

Химическая физика, 2021, T. 40, № 6, стр. 54-65
Кинетика разложения 1,1-диамино-2,2-динитроэтилена (FOX-7). 1. Природа первой стадии разложения
Г. М. Назин 1, В. В. Дубихин 1, Т. К. Гончаров 1, А. И. Казаков 1, *, А. В. Набатова 1, А. В. Шастин 1
1 Институт проблем химической физики Российской академии наук
Черноголовка, Россия
* E-mail: akazakov@icp.ac.ru
Поступила в редакцию 27.12.2019
После доработки 15.06.2020
Принята к публикации 20.07.2020
Аннотация
Методами дифференциальной сканирующей калориметрии и манометрии измерена скорость разложения 1,1-диамино-2,2-динитроэтилена (FOX-7) при 200 °С в широком диапазоне изменения условий проведения опыта и способов подготовки образца. Установлена природа первой стадии разложения и найдена причина остановки реакции на этой стадии.
ВВЕДЕНИЕ
Экспериментальное исследование термического разложения 1,1-диамино-2,2-динитроэтилена FOX-7 (I) проводили в основном методом ДСК в потоке инертного газа (см. обзор [1]). При этом было обнаружено, что разложение протекает в две стадии, которым на ДСК-кривых соответствуют два сильно различающихся по температуре (220 и 270 °С) пика тепловыделения. В работе [2] было доказано, что появление двух пиков не связано с какими-либо примесными реакциями или с образованием продуктов, которые могли бы распадаться при повышенных температурах. Ранее такой “двойной” характер реакции при разложении взрывчатых веществ никогда не наблюдался, даже в случае полифункциональных соединений, имеющих сложное строение [3–6]. В самой работе [2] первый пик (в котором выделяется 60% тепла) был объяснен наличием в кристаллах вещества I аморфного вещества, имеющего большую скорость разложения. Однако в более ранней работе [7] было показано, что в закрытых сосудах остановка реакции зависит от отношения массы навески m к объему сосуда V и может наблюдаться на малых глубинах превращения. Сам эффект торможения объяснен в [7] обратимостью первой стадии разложения, которая заключается в расщеплении I на HNO3 и соответствующий нитрилоксид. Можно отметить, однако, что сама возможность присоединения HNO3 к нитрилоксидам, тем более в парах и при высоких температурах, не доказана. В работе [8] обнаружен новый эффект: после кратковременного (~20 мин) прогрева при температуре около 200 °С в потоке аргона кристаллы соединения I приобретают новые свойства. После их охлаждения до комнатной температуры и последующего нагревания не наблюдается, как обычно, фазовых переходов α → β и β → γ, а первый пик разложения отсутствует. В [8] предположено, что во время предварительного нагревания происходит полиморфный переход с образованием новой, δ-модификации, термически- и фазово-устойчивой при температурах от комнатной до 270 °С. В этой же работе было установлено, что вещество I летуче и даже при атмосферном давлении легко возгоняется при 200 °С с подложки микроскопа, а нелетучий остаток (предполагаемая δ‑модификация) меняет цвет. Изменение цвета может быть следствием химического превращения. Однако возможная связь сублимации, реакции разложения и изменения свойства кристаллов в [8] не обсуждается, хотя именно в этой работе была впервые обнаружена одновременность этих явлений. Отметим, что еще до появления работы [8] было показано [2], что точно такие же свойства, т.е. исчезновение первого пика тепловыделения при 220 °С и отсутствие перехода в α-модификацию при охлаждении, имеет продукт, образовавшийся после 3 сут прогрева I в потоке аргона при 185 °С (40% потери веса), и структура этого продукта соответствует γ-фазе.
Никаких тепловых эффектов, которые должны сопровождать фазовый переход, в работе [8] не было замечено. Таким образом, предположение о фазовом переходе γ → δ не имеет подтверждений. Но даже если допустить, что δ-модификация существует, она не может быть причиной остановки распада. Невозможно объяснить, почему высокотемпературная модификация, образование которой идет с поглощением тепла, т.е. с увеличением теплосодержания вещества, становится, судя по разности температур появления пиков, в десятки, если не в сотни, раз более стабильной, чем предшествующая γ-фаза. Непонятно также, почему соединение, сильно летучее при 200 °С, после полиморфного перехода полностью теряет способность к возгонке даже при нагревании до 270 °С.
В то же время такие неоспоримые факты, как высокая летучесть I, подтвержденная в обстоятельной работе [9], и неизбежная быстрая реакция разложения вещества в газовой фазе, требуют более детального рассмотрения как вероятные причины появления первой стадии. Поэтому неудивительно, что в работе [10] в качестве одного из возможных объяснений первой стадии высказана следующая гипотеза: “Разложение I протекает главным образом в газовой фазе. Причиной уменьшения скорости термического разложения является образование плотной пленки на внешней поверхности кристаллов в результате каких-то вторичных реакций с участием продуктов газофазного распада. Пленка препятствует сублимации”. Однако в этой работе данная гипотеза была отклонена, и предпочтение отдано фазовому переходу.
В связи с малой вероятностью предложенных в литературе объяснений первой стадии в данной работе предпринято дальнейшее изучение разложения вещества I с целью проверки прежде всего гипотезы о самотормозящемся распаде в парах как причине первой стадии. В отличие от большинства других работ исследование проведено не в открытых системах в потоке инертного газа, а в закрытых сосудах и в основном в изотермических условиях. По сравнению с изотермическими исследованиями, выполненными в работах [2, 7], время наблюдения за реакцией было увеличено в десятки и сотни раз, что дало возможность изучить обе стадии при одной температуре. Такие методические изменения позволили получить новую информацию о разложении соединения I и определить в конечном итоге природу первой стадии разложения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез соединения I проведен по методике, описанной в работе [11]. Использовались два образца – “мелкий” с размером частиц ~0.1 мм и “крупный” с частицами ~1 мм. В обоих случаях перекристаллизация проводилась из ацетонитрила.
Калориметрические измерения в сканирующем режиме сделаны на разработанном в ИПХФ РАН приборе, который позволяет проводить точные измерения тепловыделения в запаянных ампулах объемом ~2.0 cм3 при скорости нагревания от 1 до 5 град/мин.
Манометрические измерения сделаны с помощью установки, рассчитанной на работу при высоких давлениях. Стеклянная ампула (0.5–5.0 см3), снабженная серповидной мембраной объемом 0.3 см3 со стрелкой, помещалась в разъемную металлическую капсулу с окном для наблюдения за отклонением стрелки и крышкой, которая с помощью медных трубок и вентилей соединялась с насосом, манометром и баллоном со сжатым воздухом. Давление в ампуле измеряли компенсационным методом.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В соответствии с поставленной задачей после описания наблюдаемых фактов или явлений сразу же приводится анализ их связи с газофазной или твердофазной реакциями, а также обсуждается их соответствие с механизмом разложения вещества I и общими закономерностями реакций в газовой и твердой фазах.
Исследование методом дифференциальной сканирующей калориметрии (ДСК)
Hа ДСК-кривых разложения в запаянных ампулах наблюдаются, как и при разложении в потоке инертного газа, два пика тепловыделения (рис. 1). Однако между данными, полученными в открытых и закрытых условиях, имеются два существенных различия. Пики тепловыделения в запаянной ампуле сильно сближены главным образом из-за смещения второго пика в сторону низких температур. Максимумы тепловыделения первого и второго пиков лежат в запаянной ампуле соответственно при 228 и 250 °С вместо 220 и 270 °С, как в открытых системах. Этот факт можно объяснить тем, что распад I во второй стадии является автокаталитическим процессом, скорость которого зависит от давления газообразных продуктов. Кроме того, в замкнутом объеме в первой стадии разложения выделяется не более 2% от общего тепла, вместо 40–60% в потоке.
Рис. 1.
ДСК-кривые разложения соединения I в запаянных ампулах. Скорость подъема температуры – 1 град/мин, объем ампул – 2.5 см3. Навеска: 1 – 7.65 мг, 2 – 4.2 мг. Тепловыделение в первом и втором пиках (мДж): 1 – Q1 = 250, Q2 = 28 340; 2 – Q1 = 314, Q2 = 16 780.
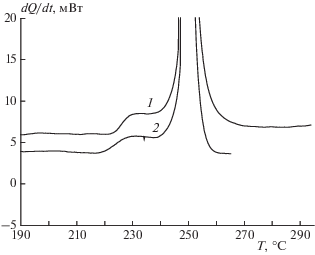
В закрытых сосудах наблюдается также, что тепловыделение в первой стадии почти не зависит от массы взятого вещества. Этот результат означает, что первая стадия не связана с какой-либо реакцией, идущей с участием твердого вещества. Следовательно, реакция идет в газовой фазе.
Второе наблюдение, показывающее газофазный характер реакции в первой стадии, касается изменения цвета образца. При нагревании пробирки, содержащей 200 мг вещества I, до 200 °С при атмосферном давлении визуально наблюдается быстрое окрашивание торцовой поверхности образца в бежевый цвет. Окраска медленно распространяется в глубь и вдоль стенок пробирки. Нижние слои вещества остаются желтыми длительное время. Такого рода наблюдения проведены многократно при описанном ниже изучении термораспада в закрытых сосудах. При небольших (до 5 мг) навесках, когда кристаллы лежат на дне сосуда тонким слоем, все они теряют желтую окраску сразу же после прогрева до 200 °С. При растирании таких потемневших кристаллов образуется порошок желтого цвета. Это означает, что изменение окраски затрагивает только тонкий внешний слой кристалла. В образцах с массой 20–30 мг, прогретых в сосуде объемом 5 cм3 при 200 °С в течение 6 ч, всегда остается часть кристаллов, которые не меняют свой первоначальный цвет.
Окрашивание только поверхности кристалла и только внешней поверхности образца свидетельствует о протекании химической реакции, но не в твердой фазе, а в парах. Для объяснения остановки этой реакции можно допустить, как предположено в работе [10], что при разложении паров образуются труднолетучие соединения, которые осаждаются на поверхности кристаллов в виде пленки. Эта пленка модифицирует поверхность кристалла и препятствует сублимации. Вышедшие в объем сосуда пары быстро разлагаются, и реакция прекращается.
Образованием пленки может быть объяснена не только остановка реакции, но и вторая особенность прогретого образца, послужившая основанием для выдвижения в [8] гипотезы о полиморфном переходе γ → δ. Эта особенность заключается, как уже отмечалось, в отсутствии быстрого перехода γ → α при охлаждении прогретого образца до комнатной температуры. Фазовый переход требует больших флуктуаций свободного объема для освобождения поступательного движения молекул. Поэтому он начинается обычно на крупных дефектах, лежащих на внешней поверхности кристалла. Закрывая поверхность, пленка дезактивирует ее активные участки и тормозит самую медленную стадию фазового перехода – образование зародышей новой фазы, задерживая, по крайней мере на некоторое время, фазовый переход. Возможно, поэтому гипотетическая высокотемпературная δ-модификация есть просто γ-форма, покрытая пленкой продуктов.
Образование защитной пленки является уникальным свойством вещества I, связанным с особенностями его строения и механизмом разложения. Чтобы понять, как образуется пленка, необходимо рассмотреть механизм реакции.
Механизм реакции
Наиболее вероятным механизмом разложения соединения I является последовательность элементарных процессов
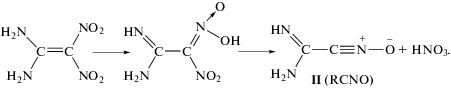
Согласно квантовохимическим расчетам [12], из пяти возможных первичных стадий разложения соединения I (рис. 2) путь d – изомеризация в аци-форму (нитроновую кислоту) через 1,5-сигматропный сдвиг водорода имеет самую низкую энергию активации. Эта закономерность сохранятся у всех непредельных мононитросоединений, имеющих в цис-положении к нитрогруппе водородсодержащие заместители, такие как CH3, OH, NH2. Правда, в случае вещества I еще одна первичная реакция, а именно внутримолекулярное окисление с образованием структуры типа оксазет (путь c), имеет такую же энергию активации, как и реакция d.
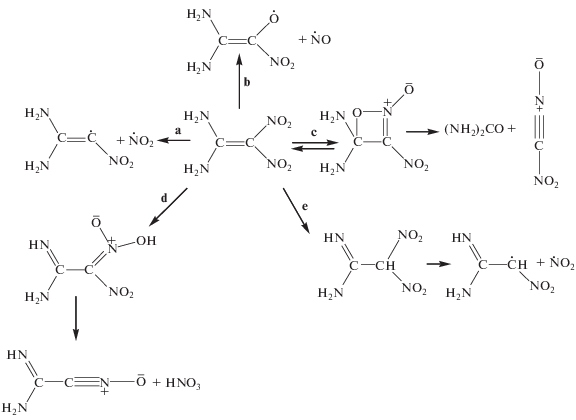
Получающаяся из I нитроновая кислота в газовой фазе отличается высокой стабильностью. В работе [12] не найдено ни одного пути превращения нитроновой кислоты с энергией активации меньшей, чем у первой стадии. Поэтому в этой работе предпочтение отдано разложению через переходное состояние типа оксазет. Однако в работе [13] показано, что эта реакция также не может идти, так как является полностью обратимой. Энергия активации обратного процесса составляет всего 10.5 кДж/моль против 125.5 кДж/моль для прямой реакции. Поэтому в [13] был предложен другой механизм первой стадии – структурная енамино-иминовая перегруппировка (путь e), идущая с энергией активации около 200 кДж/моль. Несмотря на высокое значение последней, этот путь распада считается наиболее вероятным. По-видимому, для гомогенной реакции в газовой фазе этот вывод является правильным. Однако следует учесть, что многие вещества в жидкой фазе разлагаются по другому механизму и значительно легче, чем в паровой фазе.
Из литературных данных (краткие обзоры см. в [3, 14]) известно, что нитроновые кислоты в конденсированном состоянии являются сравнительно неустойчивыми соединениями. Нитроновые кислоты ароматического и гетероциклического типов, выделенные в свободном состоянии, разлагаются при температуре 100 °С. Из-за возможности быстрой изомеризации в аци-форму скорость разложения жидкого нитрометана на 5 и более порядков выше, чем в паровой фазе [15].
При большой разнице скоростей реакций в жидкой и газовой фазах распад паров, как правило, идет гетерогенно, в слоях, адсорбированных на стенках реакционного сосуда. В этом случае скорость реакции, пропорциональная количеству адсорбата, будет зависеть от давления паров, адсорбционной способности поверхности и ее размеров.
Таким образом, учитывая большую вероятность быстрого гетерогенного распада нитроновых кислот, можно принять, что разложение соединения I идет как в паровой фазе, так и в твердом состоянии по реакции (1).
Отметим, что впервые механизм разложения (1) был предложен в экспериментальной работе [7], в которой на его основе были объяснены закономерности горения I. В этой же работе представлен анализ масс-спектрометрических исследований вещества I, выполненных в работе [16], и показано, что нитрилоксид II является одним из первичных продуктов фрагментации I при электронном ударе.
Основываясь на приведенных в работе [17] сведениях о превращении нитрилоксидов, можно представить вторичные процессы, идущие после реакции (1). При высоких температурах для нитрилоксидов характерна мономолекулярная изомеризация в изоцианат:
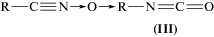
В присутствии аминосоединений изоцианаты быстро присоединяются к аминогруппам с образованием производных карбамида. Поэтому между соединениями II и III возможна реакция
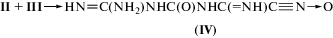
с образованием более тяжелого соединения IV, способного к тому же к дальнейшим превращениям с участием нитрилоксидной группы. Кроме изомеризации (2), при высоких температурах возможно отщепление атома кислорода с образованием нитрила:
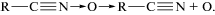
В присутствии нитрила образуется оксадиазол:
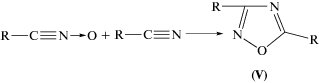
Скорость реакции (4) намного выше, чем реакций (2) и (3), поэтому может оказаться, что соединение V будет одним из продуктов разложения.
Соединения типа IV и V, имея малую летучесть, будут осаждаться на поверхность кристаллов I, возможно, закрепляясь на ней за счет образования водородных связей. Эти соединения содержат сопряженные связи C=N, поэтому образованная ими пленка имеет окраску. Отметим, что соединение с сопряженными связями C=N было обнаружено в работе [10] методом ИК-спектроскопии в конечных продуктах разложения соединения I при 215 °С. Таким образом, в рамках механизма (1) образование пленки находит естественное объяснение.
Следует отметить, что пленка, модифицируя поверхность, препятствует только сублимации паров. Продукты разложения, так же как и газы, десорбирующиеся из внутренних частей кристалла, могут свободно проходить сквозь пленку через микропоры, которые образуются под давлением этих газов.
Труднолетучие соединения, осаждающиеся на поверхность, образуются, по-видимому, в небольшой концентрации. Основными продуктами разложения в паровой фазе являются СO2, HCN, N2O, NO2, HOCN и NO [1], возникающие в результате окисления самого соединения I и продуктов его распада диоксидом азота. Последний образуется из HNO3 по реакции
Константа скорости разложения 100%-ной НNO3 в 109 раз больше константы скорости гомогенной реакции в газовой фазе. По этой причине разложение паров НNO3 протекает, подобно нитроновым кислотам, гетерогенно на стенках реакционного сосуда [3]. По аналогии с окислением аминов кислородом можно предположить, что реакция с NO2 идет по атому азота, и скорость ее тем выше, чем больше основность амина. По этой причине продукты распада будут реагировать с NO2 быстрее, чем с I.
Описанный механизм образования пленки позволяет объяснить и предвидеть некоторые явления, которые наблюдались или могут наблюдаться в эксперименте. Сублимация вещества идет с поверхности кристаллов, независимо от того, где они расположены – сверху или внутри насыпного образца. Однако свободный объем между кристаллами внутри навески слишком мал, и нелетучих продуктов, образующихся при разложении паров (с небольшим, как отмечено выше, выходом), недостаточно для образования пленки. Дефицит тяжелых продуктов может быть причиной того, что кристаллы, находящиеся внутри насыпного образца, не покрываются пленкой продуктов и не меняют свой первоначальный желтый цвет длительное время.
Эти кристаллы будут постоянным источником паров вещества. Однако выход паров в объем сосуда может быть сильно затруднен. Течение паров в узких проходах между кристаллами должно идти по законам капиллярной диффузии, и скорость этого процесса будет зависеть, прежде всего, от давления паров Рупр, давления газов в сосуде, а также от массы и формы образца.
При конденсация нелетучих продуктов из газовой фазы на поверхность кристаллов выделяется большое количество тепла. Поэтому возможен местный разогрев поверхности. Следствием разогрева могут быть самые разные явления, в частности он будет способствовать увеличению скорости таких процессов, как сублимация, распад в газовой фазе и образование пленки.
Манометрические измерения
При манометрических измерениях была замечена высокая адсорбционная способность соединения I, которая проявляется в сильном удерживании кристаллами атмосферных газов и воды. При нагревании сосуда, откачанного в стандартных условиях (15 мин при комнатной температуре), до температуры опыта эти газы создают начальное давление Р0, которое фиксируется в момент прогрева. Величина Р0 плохо воспроизводится и слабо зависит от m/V. Так, в случае нагревания до 200 °С при m/V = 10–3 г/см3 величина Р0 изменяется в пределах 50–100 Торр, а при m/V = 0.10 г/cм3 она обычно лежит в интервале 150–300 Торр. Предварительное откачивание при 110 °С в течение 15 мин мало влияет на результат. При нагревании до 180–200 °С вместе с примесными газами в сосуд выходят пары́ самого вещества. При охлаждении они конденсируются одновременно с водой и адсорбцией других газов, поэтому точно определить упругость паров из манометрических измерений невозможно. С уверенностью можно только сказать, что при 200 °С значение Рупр не превышает 50 Торр. После прогрева в вакууме при температуре 180–200 °С в течение 10–20 мин наблюдается потеря веса вещества, но Р0, т.е. десорбция адсорбированных газов, может даже увеличиться. По-видимому, полиморфные переходы способствуют десорбции примесей из внутренних слоев кристалла. Таким образом, разложение соединения I вынужденно проводилось в присутствии небольшого количества воздуха и паров воды. Во избежание перекрещивания кривых разложения в дальнейшем на рисунках начальное значение Р0 не приводится.
Разложение при температуре 200 °С
Температура, равная 200 °С, является оптимальной для изучения распада соединения I в изотермических условиях. При этой температуре можно снять полную кинетическую кривую с хорошим разделением первой и второй стадий и минимальным воздействием вторичных процессов, которые приводят к увеличению скорости разложения твердого вещества.
На рис. 3 в координатах Vg – t (Vg – приведенный к нормальным условиям объем газообразных продуктов, выделившихся из 1 г вещества) показаны кинетические кривые полного разложения, полученные при навесках 2–3 мг и m/V ∼ 10–3 г/см3. Кривая 1 получена при стандартных условиях подготовки опыта. В самом начале наблюдается быстрое газовыделение, которое идет с уменьшающейся скоростью и прекращается через несколько часов. Этот участок соответствует первому пику тепловыделения в условиях линейного нагрева. Глубина превращения на первом участке составляет 20% от общего газовыделения (кривая 1 рис. 2) и уменьшается при увеличении m/V (рис. 4), как это и должно быть в случае распада определенной порции вещества, вышедшего в газовую фазу.
Рис. 3.
Кинетические кривые полного разложения соединения I при 200 °С: 1 – исходный образец, m/V = = 7.2 ⋅ 10–4 г/cм3; 2 – образец, прогретый в вакууме 0.1 Торр в течение 20 мин при 200 °С, m/V = 8.2 ⋅ ⋅ 10–4 г/cм3.
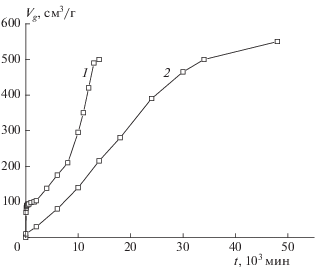
Рис. 4.
Начальные участки кинетических кривых разложения соединения I при 200 °С и следующих значениях m/V (г/см3): 1 – 7.2 ⋅ 10–4, 2 – 3.0 ⋅ 10–2, 3 – 8.2 ⋅ 10–4 (после прогрева в вакууме 0.1 Торр в течение 20 мин при 200 °С), 4 – 0.16.
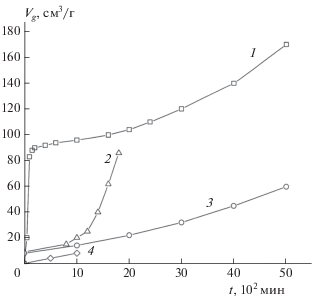
Затем следует ускоряющийся процесс с максимумом скорости, сдвинутым к концу реакции. Эта стадия соответствует второму пику тепловыделения на ДСК-кривых. На начальном этапе второй стадии (1–2% разложения) скорость не зависит от m/V в интервале значений m/V (1÷30) ⋅ 10–3 г/см3. Такая независимость характерна для твердофазного процесса. Небольшое увеличение наклона линейного участка с самого начала наблюдается только при m/V = 0.16 г/см3. По-видимому, при таких больших значениях m/V ускорение твердофазного распада начинается на очень ранней стадии реакции. На этапе ускорения скорость зависит от отношения m/V, возрастая при его увеличении (рис. 4). Это означает, что ускорение вызвано действием газообразных продуктов.
Другой вид имеет кривая 2 рис. 3, полученная после предварительного прогрева образца в вакууме при 200 °С в течение 20 мин. После такого прогрева ампулу с веществом охлаждали до комнатной температуры, запаивали под вакуумом и затем нагревали до температуры опыта. Первая стадия разложения на этой кривой практически отсутствует (выход продуктов на этой стадии уменьшается примерно в 10 раз), что соответствует исчезновению первого пика на ДСК-кривых разложения соединения I в потоке аргона после предварительного прогрева в течение 20 мин при температуре около 200 °С [8]. Результат не изменяется, если время прогрева в вакууме сократить до 5 мин или повторить цикл охлаждение–нагревание под вакуумом.
При нагревании в вакууме пленка образоваться не может. После предварительного прогрева наблюдается потеря веса вещества, но кристаллы остаются желтыми. Значит, пленка образуется в то время, когда охлажденный и запаянный под вакуумом сосуд нагревается до температуры опыта.
Возникает вопрос, почему предварительный прогрев в вакууме подавляет первую стадию или по крайней мере снижает ее глубину. За время прогрева кроме частичной сублимации происходит удаление летучих примесей с поверхности образца. Можно допустить, что адсорбированные примеси (воздух и особенно вода) препятствуют контакту конденсирующихся продуктов с поверхностью кристаллов и мешают формированию пленки. Поэтому очистка поверхности от адсорбированных газов облегчает образование пленки, которое может произойти еще до достижения температуры опыта и установления соответствующей ей упругости паров.
Как видно из рис. 3, прирост давления во времени у прогретого образца выражен слабее, чем у исходного. Явной причиной этого является отсутствие первой стадии распада у прогретого образца. Возможно, что за время первой стадии, которая наблюдается только у непрогретого образца, под действием окислительных газов происходит частичное разложение твердого вещества и накапливаются продукты, повышающие дефектность кристаллов, а значит, и скорость разложения.
Необходимо отметить, что эффективное подавление газофазной реакции с помощью предварительного прогрева в вакууме наблюдается только при небольших значениях m и m/V. Поэтому в общем случае предварительный прогрев нельзя использовать для устранения первой стадии.
Из анализа кривой 2 рис. 3 следует, что полное газовыделение ∆Vg ∞ во второй стадии разложения, т.е. при твердофазном распаде, составляет 500 см3/г или 3.3 моля на моль I. Это значение ∆Vg ∞ является приближенным, так как в опытах с навеской вещества 2–3 мг точность определения m/V не превышала 20%.
При температуре 200 °С было проведено испытание влияние размера частиц на кинетику разложения соединения I. Кривая 1 рис. 3 воспроизводится и на образце с размером частиц ~1 мм. Только стадия ускорения в этом случае выражена несколько слабее. Образцы, растертые в ступке для уменьшения размеров частиц, при нагревании до 200 °С дают вспышку при m ~ 20 мг. Воспламенение происходит в паровой фазе. Все вещество сгорает, и продукты осаждаются на стенках сосуда в виде черной пыли. Вспышки не наблюдается, если навеска измельченных образцов не превышает 5 мг. В этом случае в течение первых суток реакция идет в соответствии с кривой 2 рис. 3, т.е. имеет такую же слабовыраженную первую стадию, как у прогретого в вакууме образца. Поскольку вспышка и, наоборот, практически полное отсутствие реакции в газовой фазе связаны с измельчением кристаллов и массой образца, можно предположить, что причина этих явлений – разогрев поверхности кристаллов при конденсации твердых продуктов газофазного распада. Когда кристаллов много и общая их поверхность велика, тепла от конденсации достаточно, чтобы стимулировать сублимацию и быстро прогреть пары до температуры вспышки. Когда же навеска мала, эффект ограничивается только повышением давления паров и их невзрывным разложением, заканчивающимся быстрым образованием пленки.
У образцов, взятых до или после растирания, первая стадия вообще выражена слабо, если проводить разложение при атмосферном давлении воздуха. Простое объяснение этого факта заключается в том, что из-за уменьшения коэффициента диффузии в воздухе пары́ соединения I за время образования пленки успевают заполнить сосуд лишь частично, до давления, много меньшего Рупр.
Влияние перемешивания
Как было упомянуто выше, гипотеза об образовании защитной пленки допускает прямую проверку: реакция, замедлившаяся в конце первой стадии, должна возобновиться после растирания кристаллов или после перемешивания образца, достаточно представительного по массе. Ввиду отмеченного выше неоднозначного поведения измельченных кристаллов использован второй способ – перемешивание образца с m ~ 20 мг. Чтобы провести полный цикл исследования и не попасть в область ускорения, которое при m/V = = 0.01 г/см3 наступает уже через 1000 мин, реакцию останавливали не после полного завершения первой стадии (~300 мин), а раньше, через 150 мин. Перемешивание проводили простым встряхиванием охлажденного сосуда. Результаты показаны на рис. 5. После первого и второго перемешиваний наблюдается воспроизведение первой стадии, но с несколько уменьшенным газовыделением. После двух перемешиваний остаток вещества был растерт в ступке и загружен в такой же сосуд в количестве 6 мг. Разложение прошло за такое же время, как при первом нагревании, но с незначительным газовыделением. Аналогичный результат, т.е. малый прирост давления, наблюдался ранее для малых навесок растертого образца.
Рис. 5.
Влияние перемешивания образца после первой стадии распада на ход реакции разложения соединения I при 200 °С. Навеска m = 20 мг, m/V = 0.01 г/см3. Участки: а – начальная стадия; b – после первого встряхивания; с – после второго встряхивания; d – после растирания, m/V = 0.003 г/см3.
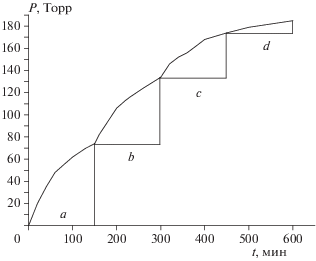
Таким образом, можно с большим основанием считать, что первая стадия – это газофазный распад I.
Кинетический анализ первой и второй стадий
Из рис. 3 видно, что при 200 °С и m/V ∼ 10–3 г/см3 первая и вторая стадии хорошо разделяются, что делает возможным проведение независимого кинетического анализа каждой из них. Цель такого анализа заключалась в сравнении скоростей реакций в газовой и твердой фазах и, самое главное, в выяснении возможности определения константы скорости k1 первой некаталитической стадии разложения в твердом состоянии. Можно полагать, что ниже 100 °С, т.е. в реальных условиях применения взрывчатых веществ, первая стадия исчезает и термическая стабильность I всецело будет определяться величиной k1.
На рис. 6 кривые 1 и 2, взятые из рис. 3, представлены в исходных координатах “давление–время”. В этих координатах удобно проанализировать первую стадию распада, представив ее в увеличенном масштабе. Соответствующий первой стадии распада начальный участок кривой 2 рис. 6 показан (наряду с другими данными) в увеличенном масштабе на рис. 7 (кривая 5). Эта кривая строго описывается уравнением 1-го порядка с периодом полураспада τ1/2 = 40 мин, что соответствует константе скорости kгаз = 2.9 ⋅ 10–4 c–1. Прирост давления в первой стадии (∆Р1) составляет 85 Торр. Близкие значения kгаз и ∆Р1 получаются при распаде образца с крупным размером частиц и образца, очищенного путем возгонки в вакууме (кривые 4 и 6 рис. 7). Строгий 1-й порядок реакции свидетельствует о том, что поступления паров вещества в сосуд во время разложения не происходит. Это согласуется со сделанным ранее выводом, что пленка отсекает пары уже к моменту окончания прогрева. Постоянство ∆Р1 на кривых 4–6 свидетельствует об одинаковом начальном давлении паров. Скорее всего, сосуды заполняются парáми до давления, равного Рупр. По величине ∆Р1 можно сделать приблизительную оценку Рупр при 200 °С. Если предположить, что стехиометрический коэффициент по газовыделению, f, при разложении паров равен трем, то Рупр ≈ 42 Торр, если f = 4, то Рупр ≈ 28 Торр.
Рис. 6.
Разложение соединения I при 200 °С. Полные кинетические кривые 2 и 4 в координатах Р–t, соответствующие кривым 1 и 2 рис. 3, и вторые участки этих кривых (1 и 3) в координатах η–t.
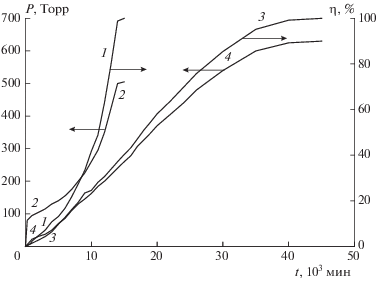
Рис. 7.
Первая стадия разложения соединения I при 200 °С и следующих значениях m/V (г/см3) и условиях предварительного прогрева (°С/мин): 1 – 0.1, без прогрева (б/п); 2 – 0.03, б/п; 3 – 0.01, б/п; 4(△) – 0.005 (кристаллы размером 0.1 мм), б/п; 5(⚪) – 0.00072, б/п; 6(◻) – 0.0023 (возгнанный образец), б/п; 7(▷) – 0.0075, 180/30; 8(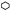 ) – 0.00082, 200/20.5; 9(☆) – 0.001 (давление воздуха – 1 атм), б/п.
) – 0.00082, 200/20.5; 9(☆) – 0.001 (давление воздуха – 1 атм), б/п.
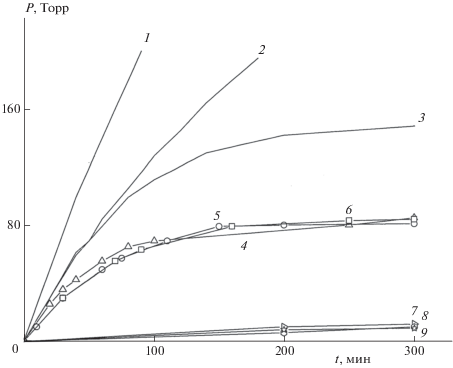
Из рис. 7 видно, что при увеличении m/V 1-й порядок начальной стадии искажается. Происходит это из-за вклада от твердофазного процесса и увеличения длительности первой стадии вследствие диффузии паров из внутренних слоев образца. Как отмечалось выше, внутри образца кристаллы покрываются пленкой не так быстро, как снаружи. Уже при m/V = 0.01 г/см3 точное определение kгаз становится невозможным.
У образцов, прогретых в вакууме при повышенных температурах (кривые 7 и 8 рис. 7), первая стадия разложения выражена слабо. Величина ∆Р1 не превышает 10 Торр. То же самое наблюдается, если разложение проведено при давлении воздуха, равном 1 атм. В обоих случаях, как уже отмечалось, пленка образуется раньше, чем пары заполняют объем сосуда до давления Рупр.
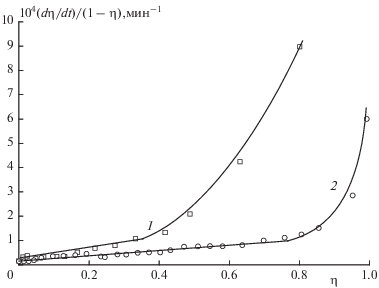
Для рассмотрения второй стадии разложения нужно сначала отделить ее от первой стадии. С этой целью из кривых 2 и 4 рис. 6 вычитаем прирост давления ∆Р1, полученный в первой стадии. Значения ∆Р1 (85 и 10 Торр для кривых 2 и 4 соответственно) найдены путем экстраполяции к t = 0 линейных участков кривых 2 и 4 рис. 6, описывающих реакцию после окончания первой стадии. Полученные после вычитания ∆Р1 кривые отражают изменение давления Ртв за счет разложения в твердой фазе. В координатах η–t (η = Ртв/Ртв ∞) они приведены на рис. 6. Кривые 1 и 3 рис. 6 получены соответственно в опытах без предварительного прогрева и с прогревом. Они практически сливаются в самом начале, когда доминирует некаталитическая реакция. Затем, когда начинается ускорение процесса, эти кривые расходятся. Максимум скорости при твердофазном распаде сдвинут к концу реакции. Кривые не описываются каким-либо простым уравнением. Однако и при неизвестном законе скорости значение k1 может быть определено при наличии полной кинетической кривой из зависимости удельной скорости Wуд = (dη/dt)/(1 – η) от η путем экстраполяции Wуд к значению η = 0.
На рис. 8 показана зависимость Wуд от η для кривых 1 и 3 рис. 6. Эта зависимость имеет линейный характер до η = 40% в случае исходного образца и до η = 80% у прогретого образца. Затем происходит резкое увеличение Wуд. Наличие линейных участков на кривых зависимости Wуд(η) позволяет провести надежную экстраполяцию и найти константу скорости начальной стадии k1. Для исходного образца k1 = 2.6 ⋅ 10–7 с–1. У прогретого образца она имеет почти такое же значение. Сравнение констант скорости в газовой и твердой фазах дает значение отношения kгаз/kтв ≈ 1000.
Рис. 8.
Зависимость удельной скорости от глубины разложения η для второй стадии разложения соединения I при 200 °С и следующих значениях m/V (г/см3): 1 – 0.00072 (исходный образец), 2 – 0.0008 (образец, прогретый в вакууме 0.1 Торр в течение 20 мин при 200 °С). Кривые 1 и 2 соответствуют кривым 1 и 3 на рис. 6.
Таким образом, концепция протекания реакции в газовой фазе позволяет объяснить многообразие фактов, обнаруженных при изучении первой стадии: реально наблюдаемое образование пленки; торможение первой стадии; независимость приращения давления продуктов в этой стадии от размеров кристаллов, способа получения и очистки образца при стандартном начале опыта; резкое уменьшение газовыделения в первой стадии после предварительного прогрева в вакууме или при атмосферном давлении воздуха; вспышка при измельчении; возобновление реакции после перемешивания образца; очень большая разница в скоростях первой и второй стадий.
ЗАКЛЮЧЕНИЕ
Разложение I в закрытых сосудах протекает в две стадии, так же как и в потоке инертного газа. Первая стадия – это сопровождающий твердофазную реакцию побочный процесс, включающий сублимацию и распад вещества в газовой фазе. Упругость паров I при 200 °С составляет несколько десятков Торр, а скорость разложения в газовой фазе примерно в тысячу раз выше, чем в твердой. Особенностью первой стадии является ее способность к самоторможению. При разложении в парáх образуются нелетучие соединения, которые осаждаются на поверхности кристаллов в виде плотной пленки. Пленка, препятствуя сублимации, прерывает источник побочной реакции. При 200 °С найдены условия, при которых первая и вторая стадии хорошо разделяются и могут быть проанализированы независимо.
Авторы выражают искреннюю благодарность В.Г. Прокудину за исследование разложения FOX-7 методом ДСК в закрытых сосудах.
Работа выполнена на средства ИПХФ РАН по теме 0089-2019-0005 “Фундаментальные и проблемно-ориентированные исследования в области создания энергетических конденсированных систем (ЭКС) различного назначения” (регистрационный номер АААА-А19-119100800130-0) при финансовой поддержке программой Президиума РАН “Перспективные физико-химические технологии специального назначения”.
Список литературы
Bellamy A.J. // Struct Bond. 2007. V. 125. P. 1;https://doi.org/10.1007/430_2006_054
Ticmanis U., Kaiser M., Pantel G. et al. // Proc. 35th Intern. Annual Conf. ICT. Karlsruhe: ICT, 2004. P. 70/1.
Манелис Г.Б., Назин Г.М., Рубцов Ю.И. и др. Термическое разложение и горение взрывчатых веществ и порохов. М.: Наука, 1996.
Неделько В.В., Захаров В.В., Корсунский Б.Л. и др. // Хим. физика. 2013. Т. 32. № 3. С. 25; https://doi.org/10.7868/S0207401X13030060
Неделько В.В., Захаров В.В., Корсунский Б.Л. и др. // Хим. физика. 2015. Т. 34. № 12. С. 39; https://doi.org/10.1134/S1990793115060172
Захаров В.В., Чуканов Н.В. Зюзин И.Н. и др. // Хим. физика. 2019. Т. 38. № 2. С. 3; https://doi.org/10.1134/S0207401X19020146
Sinditskii V.P., Levshenkov A.I., Egorshev V.Yu. et al. // Proc. 30th Intern. Pyrotechnics Sem. Saint-Malo, 2003. V. 1. P. 299.
Garmasheva N.V., Chemagina I.V., Filin V.P. et al. // Proc. 36th Intern. Annual Conf. ICT. Karlsruhe: ICT, 2005. P. 87/1.
Волкова Н.Н, Шастин А.В., Алиев З.Г. и др. // Успехи в специальной химии и химической технологии. Тр. Всерос. науч.-техн. конф., посвященной 80-летию основания Инженерного химико-технологического факультета РХТУ им. Д.И. Менделеева. М.: РХТУ им. Д.И. Менделеева, 2015. С. 142.
Zakharov V.V., Chukanov N.V., Dremova N.N. et al. // Propellants Explos. Pyrotech. 2016. V. 41. P. 1006; https://doi.org/10.1002/prep.201600071
Астратьев А.И. Дашко Д.В., Мершин А.Ю. и др. // Журн. орган. химии. 2001.Т. 37. № 5. С. 766; https://doi.org/10.1023/A:1012568305472
Храпковский Г.М., Шамов А.Г., Николаева Е.В. и др. // Успехи химии. 2009. Т. 78. № 10. С. 980; https://doi.org/10.1070/RC2009v078n10ABEH004053
Kiselev V.G., Gritsan N.P. // J. Phys. Chem. A. 2014. V. 118. № 36. P. 8002;https://doi.org/10.1021/jp507102x
Фюйер Г. Химия нитро- и нитрозо-групп. М.: Мир, 1972.
Храпковский Г.М., Столяров П.Н., Дорожкин В.П. и др. // Хим. физика. 1990. Т. 9. № 5. С. 648.
Oestmark H., Bergman H., Baum U. et al. // Proc. 32th Intern. Annual Conf. ICT “Energetic Materials: Ignition, Combustion and Detonation”. Karlsruhe: Inst. of ICT, 2001. P. 26/1.
Хмельницкий Л.И., Новиков С.С., Годовикова Т.И. Химия фуроксанов. Реакции и применение. M.: Наука, 1996.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика