

Химическая физика, 2021, T. 40, № 7, стр. 16-23
Кинетика разложения 1,1-диамино-2,2-динитроэтилена (FOX-7). 2. Реакция в твердом состоянии
Г. М. Назин 1, В. В. Дубихин 1, Т. К. Гончаров 1, А. И. Казаков 1, *, А. В. Набатова 1, А. В. Шастин 1
1 Институт проблем химической физики Российской академии наук
Черноголовка, Россия
* E-mail: akazakov@icp.ac.ru
Поступила в редакцию 27.12.2019
После доработки 15.06.2020
Принята к публикации 20.07.2020
Аннотация
Исследована кинетика термического разложения 1,1-диамино-2,2-динитроэтилена (FOX-7) в твердом состоянии манометрическим и калориметрическим методами. Найдены условия, при которых твердофазная реакция и сопровождающие ее быстрые побочные процессы сублимации вещества и распада паров разделены во времени, а вклад вторичных реакций в начальную скорость твердофазного разложения невелик. В этих условиях определена константа скорости некаталитической стадии разложения вещества в твердой фазе.
ВВЕДЕНИЕ
Цель данной работы заключалась в изучении термического разложения 1,1-диамино-2,2-динитроэтилена – FOX-7 (I) в твердом состоянии и определении в конечном итоге кинетических параметров начальной некаталитической стадии реакции. Скорость разложения на этой стадии определяет верхнюю границу термической стабильности вещества в условиях хранения и практического применения. Как показано в работе [1], трудность изучения твердого соединения I заключается в том, что из-за высокой летучести этого соединения твердофазный распад сопровождается сублимацией и быстрым разложением вещества в паровой фазе. В принципе, конкуренцию газовой реакции можно исключить, повысив величину отношения массы навески m к объему сосуда V, т.е. создав условия, когда скорость реакции в газовой фазе, которую можно представить как произведение массы паров на константу скорости распада, mгkг, будет много меньше скорости в твердой фазе, mтвkтв, т.е. когда kг/kтв$ \ll $ mтв/mг. В литературе описано успешное применение этого способа нахождения kтв в случае гексогена [2]. Упругость паров гексогена при 180 °С равна 1 Торр [3], kг/kтв = 100 [2], и газовая реакция становится незаметной уже при m/V < 0.01 г/см3. Однако в случае соединения I величины Рупр (давление паров) и kг/kтв имеют [1] на порядок бóльшие значение, чем у гексогена [1], и при свободном поступлении паров в сосуд вклад газовой реакции при температурах исследования может оказаться значительным при любом значении m/V. Поэтому для устранения газовой реакции в работе [1] предложено не повышать величину m/V, а воспользоваться уникальной особенностью соединения I, которая заключается в способности газофазной реакции к самоторможению. Последнее связано с образованием при газофазном распаде труднолетучих соединений, которые конденсируются на поверхности кристаллов в виде пленки, препятствующей сублимации. Отметим, что в литературе описаны случаи, когда при разложении взрывчатых веществ образуются продукты с большим молекулярным весом [4–7]. Однако происходит это не в паровой, а в конденсированной фазе, и продукты находятся в смеси с исходным веществом, а не осаждаются на его поверхности.
В закрытых сосудах образование пленки идет быстро, при температуре 200 °С – фактически за время прогрева. Пары́, вышедшие в сосуд, разлагаются за короткое время, и газовая реакция заканчивается. Таким образом, защитная пленка решает проблему устранения побочной реакции. Однако на полноту эффекта влияют ряд ограничений, касающихся массы вещества, объема реакционного сосуда и предварительной обработки вещества. Все закономерности газовой реакции и ее ингибирования описаны в работе [1].
Разложение твердого соединения I идет с самоускорением. Поэтому для измерения kтв необходимо найти условия эксперимента, при которых начальный некаталитический процесс хорошо отделяется не только от побочной газовой реакции, но и от последующей каталитической стадии. Поиск этих условий облегчает использование результатов детального кинетического исследования распада I при 200 °С, проведенного в [1].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использованные в данной работе образцы FOX-7 и аппаратура для манометрических измерений при повышенных давлениях описаны в работе [1]. Для исследования реакции в изотермическом режиме использовался также дифференциальный автоматический калориметр ДАК-1-2 [8] с чувствительностью 10–6 Вт. Эксперименты выполняли в запаянных стеклянных предварительно вакуумированных ампулах, выдерживающих при объеме ампулы в 2 см3 давление до 20 атм.
РЕЗУЛЬТАТЫ
В работе [1] показано, что при 200 °С и m/V ∼ ∼ 10–3 г/см3 первая и вторая стадии хорошо разделяются. Можно было надеяться, что такое разделение сохранится и при некоторых более высоких температурах.
Разложение при температурах выше 200 °С
На рис. 1 приведены полные кинетические кривые разложения образца FOX-7 с размером кристаллов ~1 мм при m/V = 1.25 · 10–3 г/cм3 (m = = 5 мг) и температурах 200–230 °С, а также распад при 200 °С “мелкого” образца FOX-7 с размером кристаллов ~0.1 мм. Для удобства сравнения использованы координаты η – t, где η = Ртек/Р∞ – степень превращения, равная отношению значений текущего давления к конечному. Полное газовыделение в этих опытах в среднем составляет 450 см3/г, т.е. три моля на один моль I. Из рис. 1 видно, что вплоть до 220 °С в области 20%-ного разложения наблюдаются изломы, соответствующие завершению 1-й стадии. При 230 °С газофазный и твердофазный процессы описываются одной кривой, которая имеет небольшой изгиб при 50%-ном разложении. Таким образом, возможности кинетического анализа как первой, так и второй стадий ограничены температурой в 220 °С.
Рис. 1.
Кинетические кривые разложения соединения I при температурах 230 °С (1), 220 °С (2 и 3), 215 °С (4 и 5) и 200 °С (6–9); кривые 1–7 – крупные кристаллы размером ~1 мм, m/V = 1.2 · 10–3 г/см3; кривые 8, 9 – кристаллы размером ~0.1 мм, m/V = 7.2 · 10–4 г/см3; 1, 2, 4, 6, 8 – полные кинетические кривые; 3, 5, 7, 9 – вторые стадии кривых 2, 4, 6, 8.
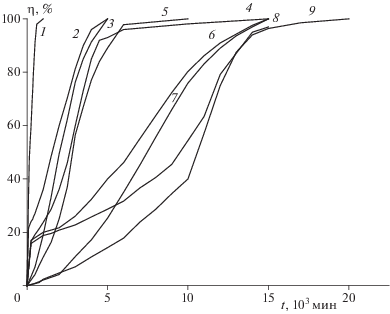
Большая разница в скоростях первой и второй стадий при температурах 200–220 °С позволяет разделить эти стадии и из полной кинетической кривой выделить кривую, описывающую только твердофазную реакцию. Как показано в работе [1], такое разделение проще всего провести, используя исходную кинетическую кривую газовыделения, записанную в координатах Р – t. Сначала путем экстраполяции к t = 0 линейного участка кривой, описывающей реакцию после окончания первой стадии, находится прирост давления ∆Р1, полученный в первой стадии. Затем ∆Р1 вычитается из полной кинетической кривой, и находится зависимость Ртв от t, отражающая изменение давления Ртв за счет разложения в твердой фазе. Такое преобразование сделано для кривых 2, 4, 6, 8, показанных на рис. 1. Полученные кривые 3, 5, 7, 9 твердофазного распада в координатах η–t (η = = Ртв/Ртв ∞) также приведены на рис. 1.
На рис. 2 показана зависимость удельной скорости Wуд = (dη/dt)/(1 – η) от степени превращения η для кривых 3, 5, 7, 9 рис. 1, которые описывают разложение в твердой фазе. Из рис. 2 видно, что линейный рост Wуд наблюдается в случае “крупного” образца при всех температурах до 80% глубины разложения I, а в случае “мелкого” образца при 200 °С – не более чем до 40%. Далее следует резкий подъем удельной скорости распада. Максимум скорости сдвинут к концу реакции.
Рис. 2.
Зависимость удельной скорости от глубины разложения твердого соединения I по данным рис. 1. Значения температуры (°С) и константы скорости kтв (с–1): 1 – 220 и 2.3 ⋅ 10–6, 2 – 215 и 1.0 ⋅ 10–6, 3 и 4 – 200 и 3.1 ⋅ 10–7. Размер кристаллов ~1 мм для кривых 1, 2, 4 и ~0.1 мм для кривой 3.
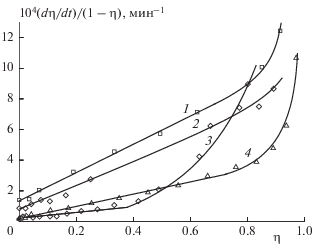
Наличие линейных участков на кривых зависимости Wуд (η) облегчает экстраполяцию Wуд к η = 0. Из рис. 2 по отрезкам, отсекаемым линиями на оси Y, определены константы скорости kтв начальной стадии твердофазного распада. Эти данные приведены в подписи к рис. 2.
В работе [1] было обнаружено, что при навеске 20 мг после окончания 1-й стадии внутри образца остаются не покрытые пленкой кристаллы. Это происходит потому, что свободный объем между кристаллами внутри навески слишком мал, и нелетучих продуктов, образующихся при разложении паров, недостаточно для образования пленки. Такие кристаллы являются источником паров, которые могут диффундировать в объем сосуда. В результате газовая реакция может проявляться неопределенно долгое время. Дополнительные визуальные наблюдения за изменением цвета кристаллов, сделанные при 200 °С, показали, что при навеске 20 мг все кристаллы теряют свой первоначальный желтый цвет только через 24 ч прогревания.
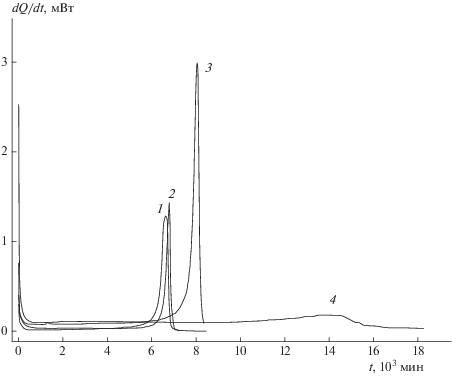
Для выявления возможного влияния газовой реакции на распад соединения I в твердой фазе он был изучен калориметрическим методом при 214.5 °С при разных значениях m и m/V. На рис. 3 показаны скоростные кривые тепловыделения при 214.5 °С для двух опытов с m = 20 мг при разных значениях m/V и двух опытов с меньшими навесками. На этом рисунке видно хорошее разделение первой и второй стадий и острые пики тепловыделения во время твердофазного распада.
Рис. 3.
Зависимость скорости тепловыделения при разложении мелких кристаллов I от времени при 214.5 °С. Значения m (мг) и V (см3): 1 – 6.8 и 0.47, 2 – 11.6 и 1.21, 3 – 20.7 и 1.93, 4 – 19.4 и 7.47.
Кинетические кривые, соответствующие второй стадии тепловыделения, при 214.5 °С в координатах η–t показаны на рис. 4. Представлены два опыта с m = 20 мг, но при разных значениях m/V, и опыт с малыми навеской и объемом сосуда. На этих кривых длительные, почти линейные участки сменяются участками с резким повышением скорости. Положение максимума скорости зависит от величины отношения m/V. Но на начальной стадии кривые отличаются друг от друга незначительно. Наклоны кривых при объеме сосудов 0.47 и 7.47 см3 практически совпадают. Из рис. 4 видно, что скорость разложения на линейных участках не зависит от m/V. Значит, постоянство скорости поддерживается чисто твердофазной реакцией. При этом было установлено, что конечный продукт распада, представляющий собой пылевидный порошок, каталитической активности не имеет.
Рис. 4.
Кинетические кривые тепловыделения в координатах η–t для второй (твердофазной) стадии процесса при 214.5 °С. Значения m (мг), V (см3) и константы скорости k1 ⋅ 106 (c–1): 1 – 6.8, 0.47 и 1.0; 2 – 20.7, 1.93 и 0.87; 3 – 19.4, 7.47 и 1.2.
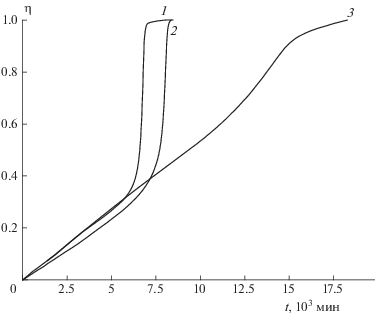
Для кривых, изображенных на рис. 4, из зависимости Wуд от η найдены константы скорости kтв. Их значения приведены в подписи к рис. 4. Во всех трех опытах эти константы имеют близкие значения. Средняя величина, равная 10.4 ⋅ 10–7 с–1, совпадает с найденной манометрическим методом (рис. 2) для крупных кристаллов – 10.0 ⋅ 10–7 с–1.
Таким образом, при 214.5 °С и m = 20 мг на стадии твердофазного распада нет дополнительного вклада от газовой реакции. Значит выход паров из внутренних частей образца в объем отсутствует.
Разложение при температурах ниже 200 °С
Чтобы не выходить из области фазовой устойчивости γ-модификации (ниже 165 °С), исследование проведено при температурах 170–190 °С. При этих температурах реакцию нельзя довести до конца из-за большой длительности опытов. Зато можно, увеличив чувствительность методики за счет повышения величины отношения m/V, точно измерить скорость на самом раннем этапе разложения. Задав определенное значение стехиометрического коэффициента по газовыделению, f, по измеренному значению скорости можно определить kтв.
Из механизма разложения I следует, что на начальной стадии единственным первичным продуктом, который выходит в газовую фазу, является азотная кислота. Разложение паров HNO3 протекает гетерогенно по реакции
(1)
$2{\text{НN}}{{{\text{O}}}_{3}} \rightleftarrows 2{\text{N}}{{{\text{O}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} + 0.5{{{\text{O}}}_{2}},$свойственной жидкой фазе. Скорость гетерогенной реакции пропорциональна количеству адсорбата и зависит от давления паров HNO3. Поэтому можно полагать, что на раннем этапе разложения, когда HNO3 только накапливается и является единственным газообразным продуктом, коэффициент f = 1. По мере разложения I давление HNO3 и скорость ее распада увеличиваются, в парáх появляется NO2, и начинается окисление твердого вещества. В результате окислительного разложения появляются легкие газы и коэффициент f увеличивается до f = 3 – величины, которая была определена в опытах при 200–220 °С (рис. 1). Степень превращения, для которой f = 1, по-видимому, очень мала, и при повышенных температурах ее зафиксировать невозможно. Но при низких температурах из-за уменьшения скорости разложения HNO3 участок кривой, соответствующий f = 1, должен увеличиться и может стать доступным для измерения.
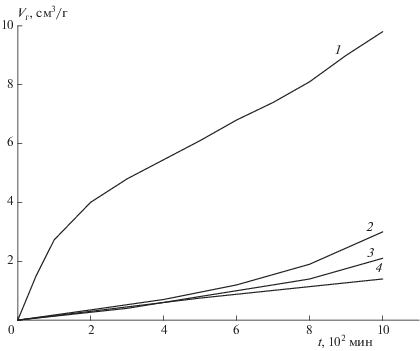
На рис. 5 показаны результаты опытов при 190 °С. При m/V = 0.16 г/cм3 на глубине превращения около 1% (4.5 см3/г) наблюдается побочная реакция, которая постепенно замедляется, но уже через 5 ч переходит в стадию ускорения. Побочный процесс на столь ранней стадии может быть вызван разложением как паров, так и содержащихся в соединении I нестабильных примесей. Для устранения этих процессов был использован предварительный контролируемый прогрев при 200 °С. Одновременно, для того чтобы не попасть в стадию ускорения, в пять раз была уменьшена величина m/V. После шестичасового прогрева при 200 °С, когда реакция вышла на постоянную скорость, опыт был проведен при 190 °С. После этого разложение идет уже без побочной стадии (кривая 3 рис. 5). Заметное ускорение процесса наблюдается только через 8–9 ч после начала опыта. Близкие результаты получаются после 50 ч предварительного прогрева при 180 °С или шестичасового прогрева при 210 °С (кривые 2 и 4 рис. 5). В последнем случае опыт был проведен при m/V = = 0.005 г/cм3, и ускорения распада не наблюдалось даже через 3 сут.
Рис. 5.
Начальные участки разложения I при 190 °С (мелкие кристаллы). Значения m/V (г/cм3) и условия предварительного прогрева (°С/ч): 1 – 0.16, без прогрева; 2 – 0.025, 180/50; 3 – 0.035, 200/6; 4 – 0.005, 210/6.
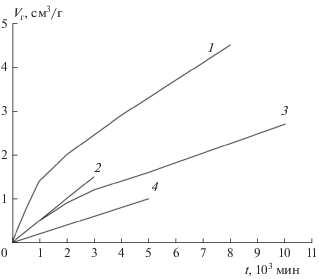
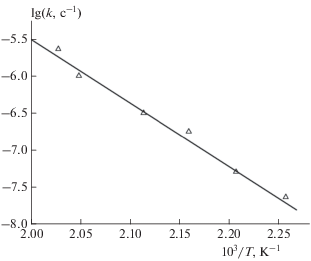
На рис. 6 показано разложение I при 180 и 170 °С. При m/V = 0.1–0.16 г/cм3 сначала проявляются, как и при 190 °С, побочные процессы, которые заканчиваются через 2–3 сут на глубине разложения менее 0.5%. После этого наблюдается длительный, хорошо выраженный линейный участок, по которому можно определить скорость начальной, не осложненной катализом стадии твердофазной реакции. После предварительного прогрева при 200 °С (при меньших значениях m/V) примесные реакции исчезают, а скорости на линейных участках воспроизводятся. По данным рис. 5 и 6 время выделения 1 см3/г газовых продуктов при 190, 180 и 170 °С в среднем равно соответственно 625, 2150 и 4750 мин. Константы скорости kтв для этих температур, вычисленные в предположении, что для начальной стадии конечное газовыделение Vг ∞ = = 150 см3/г, т.е. f = 1, соответственно равны 1.8 ⋅ 10–7, 5.1 ⋅ 10–8 и 2.3 ⋅ 10–8 с–1. Эти данные наряду со значениями kтв, полученными при температурах 200–220 °С (рис. 2), использованы для построения аррениусовской зависимости (рис. 7). Активационные параметры начальной стадии твердофазного разложения Ea = (161 ± 1.8) кДж/моль, lg(A, c–1) = = 11.3 ± 0.9. Значения lgkтв, найденные при высоких и низких температурах, лежат на одной прямой, что подтверждает правильность выбранных способов их определения.
ОБСУЖДЕНИЕ РЕЗУЛЬТОВ
Результаты проведенных исследований в сочетании с механизмом распада I, рассмотренным в работе [1], позволяют составить представление о том, как происходит разложение этого соединения в твердой фазе. Отметим, что при температурах, которые используются для изучения распада I, реакцию в твердой фазе можно наблюдать только “под пленкой”, т.е. после завершения быстрых побочных процессов сублимации и разложения паров, в результате которых кристаллы соединения I покрываются пленкой, препятствующей сублимации.
Согласно [1], первым актом разложения I является изомеризация в ациформу, которая быстро разлагается на HNO3 и нитрилоксид RC≡N+−O–, где R=HN=C(NH2)−. Нитрилоксид изомеризуется в изоцианат RN=C=O, и оба соединения остаются в твердой фазе как конденсированные продукты распада. В газовую фазу выходят только пары́ HNO3. Разложение паров HNO3 протекает гетерогенно по бимолекулярной реакции (1), скорость которой сильно зависит от давления паров HNO3 и температуры. Поэтому на малых степенях превращения I, когда HNO3 будет единственным газообразным продуктом, возможно вычисление kтв по скорости процесса в предположении, что f = 1. В опытах при пониженных температурах эта возможность реализована. По мере разложения HNO3 начнется процесс окисления твердых соединений посредством NO2, и в продуктах появляются легкие газы. Стехиометрический коэффициент f возрастает до трех. Изменение f зависит главным образом от скорости разложения HNO3. При повышенных температурах оно происходит на малых глубинах разложения, т.е. уже за время прогрева.
С NO2 реагируют только продукты распада, которые содержат легко окисляющиеся группы R. Само же соединение I посредством NO2 не окисляется. Этот вывод следует из того, что на начальных участках кинетических кривых скорость от m/V не зависит (рис. 4).
Для разложения I характерно наличие на кинетических кривых линейных участков, свидетельствующих о постоянной скорости реакции. Особенно хорошо это видно на кривых тепловыделения (рис. 4). Постоянство общей скорости разложения означает рост удельной скорости. Разложение твердого соединения I не сопровождается появлением жидкой фазы, отсутствуют также автокатализ твердыми продуктами и прямое взаимодействие I с NO2. Поэтому единственной причиной ускорения можно считать обычный для твердых веществ топохимический процесс, действующий по механизму размножения дислокаций.
Особенность твердофазного разложения I заключается в том, что спокойное течение реакции на линейных участках внезапно сменяется очень сильным ускорением. За короткое время скорость увеличивается в десятки раз, после чего реакция быстро заканчивается. Линейные участки кинетических кривых часто встречаются при изучении топохимических реакций разложения. Во многих случаях после этих линейных участков наблюдается значительное ускорение, но максимум скорости обычно приходится примерно на середину процесса. Сдвиг максимальной скорости к концу реакции в процессах, идущих без образования жидкой фазы, наблюдается только для соединения I. Возможно, что в основе этого явления лежит та же причина, из-за которой в реакциях типа “твердое → твердое + газ” (к числу которых принадлежит и разложение I) продукты всегда образуются в высокодисперсном состоянии. Сама эта причина, несмотря на широкое распространение и даже использование таких реакций для промышленного получения высокодисперсных материалов [9], обычно не обсуждается. Возможно, она связана с одной из основных закономерностей разложения твердых веществ, которая впервые была сформулирована в работе [10]. Эта закономерность заключается в том, что в реальном кристалле реакции идут в основном на поверхности микроблоков (внутренней поверхности кристалла). По мере выгорания с поверхности объем микроблоков уменьшается, связь между ними ослабевает, и после достижения некоторой критической глубины разложения ηкр кристалл рассыпается на отдельные микроблоки, хотя реакция в них может быть еще незаконченной.
Само по себе превращение внутренней поверхности во внешнюю не приводит к существенному изменению скорости распада вещества. Однако в случае соединения I появление свежей поверхности сопровождается возобновлением таких специфических для этого вещества процессов, как сублимация, газофазный распад, разогрев, связанный с конденсацией продуктов и стимулирующий сублимацию и газофазный распад. Так, во время твердофазного разложения I из-за разрушения защитной пленки возобновляются процессы, характерные для первой, побочной стадии разложения.
Как уже отмечалось, после начала ускорения скорость растет очень быстро. За короткий промежуток времени достигается максимальное значение скорости, после чего реакция быстро заканчивается. Ввиду гетерогенного характера реакции окисления разрушению подвергаются прежде всего поверхностные и приповерхностные слои кристалла. Чем больше m/V, тем тоньше разрушенный слой и тем меньше степень превращения вещества, при которой начинается ускорение.
В принципе, при низких температурах из-за уменьшения упругости паров соединения I конкуренция газофазной реакции с разложением твердого вещества должна исчезнуть. Но при повышенных температурах она может проявляться не только в виде первичного побочного процесса, но и как причина ускорения на глубоких стадиях распада кристаллов, покрытых защитной пленкой.
ЗАКЛЮЧЕНИЕ
При разложении FOX-7 проявляются закономерности, характерные для реакций ”твердое → → твердое + газ”, идущих без автокатализа конденсированными или газообразными продуктами. Реакция идет с ускорением, связанным с размножением дислокаций на частицах новой фазы. Кинетические параметры для начальной стадии разложения равны: Ea = 161 кДж/моль, A = 1011.3 с–1. Судя по этим параметрам, FOX-7 и гексоген близки по термической стабильности. На глубоких стадиях реакции кристаллы разрушаются, возобновляются процессы сублимации и разложения вещества в паровой фазе, скорость разложения сильно увеличивается.
Работа выполнена на средства ИПХФ РАН по теме 0089-2019-0005 “Фундаментальные и проблемно-ориентированные исследования в области создания энергетических конденсированных систем (ЭКС) различного назначения” (регистрационный номер АААА-А19-119100800130-0) при финансовой поддержке программой Президиума РАН “Перспективные физико-химические технологии специального назначения”.
Список литературы
Назин Г.М., Дубихин В.В., Гончаров Т.К. и др. // Хим. физика. 2021. Т. 40. № 6.
Энергетические конденсированные системы. Кр. энцикл. словарь / Под ред. Жукова Б.П. М.: “Янус-К”, 1999.
Максимов. Ю.Я. // Теория взрывчатых веществ. Сб. статей / Под ред. Андреева К.К. M.: Высш. шк., 1967. С. 73.
Чарский В.В., Дубихин В.В., Назин Г.М. // Хим. физика. 2004. Т. 23. № 10. С. 38.
Чуканов Н.В., Чапышев С.В., Неделько В.В. и др. // Хим. физика. 2018. Т. 37. № 2. С. 19; https://doi.org/10.7868/S0207401X18020036
Неделько В.В., Чуканов Н.В. Корсунский Б.Л. и др. // Хим. физика. 2018. Т. 37. № 11. С. 36; https://doi.org/10.1134/S0207401X18110092
Wang Y.L., Zhao F.Q., Ji Y.P. et al. // J. Anal. Appl. Pyrol. 2012. V. 98. P. 231; https://doi.org/10.1016/j.jaap.2012.08.014
Гальперин Л.Н., Колесов Ю.Р., Машкинов Л.Б. и др. // Тр. 4-й Всесоюз. конф. по калориметрии. Тбилиси: Мецниереба, 1973. С. 539.
Мурашкевич А.Н. Химическая технология материалов и изделий электронной техники. Минск: БГТУ, 2013.
Химия твердого состояния / Под ред. Гарнера В.Е. М.: Изд-во иностр. лит., 1961.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика