ \end{gathered} $Расход озона определяется реакционным потоком
Из представления γ через реакционный поток аналогично сделанному выше (уравнения (5) и (8)), мы получаем амплитудную величину γa коэффициента захвата, выраженную через элементарные параметры:
(14)
${{\gamma }_{a}} = \gamma _{a}^{0}\frac{{K_{L}^{2}[{{{\text{O}}}_{3}}]}}{{{{{(1 + {{K}_{L}}[{{{\text{O}}}_{3}}])}}^{2}}}}.$Здесь $\gamma _{a}^{0}$ = ${{{\text{4}}{{k}_{r}}{{{\left[ {{{{\text{z}}}_{0}}} \right]}}^{2}}} \mathord{\left/ {\vphantom {{{\text{4}}{{k}_{r}}{{{\left[ {{{{\text{z}}}_{0}}} \right]}}^{2}}} {{{c}_{{{{{\text{O}}}_{3}}}}}}}} \right. \kern-0em} {{{c}_{{{{{\text{O}}}_{3}}}}}}}.$ Все остальные параметры определены выше (см. формулы (11)–(13)). Аппроксимация экспериментальных данных по зависимости амплитудной величины γa от [O3] по формуле (14) с параметрами $\gamma _{a}^{0}$ и KL из табл. 6 приведена на рис. 3 штриховой линией. Точечной кривой на этом же рисунке показано аналитическое продолжение в область малых концентраций. Из отношений параметров $\gamma _{a}^{0}$(295 К)/$\gamma _{a}^{0}$(256 К) и KL(295 К)/KL(256 К), приведенных в табл. 6 в предположении аррениусовской зависимости констант скорости десорбции kd и константы скорости бимолекулярной реакции kr оценены теплота адсорбции Qad и энергия активации Ea.
Таблица 6.
Механизм Лэнгмюра–Хиншельвуда. Параметры аппроксимации по формуле (14) амплитудного коэффициента захвата О3 на свежем сажевом покрытии
| T, K | KL, 10–12 см3 | $\gamma _{r}^{0},$ 109 см–3 | Qad, кДж/моль | Ea, кДж/моль |
|---|---|---|---|---|
| 295 | 0.35 ± 0.03 | 7.1 ± 0.3 | 9.4 ± 5.0 | 12.4 ± 0.8 |
| 256 | 0.59 ± 0.19 | 3.53 ± 0.07 |
Из сравнения описания данных на рис. 3 двумя альтернативными механизмами захвата видно, что в диапазоне наших рабочих концентраций озона невозможно отдать предпочтение какому-либо из них. Аналитические зависимости принципиально различаются только в пределе малых концентраций. Для механизма Лэнгмюра–Хиншельвуда такими концентрациями является [O3] $ \ll $ 1/KL, т.е. $ \ll $2.9 ⋅ 1012 см–3 при Т = 295 К и $ \ll $1.7 ⋅ 1012 см–3 при Т = 256 К. В этом диапазоне концентраций амплитудная величина γa при механизме мономолекулярного распада остается постоянной и равной $\gamma _{r}^{0}$ и $\gamma _{s}^{0}$ из табл. 4, а при механизме Лэнгмюра–Хиншельвуда она становится прямо пропорциональной концентрации О3.
В литературе есть две работы, согласно которым можно сделать однозначный выбор в пользу одного из механизмов. Амплитудная величина γa захвата озона на свежей метановой саже в пересчете на ВЕТ-поверхность, измеренная в работе [36] при [O3] = 3 ⋅ 1010 см–3 и Т = 298 К, составляет ∼1 ⋅ 10–3, что полностью согласуется с нашими данными по $\gamma _{r}^{0}$ из табл. 4 для механизма мономолекулярного распада. В работе [42] исследовался захват озона на ряде органических мономолекулярных покрытий при Т = 220–298 К. В работе [42] отмечается, что амплитудная величина γa ≈ 2 ⋅ 10–4 не зависит от концентрации озона в диапазоне 1 ⋅ ⋅ 109–1 ⋅ 1011 см–3, что также соответствует представлению о механизме мономолекулярного распада.
ЗАКЛЮЧЕНИЕ
Практически во всех работах по захвату озона на органических покрытиях анализировался канал гетерогенного стока озона в тропосфере и был сделан вывод о его малом влиянии. С другой стороны, озон является только одним из многих других малых газовых составляющих, для которых этот канал может быть и определяющим. Для оценки вклада этого канала в условиях конкурентной адсорбции необходимо знать изотермы адсорбции конкурирующих газов-реагентов и их температурные зависимости. Лэнгмюровская константа KL и теплота адсорбции Qad, которые определяют температурную зависимость изотермы адсорбции, зависят от механизма, на базе которого аппроксимируются экспериментальные данные. Примером служит сравнение этих параметров из табл. 4–6.
Механизм мономолекулярного распада, который в литературе ошибочно называют механизмом Лэнгмюра–Хиншельвуда, является, по-видимому, общим для описания захвата озона на органических покрытиях. Такой вывод сделан в работе McCabe и Abbatt [38], в которой проанализированы данные по захвату озона на ряде поверхностей, включая как органические, так и неорганические.
Включение в схему захвата последующей, медленной стадии, которая протекает на тех же центрах адсорбции, позволяет объяснить сложную временнýю зависимость коэффициента захвата. В частности, становится понятной разница в плотности центров адсорбции на одних и тех же покрытиях, измеренной при захвате разных адсорбатов. Из аналитического описания временнóй зависимости γ(t) получена оценка константы скорости мономолекулярного распада, которая в пределах статистической ошибки совпадает с тем же параметром, полученным из зависимости амплитудной величины γa от концентрации озона. Малая разница между теплотой адсорбции и энергией активации константы скорости в совокупности с видом зависимости γ = f([O3]), представленным формулой (11), объясняет, почему в ряде работ по захвату озона на органических покрытиях не наблюдалась выраженная зависимость коэффициента захвата от температуры.
Работа выполнена по программе фундаментальных исследований РАН (регистрационный номер АААА-А18-118112290069-6).
Список литературы
Zhang L., Lin M., Langford A.O. et al. // Atmos. Chem. Phys. 2020. V. 20. P. 10379; https://doi.org/10.5194/acp-20-10379-2020
Kelley M., Schmidt G.A., Nazarenko L.S. et al. // J. Adv. Modeling Earth Systems 2020. V. 12. P. e2019MS002025; https://doi.org/10.1029/2019MS002025
Smith D.M., Chughtai A.R. // J. Geophys. Res. 1996. V. 101D. P. 19607;https://doi.org/10.1029/95JD03032
Blanchard C.L., Shaw S.L., Edgerton E.S., Schwab J.J. // Atmos. Environ. X3 (2019) 100033; https://doi.org/10.1016/j.aeaoa.2019.100033
Gore C., Chiao S. // Atmos. Environ. X7 (2020) 1009085; https://doi.org/10.1016/j.aeaoa.2019.100085
Hembeck L., He H., Vinciguerra T.P., Canty T.P., Dicker-son R.R. et al. // Atmos. Environ. X2 (2019) 100017; https://doi.org/10.1016/j.aeaoa.2019.100017
Moeini O., Tarasick D.W., McElroy C.T., Liu J., Osman M.K. et al. // Atmos. Environ. X7 (2020) 100078; https://doi.org/10.1016/j.aeaoa.2020.100078
Stadler D., Rossi M.J. // Phys. Chem. Chem. Phys. 2000. V. 2. P. 5420; https://doi.org/10.1039/b005680o
Cohen J.B., Wang C. // J. Geophys. Res. Atmos. 2014. V. 119. P. 307; https://doi.org/10.1002/2013JD019912
Öztürk F., Bahreini R., Wagner N.L. et al. // J. Geophys. Res. Atmos. 2013. V. 118. P. 13591; https://doi.org/10.1002/2013JD019923
Kostenidou E., Florou K., Kaltsonoudis C. et al. // Atmos. Chem. Phys. 2015. V. 15. P. 11355; https://doi.org/10.5194/acp-15-11355-2015
Akhter M.S., Chughtai A.R., Smith D.M. // Appl. Spectrosc. 1985. V. 39. P. 143.
Salgado M.S., Rossi M.J. // Intern. J. Chem. Kin. 2002. V. 34. P. 620; https://doi.org/10.1002/kin.10091
Knopf D.A., Wang B., Laskin A. et al. // Geophys. Res. Lett. 2010. V. 37. P. L11803; https://doi.org/10.1029/2010GL043362
Bond T.C., Doherty S.J., Fahey D.W. et al. // J. Geophys. Res. Atmos. 2013. V. 118. P. 5380; https://doi.org/10.1002/jgrd.50171
Laborde M., Crippa M., Tritscher T. et al. // Atmos. Chem. Phys. 2013. V. 13. P. 5831; https://doi.org/10.5194/acp-13-5831-2013
Decesari S., Facchini M.C., Matta E. et al. // Atmos. Environ. 2002. V. 36. P. 1827.
He X., Pang S., Ma J., Zhang Y. // Atmos. Environ. 2017. V. 165. P. 198; https://doi.org/10.1016/j.atmosenv.2017.06.049
Schurath U., Naumann K.-H. // Pure and Appl. Chem. 1998. V. 70. P. 1353.
Arens F., Gutzwiller L., Baltensperger U., Gaggeler H.W., Ammann M. // Environ. Sci. Technol. 2001. V. 35. P. 2191; https://doi.org/10.1021/es000207s
Rudich Y., Donahue N.M., Mentel T.F. // Annu. Rev. Phys. Chem. 2007. V. 58. P. 321; https://doi.org/10.1146/annurev.physchem.58.032806. 104432
Kirchner U., Scheer V., Vogt R. // J. Phys. Chem. A. 2000. V. 104. P. 8908; https://doi.org/10.1021/jp0005322
Li Q., Shang J., Zhu T. // Atmos. Environ. 2013. V. 81. P. 68; https://doi.org/10.1016/j.atmosenv.2013.08.043
Setyan A., Sauvain J.-J., Guillemin M. et al. // Chem. Phys. Chem. 2010. V. 11. P. 3823; https://doi.org/10.1002/cphc.201000490
Chughtai A.R., Jassim J.A., Peterson J.H., Stedman D.H., Smith D.M. // Aer. Sci. Technol. 1991. V. 15. P. 112; https://doi.org/10.1080/027868291089859518
Daly H.M., Horn A.B. // Phys. Chem. Chem. Phys. 2009. V. 11. P. 1069; https://doi.org/10.1039/b815400g
Liu Y., Liu C., Ma J., Ma Q., He H. // Ibid. 2010. V. 12. P. 10896; https://doi.org/10.1039/c0cp00402b
Smith D.M., Akhter M.S., Jassin J.A. et al. // Aer. Sci. Technol. 1989. V. 10. P. 311; https://doi.org/10.1080/02786828908959267
Smith D.M., Chughtai A.R. // J. Atmos. Chem. 1997. V. 26. P. 77.
Zhu J., Shang J., Chen Y., Kuang Y., Zhu T. // Environ. Sci. Technol. 2020. V. 54. P. 8558; https://doi.org/10.1021/acs.est.0c01150
Stephens S., Rossi M.J., Golden D.M. // Intern. J. Chem. Kin. 1986. V. 18. P. 1133; https://doi.org/10.1002/kin.550181004
Disselkamp R.S., Carpenter M.A., Cowin J.P. et al. // J. Geophys. Res. 2000. V. 105D. P. 9767.
Fendel W., Matter D., Burtscher H., Schmidt-Ott A. // Atmos. Environ. 1995. V. 29. P. 967.
Kamm S., Möhler O., Naumann K-H. Saathoff H., Schurath U. // Ibid. 1999. V. 33 P. 4651.
Stephens S.L., Birks J.W., Calvert J.G. // Aer. Sci. Technol. 1989. V. 10. P. 326; https://doi.org/10.1080/02786828908959268
Longfellow C.A., Ravishankara A.R., Hanson D.R. // J. Geophys. Res. 2000. V. 105D. P. 24345;https://doi.org/0148-0227/00/2000JD900297
Chughtai A.R., Kim J.M., Smith D.M. // J. Atmos. Chem. 2003. V. 45. P. 231.
McCabe J., Abbatt J.P.D. // J. Phys. Chem. C. 2009. V. 111. P. 2120; https://doi.org/10.1021/jp806771q
Lelievre S., Bedjanian Yu., Pouvesle N. et al. // Phys. Chem. Chem. Phys. 2004. V. 6. P. 1181; https://doi.org/10.1039/b316895f
Bedjanian Yu., Nguyen M.L. // Chemosphere. 2010. V. 79. P. 387; https://doi.org/10.1016/j.chemosphere.2010.02.009
Pöschl U., Letzel T., Schauer C., Niessner R. // J. Phys. Chem. A. 2001. V. 105. P. 4029; https://doi.org/10.1021/jp004137n
Moise T., Rudich Y. // J. Geophys. Res. 2000. V. 105D. P. 14667;https://doi.org/0148-0227/00/2000JD900071
Kahan T.F., Kwamena N.-O.A., Donaldson D.L. // Atmos. Environ. 2006. V. 40. P. 3448; https://doi.org/10.1016/j.atmosenv.2006.02.004
Perraudin E., Budzinski H., Villenave E. // J. Atmos. Chem. 2007. V. 56. P. 57; https://doi.org/10.1007/s10874-006-9042-x
Kwamena N.-O.A., Thornton J.A., Abbatt J.P.D. // J. Phys. Chem. A. 2004. V. 108. P. 11626; https://doi.org/10.1021/jp046161x
Kwamena N.-O.A., Earp M.E., Young C.J., Abbatt J.P.D. // J. Phys. Chem. A. 2006. V. 110. P. 3638; https://doi.org/10.1021/jp056125d
Kwamena N.-O.A., Staikova M.G., Donaldson D.J., George I.J., Abbatt J.P.D. // Ibid. 2007. V. 111. P. 11050; https://doi.org/10.1021/jp075300i
Kolb C.E., Cox R.A., Abbatt J.P.D. et al. // Atmos. Chem. Phys. 2010. V. 10. P. 10561; https://doi.org/10.5194/acp-10-10561-2010
Ammann M., Pöschl U. // Ibid. 2007. V. 7. P. 6025; www.atmos-chem-phys.net/7/6025/2007/
Masel R.I. // Principles of Adsorption and Reaction on Solid Surfaces 1st ed. N.Y.: John Wiley and Sons, 1996.
Wei C.-F., Larson S.M., Patten K.O., Wuebbles D.J. // Atmos. Environ. 2001. V. 35. P. 6167.
Ammann M., Pöschl U., Rudich Y. // Phys. Chem. Chem. Phys. 2003. V. 5. P. 351; https://doi.org/10.1039/b208708a
Pöschl U., Rudich Y., Ammann M. // Atmos. Chem. Phys. 2007. V. 7. P. 5989; www.atmos-chem-phys.net/7/5989/2007/
Berkemeier T., Ammann M., Krieger U.K. et al. // Ibid. 2017. V. 17. P. 8021; https://doi.org/10.5194/acp-17-8021-2017
Зеленов В.В., Апарина Е.В., Козловский В.И., Сулименков И.В., Носырев А.Е. // Хим. физика. 2018. Т. 37. № 3. С. 72; https://doi.org/10.7868/S0207401X18030111
Karagulian F., Rossi M.J. // J. Phys. Chem. A. 2007. V. 111. P. 1914; https://doi.org/10.1021/jp0670891
Gershenzon Yu.M., Grigorieva V.M., Ivanov A.V., Remo-rov R.G. // Farad. Discuss. 1995. V. 100. P. 83.
Зеленов В.В., Апарина Е.В., Каштанов С.А., Шардакова Э.В. // Хим. физика. 2015. Т. 34. № 3. С. 87; https://doi.org/10.7868/S0207401X15030140
Laidler K.J. // Chemical kinetics, 2nd ed. N.Y.: McGraw-Hill, 1965.
Berkemeier T., Huisman A.J., Ammann M. et al. // Atmos. Chem. Phys. 2013. V. 13. P. 6663; https://doi.org/10.5194/acp-13-6663-2013
Nathanson G.M., Davidovits P., Worsnop D.R., Kolb C.E. // J. Phys. Chem. 1996. V. 100. P. 13007.
Aubin D.G., Abbatt J.P. // J. Phys. Chem. A. 2003. V. 107. P. 11030; https://doi.org/10.1021/jp036105g
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика