

Химическая физика, 2023, T. 42, № 4, стр. 12-19
Молекулярное моделирование взаимодействия кластера хромсодержащего полиакрилонитрила с газами-поллютантами
М. М. Авилова 1, Н. В. Золотарёва 2, О. В. Попова 1, *
1 Донской государственный технический университет
Ростов-на-Дону, Россия
2 Астраханский государственный университет
Астрахань, Россия
* E-mail: olvp2808@rambler.ru
Поступила в редакцию 24.03.2022
После доработки 15.05.2022
Принята к публикации 20.05.2022
- EDN: MUUHYE
- DOI: 10.31857/S0207401X23040027
Аннотация
Оценена возможность адсорбции приоритетных газов-поллютантов (диоксида азота, метана, аммиака, оксида серы (II), сероводорода, озона, монооксида углерода, оксида углерода (II), хлора) на поверхности хромсодержащего пиролизованного полиакрилонитрила (пПАН). Построена модель кластера хромсодержащего пПАН (Cr–ПАН). Методом молекулярного моделирования в работе определены термодинамические показатели систем “кластер Cr–пПАН – молекула газа”, “кластер Cr–пПАН – молекула кислорода”, “кластер Cr–пПАН – молекула воды”, “кластер Cr–ПАН – молекула кислорода” – молекула газа, кластер Cr–пПАН – молекула воды – молекула газа и проведено их сравнение. Выявлены влияние молекулы воды на процесс адсорбции газов-поллютантов на поверхности кластера Cr–ПАН и отсутствие влияния молекулы кислорода, находящихся в непосредственной близости от кластеров. Установлено, что Cr–пПАН обладает свойством селективной адсорбции следующих газов: диоксида азота, хлора и аммиака. В рамках теории функционала плотности оценены силовые параметры структуры Cr–пПАН и подтверждено увеличение зоны контактной поверхности при внедрении в нее молекулы Cr2O3.
ВВЕДЕНИЕ
Разработка датчиков газов имеет большое значение для мониторинга окружающей среды и управления производственными процессами. В качестве чувствительных материалов датчиков резистивного типа используют в основном полупроводниковые оксиды металлов, углеродные материалы на основе наночастиц и проводящие полимеры [1–3]. Среди проводящих полимеров выделяют модифицированный полиакрилонитрил [4–6], который проявляет высокую селективность и высокую чувствительность к газам NO2, Cl2, NH3 [1, 7–10], что и позволяет использовать указанный материал в качестве сенсорного элемента в датчиках газов.
При этом известно, что наилучшей газочувствительностью обладают композиты на основе модифицированных металлами пиролизованных полиакрилонитрилов (пПАН) [7–13]. К таким материалам относится хромсодержащий пиролизованный полиакрилонитрил (Cr–пПАН). Установлено, что внедрение модифицирующей добавки (хрома) в пленки пПАН усиливает их электропроводящие свойства более чем в 7–9 раз [14, 15].
Выявление факторов селективной газочувствительности Cr–пПАН предполагает установление причины адсорбции одних газов-загрязнителей на пПАН и невозможности данного процесса для других газов. Эти исследования возможно выполнить на основе методов квантовой и молекулярной механики, методов самосогласованного поля Хартри–Фока для открытых оболочек, теории функционала плотности и других, что позволяет быстро и корректно изучить сложные системы, исключить лишние ресурсные и финансовые затраты. Ранее в публикациях [16–20] методами молекулярного моделирования исследованы причины газочувствительности Cu-, Co-, Cd-, Fe-содержащих пПАН.
Цели данной работы – исследование адсорбции приоритетных газов-поллютантов на поверхности кластера Cr–пПАН методами молекулярного моделирования и оценка влияния молекулы кислорода и молекулы воды на данный процесс, а также объяснение причин улучшения полупроводниковых свойств при внедрении модифицирующей добавки в матрицу ПАН.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Модель кластера хромсодержащего ПАН получили методом молекулярной механики (ММ2), а именно методом минимизации потенциальной энергии системы в модифицированной версии силового поля. С целью формирования правильной и максимально приближенной к реальной структуре модели Cr–пПАН изучили технологию получения композитных пленок на основе электропроводящего хромсодержащего пПАН. Для получения материала используют хромкарбонильный ПАН с концентрацией хрома до 3.6 мас.% [14]. Отжиг металлсодержащего ПАН проводят при температурах 200–400 °С [14, 15]. Образцы композита представляют собой пленки толщиной 8–12 мкм [14]. По данным рентгеновской эмиссионной спектроскопии Cr2O3 в матрице пПАН содержится в виде ультрадисперсных частиц [14]. При полимеризации самого пПАН и формировании его сопряженных цепочек вследствие протекающих химических реакций в процессах ИК-отжига выделяются газы СО и Н2 [21, 22]. Принимая во внимание тот факт, что Cr2O3 является твердым тугоплавким порошком, который не вступает в реакции с СО и Н2, наличие других соединений хрома в системе кластера исключили.
Оптимальная модель структуры кластера Cr–пПАН получена путем варьирования положения молекул Cr2O3 относительно друг к друга и относительно матрицы пПАН и последующего расчета методом ММ2 минимальных стерических энергий полученных моделей Cr–пПАН. В результате определена наиболее энергетически выгодная конформация модели Cr–пПАН (рис. 1).
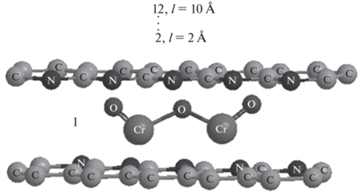
Для корректного прогнозирования структурных и электронных свойств кластерной модели “пПАН−Cr2O3” выполнены разноуровневые квантовомеханические расчеты исходных компонентов и контактной системы в целом. Переход в расчетах от классического силового поля в методе MM2 [23] к использованию нового полуэмпирического метода PM7 [24] обоснован, в первую очередь, улучшенной схемой описания дисперсионных взаимодействий и водородных связей в системе. На данном этапе вычисляли объем контактной поверхности пПАН и его модифицированных форм взаимодействия по методу COSMO [25]. Необходимо отметить, что вводимые в параметризированный метод PM7 поправки аналогичны поправкам, применяемым в теории функционала плотности (ТФП) для улучшения описания межмолекулярных контактов.
При расчете кластерной модели применяли метод самосогласованного поля Хартри–Фока с открытой электронной оболочкой: ROHF/6-31G, что позволило значительно упростить и ускорить расчеты на стадии оптимизации геометрической структуры. Рассматривали все возможные для кластерной модели значения спиновой мультиплетности. Затем выбирали структуру с наименьшей полной энергией, которую и принимали за основное состояние для данного значения энергии. Далее проводили полную оптимизацию и расчет частот колебаний в рамках ТФП на уровне B3LYP/cc-PVDZ [26]. Кроме этого, рассчитаны оптимальные межатомные расстояния и распределения точечных зарядов на атомах по Малликену в исходных молекулах и в кластерной модели (рис. 1). Методом HOMO–LUMO вычислены параметры объема и значения энергии щели, EHL, для оксида хрома (III).
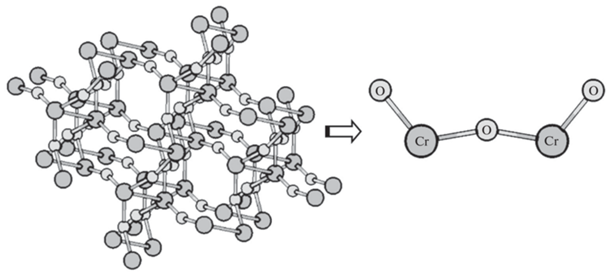
При моделировании кластера кристаллической структуры Cr2O3 (рис. 2) использованы данные из базы Crystallography Open Database (COD 1011067). Модель фрагмента кластера (молекула Cr2O3), включенная в квантовомеханический расчет, предварительно оптимизирована полуэмпирическим методом PM7 по алгоритму следования собственному вектору в рамках метода самосогласованного поля Хартри–Фока (ROHF). Использование полуэмпирического метода обосновано, в первую очередь, параметризацией относительно большинства металлсодержащих твердотельных соединений, в частности применимостью к описанию кристаллической структуры оксида хрома (III) [27].
Геометрическую оптимизацию исходных структур и кластерных моделей “пПАН−Cr2O3” проводили по алгоритму следования собственному вектору, который надежно зарекомендовал себя в поиске вероятных переходов от определенного минимума потенциальной энергии к соседним минимумам энергии [28]. Вычисленные частоты колебаний молекулы пПАН были сопоставлены с экспериментальными данными ИК-спектроскопии. Объем контактной поверхности пПАН и его форм взаимодействия вычисляли по методу COSMO [25].
При изучении возможности адсорбции газов-поллютантов на поверхности кластера Cr–пПАН рассматривали молекулы следующих газов: оксида азота (IV), аммиака, оксида серы (II), сероводорода, озона, оксида углерода (IV), оксида углерода (II), хлора, метана. Для определения стерических энергий систем “кластер Cr–пПАН – молекула газа” задавали различные позиции молекул газа: 1 – внутри кластера и 2–12 – посередине кластера на расстоянии от поверхности 2–10 Å (рис. 1). На следующем этапе оценивали и сравнивали системы, в которых адсорбция газов-поллютантов происходит при наличии молекулы воды и молекулы кислорода: “кластер Cr–пПАН – молекула кислорода”, “кластер Cr–пПАН – молекула воды”, “кластер Cr–пПАН – молекула кислорода/воды – молекула газа”. Термодинамические характеристики исследуемых систем, а именно минимальная стерическая энергия (Еmin), энергия связи (ΔE), расстояние между кластером Cr–пПАН и точкой энергетического минимума (lmin), расстояния от газов-поллютантов до молекулы кислорода/воды (${{L}_{{{{{\text{О}}}_{{\text{2}}}}}}},$ ${{L}_{{{{{\text{Н}}}_{{\text{2}}}}{\text{О}}}}}$) определены по методу ММ2.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Анализ природы химической связи в исходных структурах Cr2O3, пПАН, а также в модельных системах осуществляли на основании рассчитанных в базисе атомных орбиталей локальных характеристик (заряды на атомах по Малликену, электронные заселенности, порядки связей).
В кристаллической структуре Cr2O3 вычисленные методом PM7 значения межатомных расстояний Cr…Cr и Cr–O составляют 2.83 и 1.82 Å соответственно. Для сравнения полученные в работе [29] экспериментальные значения этих параметров Cr…Cr и Cr–O существенно меньше: (2.62 ± 0.01) и (1.95 ± 0.01) Å. В мономолекуле Cr2O3 вычисленные методом PM7 значения межатомных расстояний Cr…Cr, Cr–O для крайних атомов кислорода и Cr–O для центрального атома кислорода составляют 2.67, 1.73 и 1.84 Å соответственно.
В целях оценки энергетических параметров образующейся кристаллической структуры Сr2O3 и расчета объема контактной поверхности для Сr2O3 используются возможности вычислительных программ MOPAC 2016 [30] и GAMESS [31]. Вычисленная методом PM7 ширина HOMO–LUMO-зоны, EHL, для фрагмента кристаллической структуры оксида хрома (III) составляет 2.86 эВ, а для моноструктуры – 3.27 эВ.
Экспериментальное и рассчитанное в приближении PBE/DFT значения ширины запрещенной зоны для Сr2O3 составляют 3.2 эВ [32] 3.4 эВ [33] соответственно. Подобное повышение величины запрещенной зоны по сравнению с вычисленным значением EHL для фрагмента кристаллической структуры может быть обосновано наличием развитой поверхности в объемном материале, а также широким разбросом значений межатомных расстояний Cr–O. В результате в кристаллической структуре Cr2O3 происходит увеличение распределений зарядов на атомах Cr (1.22–1.33 а.е. заряда) и их понижение на атомах кислорода (‒0.97÷–1.01 а.е. энергии). Параметры сходимости процедуры самосогласования устанавливались на уровне 10–6 Хартри (рис. 3).
Незначительные искажения валентных углов наблюдали на концевых фрагментах HСH (максимальное отклонение угла составило 3 град). В процессе оптимизации пПАН методом DFT/cc-PVDZ был достигнут минимум на поверхности потенциальной энергии и найдены характеристические частоты колебаний отдельных функциональных групп: интенсивный сигнал в области 1700–1600 см–1 соответствует валентным колебаниям связей −N=C<, идентифицируются валентные колебания связей C−H в области 3000−2960 см–1. Слабые деформационные колебания в диапазоне 1470−1410 см–1 характерны для =CH2-групп.
По углеродному скелету точечный заряд распределен неравномерно, так для атомов углерода в группах =CH2 значения его изменяются в диапазоне от –0.17 до –0.48 а.е. заряда, тогда как на атомах углерода в группах −N=C< сосредоточено минимальное количество заряда. Вероятно, эти группы пПАН вступают в бимолекулярные взаимодействия за счет сил Ван-дер-Ваальса (стэкинг-взаимодействия).
Результаты расчета характеристических параметров: ширины HOMO-LUMO-зоны EHL, объема контактной поверхности пПАН и его модифицированных форм взаимодействия, приведены в табл. 1.
Таблица 1.
Характеристические параметры пиролизованного ПАН и его модифицированных форм
| Модель | Объем*, Å3 | EHL, эВ |
|---|---|---|
| пПАН | 312.81 | 3.05 |
| пПАН (стэкинг-взаимодействие, 3.5 Å) | 639.03 | 3.00 |
| Модель “пПАН−Cr2O3” | 703.14 | 2.89 |
Для моделирования эффектов сольватации проводится расчет объемов контактной поверхности, а также распределения зарядов. Отметим, что при сближении молекул ПАН происходит увеличение объема контактной поверхности в два раза. При этом на концах молекул минимальное межъядерное расстояние изменяется в диапазоне 2.5–2.7 Å, тогда как по центру бимолекулярного слоя этот диапазон увеличивается до 3–3.5 Å. Увеличение расстояния между молекулами только способствует процессу внедрения мономолекулы оксида хрома (III).
В молекуле Cr2O3 и в кластерной модели пПАН−Cr2O3 заряды на атомах распределены следующим образом: крайние атомы O (0.646/−0.662 а.е. заряда), центральные атомы O (−0.897/−1.109 а.е. заряда), Cr (1.001/0.888 а.е. заряда). Это свидетельствует о том, что внедрение молекулы Cr2O3 в структуру пПАН увеличивает ширину зоны контактной поверхности и сказывается на распределении точечных зарядов, а также локализации активных центров, что подтверждает результаты, представленные в работах [14, 15].
Вычисления, проведенные в рамках силового поля в методе ММ2, также подтверждают, что наиболее энергетически выгодный вариант расположения оксида хрома (III) – в центральной области внутри кластера ПАН (рис. 1). При этом расстояние между цепочками пПАН составляет 4–6 Å, расстояние от молекул Cr2O3 до цепочек пПАН – 2.5–4 Å, минимальная стерическая энергия кластера Cr2O3–пПАН (Eмин) – 1239.5 кДж/моль. Минимальная стерическая энергия системы “кластер Cr–пПАН – молекула кислорода” составляет 2318.94 кДж/моль, системы “кластер Cr–пПАН – молекула воды” – 1029.33 кДж/моль.
Исходя из изложенного выше, делаем вывод, что молекула кислорода, находящаяся в предасорбционном состоянии, не будет оказывать влияния ни на объем поверхности кластера, ни на процесс адсорбции других молекул газов (Eмин (Cr–пПАН) $ \ll $ Eмин (Cr–пПАН + О2)). При этом молекула воды, напротив, будет оказывать влияние на кластер Cr–пПАН (Eмин (Cr–пПАН) + H2О) < Eмин (Cr–пПАН)).
Далее приведены результаты молекулярного моделирования процесса адсорбции газов-поллютантов на поверхности Cr–пПАН в различных газовых средах (табл. 2 и 3, рис. 4–6). Анализ расчетов Емин систем “кластер Cr–пПАН – молекула газа”, “кластер Cr–пПАН – молекула кислорода – молекула газа” и “кластер Cr–пПАН – молекула воды – молекула газа” показал, что Cr–пПАН будет обладать чувствительностью к хлору, монооксиду углерода, диоксиду азота и аммиаку (рис. 4 и 5), так как в системах, в которых присутствуют данные газы, энергии связи систем ниже или находятся близко к значениям энергий самих кластеров. Следует отметить, что стерические энергии систем, в которых присутствует метан, также ниже энергии соответствующих кластеров. Можно предположить, что при определенном температурном режиме может также обнаруживаться чувствительность Cr–пПАН к молекуле метана.
Таблица 2.
Значения энергии связи в системах “кластер Cr–пПАН – молекула газа”, “кластер Cr–пПАН – молекула H2O – молекула газа”, “кластер Cr–пПАН – молекула O2 – молекула газа”
| Газ | ΔE, ккал/моль | ||
|---|---|---|---|
| Cr–пПАН | Cr–пПАН + Н2О | Cr–пПАН + О2 | |
| Cl2 | 19.48 | 6.14 | 18.27 |
| CO | 2.51 | 89.87 | 32.23 |
| О3 | 15.55 | 24.58 | 45.39 |
| NO2 | 6.10 | 75.59 | 7.99 |
| H2S | 2.30 | 27.25 | 16.51 |
| СО2 | 6.10 | 16.47 | 51.92 |
| SO2 | 29.30 | 15.17 | 37.96 |
| NH3 | 8.57 | 17.10 | 22.82 |
| CH4 | 6.06 | 29.59 | 1.05 |
Таблица 3.
Расстояния между молекулой газа и поверхностью кластера в системах “кластер Cr–пПАН – молекула газа”, “кластер Cr–пПАН – молекула H2O – молекула газа”, “кластер Cr–пПАН – молекула O2 – молекула газа”
| Газ | lmin, Å | ||
|---|---|---|---|
| Cr–пПАН | Cr–пПАН + Н2О | Cr–пПАН + О2 | |
| Cl2 | 2.5 | 2.5 | 3.2 |
| CO | 3.2 | 3.7 | 3.0 |
| О3 | 2.5 | 2.5 | 2.5 |
| NO2 | 6 | 3.7 | 8.0 |
| H2S | 2.5 | 3.0 | 4.0 |
| СО2 | 2.5 | 2.5 | 3.4 |
| SO2 | 3 | 3.7 | 3.7 |
| NH3 | 3.5 | 4 | 2.5 |
| CH4 | 2.5 | 2.5 | 8.0 |
Рис. 4.
Зависимости величины энергии системы “кластер Cr–пПАН – молекула газа” от расстояния R между молекулой газа и поверхностью кластера.
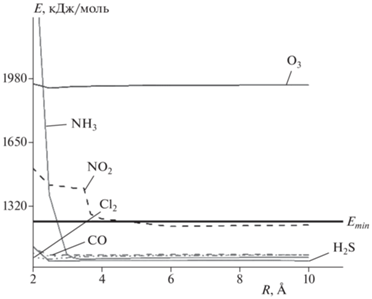
Рис. 5.
Зависимости величины энергии системы “кластер Cr–пПАН – молекула кислорода – молекула газа” от расстояния между молекулой газа и поверхностью кластера.
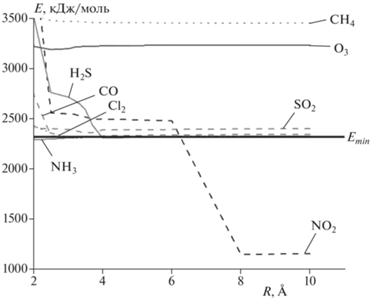
Рис. 6.
Зависимости величины энергии системы “кластер Cr–пПАН – молекула воды – молекула газа” от расстояния между молекулой газа и поверхностью кластера.
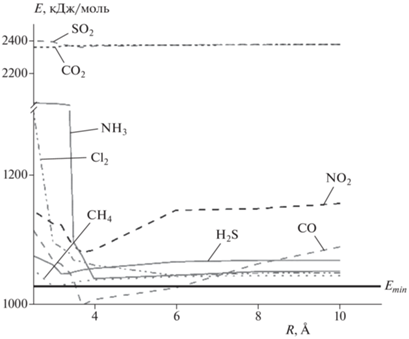
Величины энергий связи в изученных системах соответствуют межмолекулярному типу взаимодействий, а именно связям Ван-дер-Ваальса (табл. 3). Значения приведенных в табл. 4 расстояний между компонентами исследуемых систем исключают возможность образования химической связи, но подтверждают наличие межмолекулярных взаимодействий.
Кроме того, в рамках модели ММ2 при взаимодействии между молекулами газов и поверхностью кластера Cr–пПАН факт зарядопереноса не установлен, что позволяет полностью исключить возникновение в рассмотренном случае химической связи. Следует также отметить, что расстояния между молекулами газов-поллютантов и молекулой кислорода/воды (${{L}_{{{{{\text{О}}}_{{\text{2}}}}}}},$ ${{L}_{{{{{\text{Н}}}_{{\text{2}}}}{\text{О}}}}}$) превышают 2 Å, причем в большинстве случаев они находятся в диапазоне 3.2–4 Å, что позволяет говорить о наличии межмолекулярного взаимодействия между указанными компонентами системы.
ЗАКЛЮЧЕНИЕ
Проведенные методом ММ2 исследования позволили установить, что во всех рассмотренных системах: “кластер Cr–ПАН – молекула газа”, “кластер Cr–пПАН – молекула кислорода – молекула газа”, “кластер Cr–пПАН – молекула воды – молекула газа”, взаимодействие с молекулами Cl2, NO2, NH3 на поверхности указанного кластера является энергетически выгодным. Учитывая невысокие значения энергии связи, полученные для системы “кластер Cr–пПАН – молекула газа”, можно предположить, что в большинстве случаев взаимодействия между компонентами системы происходят посредствам сил Ван-дер-Ваальса.
Кроме того, в рамках ТФП и использования метода COSMO подтверждено, что полупроводниковые свойства при внедрении молекулы Cr2O3 в структуру пПАН повышаются.
Таким образом, по результатам проведенных теоретических исследований можно сделать вывод, что Cr–пПАН является перспективным электропроводящим материалом, обладающим свойством селективной адсорбции Cl2, NO2, NH3.
Список литературы
Ke F., Zhang Q., Ji L. et al. // Compos. Commun. 2021. V. 27. 100817; https://doi.org/10.1016/j.coco.2021.100817
Герасимов Г.Н., Громов В.Ф., Иким М.И., Трахтенберг Л.И. // Хим. физика. 2021. V. 40. № 11. P. 65; https://doi.org/10.31857/S0207401X21110030
Боднева В.Л., Кожушнер М.А., Посвянский В.С., Трахтенберг Л.И. // Хим. физика. 2019. V. 38. № 1. P. 75; https://doi.org/10.1134/S0207401X19010060
Wang W., Zheng Y., Jin X. et al. // Nano Energy. 2019. V. 56. P. 588; https://doi.org/10.1016/j.nanoen.2018.11.082
Efimov M.N., Sosenkin V.E., Volfkovich Yu.M. et al. // Electrochem. Commun. 2018. V. 96. P. 98; https://doi.org/10.1016/j.elecom.2018.10.016
Imanian Z., Hormozi F., Torab-Mostaedi M., Asadollahzadeh M. // Sep. Purif. Technol. 2022. V. 289. 120749; https://doi.org/10.1016/j.seppur.2022.120749
Kozlov V.V., Karpacheva G.P., Petrov V.S., Lazovskaya E.V. // Polym. Sci., Ser. A. 2001. V. 43. P. 20.
Laffont L., Monthioux M., Serin V. et al. // Carbon. 2004. V. 42. P. 2485; https://doi.org/10.1016/j.carbon.2004.04.043
Yoshida H., Sato N. // Rus. J. Phys. Chem. A. 2006. V. 110. P. 4232; https://doi.org/10.1021/jp0546397
Kozlov V.V., Kozhitov L.V., Kostishyn V.G. et al. // IOP Conf. Ser: Mater. Sci. Eng. 2009. V. 5. 012021; https://doi.org/10.1088/1757-899X/5/1/012021
Merdrignac-Conanec O., Bernicot Y., Guyader J. // Sens. Actuators, B. 2000. V. 63. P. 86; https://doi.org/10.1016/S0925-4005(00)00302-6
Ghorpade R.V., Cho D.W., Hong S.C. // Carbon. 2017. V. 121. P. 502; https://doi.org/10.1016/j.carbon.2017.06.015
Kim Ye-Na, Park Eun-Young, Lee Deuk Yong // J. Korean Ceram. Soc. 2007. V. 44. P. 194; https://doi.org/10.4191/kcers.2007.44.4.194
Ерёмин В.С., Бронштейн Л.М., Дьячкова В.П. и др. // Высокомолекуляр. соединения. А. 1993. Т. 35. № 4. С. 450.
Солодовников С.П., Бронштейн Л.М., Логинова Т.П. и др. // Высокомолекуляр. соединения. Б. 1993. Т. 35. № 1. С. 26.
Авилова М.М., Марьева Е.А., Попова О.В., Финоченко Т.А. // ЖФХ. 2020. Т. 94. № 6. С. 898; https://doi.org/10.31857/S0044453720060047
Авилова М.М., Марьева Е.А., Попова О.В., Иванова Т.Г. // Изв. вузов. Химия и химическая технология. 2020. Т. 63. № 4. С. 49; https://doi.org/10.6060/ivkkt.20206304.6008
Авилова М.М., Петров В.В. // Хим. физика. 2018. Т. 37. № 4. С. 69; https://doi.org/10.7868/S0207401X18040088
Авилова М.М., Петров В.В. // Хим. физика. 2017. Т. 36. № 7. С. 90; https://doi.org/10.7868/S0207401X17070020
Avilova M.M., Petrov V.V. // Chemosensors. 2018. V. 6. № 3. P. 39; https://doi.org/10.3390/chemosensors6030039
Gupta A.K., Paliwal D.K., Bajaj P. // J. Appl. Polym. Sci. 1995. V. 58. № 7. P. 1161; https://doi.org/10.1002/app.1995.070580710
Surianarayanan M., Vijayaraghavan R., Raghavan K.V. // J. Polym. Sci., Part A: Polym. Chem. 1998. V. 36. № 14. P. 2503; https://doi.org/10.1002/(SICI)1099-0518(199810)36: 14<2503::AID-POLA9>3.0.CO;2-T
Allinger N.L. // J. Amer. Chem. Soc. 1977. V. 99. № 2). P. 8127; https://doi.org/10.1021/ja00467a001
Stewart J.J.P. // J. Mol. Modeling. 2013. V. 19. № 1. P. 1; https://doi.org/10.1007/s00894-012-1667-x
Klamt A., Schuurmann G. // J. Chem. Soc., Perkin Trans. 2. 1993. № 5. P. 799; https://doi.org/10.1039/P29930000799
Pritchard B.P., Altarawy D., Didier B. et al. // J. Chem. Inf. Model. 2019. V. 59. № 11. P. 4814; https://doi.org/10.1021/acs.jcim.9b00725
Anandan K., Rajendran V. // Mater. Lett. 2015. V. 146. P. 99; https://doi.org/10.1016/j.matlet.2015.02.014
Baker J. // J. Comp. Chem. 1986. V. 7. № 4. P. 385; https://doi.org/10.1002/jcc.540070402
Пономарев Д.А. Дис. … канд. физ.-мат. наук. Екатеринбург: Институт физики металлов им. М.Н. Михеева УрО РАН, 2018.
MOPAC2016 / James J.P. Stewart, Stewart Computational Chemistry/ Colorado Springs, CO, USA, 2016; http://openmopac.net/
Ito S., Fedorov D.G., Okamoto Y., Irle S. // Comput. Phys. Commun. 2018. V. 228. P. 152; https://doi.org/10.1016/j.cpc.2018.01.014
Abdullah M.M., Rajab F.M., Al-Abbas S.M. // AIP Advances. 2014. V. 4. 027121; https://doi.org/10.1063/1.4867012
Skjelbred K.M., Astrand Per-Olof et al. // AIP Conference Proceedings. 2015. V. 1702. 090061; https://doi.org/10.1063/1.4938869
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика