

Химия твердого топлива, 2019, № 2, стр. 55-62
ВЛИЯНИЕ СОСТАВА УГОЛЬНО-БИОМАССНОГО ТОПЛИВА НА ЭФФЕКТИВНОСТЬ ЕГО ГАЗИФИКАЦИИ В ГАЗОГЕНЕРАТОРАХ ПОТОЧНОГО ТИПА
И. Г. Донской *
ФГБУН Институт систем энергетики имени Л.А. Мелентьева СО РАН
664033 Иркутск, Россия
* E-mail: donskoy.chem@mail.ru
Поступила в редакцию 25.10.2018
После доработки 25.10.2018
Принята к публикации 01.12.2018
Аннотация
С помощью математического моделирования исследован процесс совместной газификации смесей угля и растительной биомассы в несущем потоке парокислородного дутья. Получены расчетные зависимости характеристик процесса от управляющих параметров: состава топлива, состава дутьевого агента и стехиометрического соотношения. Для линейной зависимости условий плавления золы от ее состава выделены области параметров, в которых процесс технологически осуществим. Оценено влияние температурных ограничений на эффективность процесса газификации.
Совместное сжигание биомассы и угля, в том числе на крупных станциях, получило распространение в энергетике за счет эффектов снижения вредных выбросов (в первую очередь, оксидов серы и азота). Согласно [1, 2], мировое потребление биомассы для получения энергии достигает 1 млрд т в нефтяном эквиваленте, и по разным прогнозам увеличится в 2–3 раза к 2040 г. Проводятся интенсивные работы по замещению угля биомассой, например в Нидерландах целевой показатель достигает 30%; в США совместное сжигание угля и биомассы организовано на 80 тепловых электростанциях.
Альтернатива сжиганию – это получение из биомассы промежуточных энергоносителей, таких, как биодизель, древесный уголь, генераторный газ. Газификация угля и нефтекокса с добавлением биомассы в пылегазовом потоке реализована на промышленных энергетических установках [3, 4]. Показана возможность проведения процесса при высокой доле биомассы (до 70%). Совместная газификация на крупных станциях позволяет снизить эмиссию СО2 и утилизировать горючие отходы при небольшом снижении эффективности [5, 6]. Существуют предпосылки для развития технологий совместной газификации в странах Юго-Восточной Азии, где большие объемы сельскохозяйственных отходов могут быть использованы для частичной замены импортируемого угля [7].
Из-за небольшого содержания углерода процесс газификации биомассы требует меньшего удельного расхода дутья (кислорода и пара), а также позволяет получать генераторный газ с большим содержанием водорода. Кроме того, биомасса обладает большей реакционной способностью, поэтому может инициировать зажигание угольных частиц. С другой стороны, теплотворная способность биомассы, как правило, меньше теплотворной способности угля, поэтому их совместная переработка сопровождается снижением температурного уровня. При этом могут возникать кинетические затруднения для завершения некоторых химических реакций, в том числе в газовой фазе, что может приводить к снижению качества газа [8].
Совместная конверсия угля и биомассы часто сопровождается неаддитивными эффектами: такие важные характеристики, как состав выделяющихся газов, содержание соединений серы и азота, скорость и полнота превращения, не могут быть интерполированы по результатам, полученным для отдельных компонентов. Данные термического анализа показывают, что при низких скоростях нагрева наложения стадий пиролиза и газификации разных топлив, как правило, не происходит [9, 10]. Однако при совместном окислении неаддитивные эффекты проявляются довольно часто [11–13]. Причины таких эффектов могут быть связаны с взаимодействием продуктов пиролиза, каталитическими свойствами минеральной части [14], различной пористой структурой и сорбционной активностью топлив [15, 16]. Другим эффектом совместной газификации угля и биомассы является смещение температурных границ размягчения и плавления золы [17, 18], что особенно важно для процессов высокотемпературной газификации [19].
Теоретический анализ процессов совместной газификации угля и биомассы в научной литературе представлен намного слабее. Равновесные термодинамические модели часто применяются для анализа процесса в составе энергетических установок [20–22]. Из всей специфики совместной переработки при этом учитывается только изменение элементного состава смеси. При применении термодинамических моделей к реальным реакторам часто требуется их модификация (введение эмпирических коэффициентов и ограничений на достижимость конечного равновесного состояния [23, 24]). Поведение минеральной части оценивается либо эмпирически, либо с привлечением полуэмпирических термодинамических моделей, как например, в работе [25].
Существующие кинетические модели процессов в газогенераторе, как правило, не учитывают термохимического взаимодействия разных топлив: эффекты совместной газификации сводятся к изменению состава летучих и теплотворной способности. Тем не менее такие модели могут давать удовлетворительные результаты, например в работе [26] предложена аддитивная модель совместной газификации частиц угля и биомассы, которая дала хорошее совпадение с результатами экспериментов в реакторе кипящего слоя [27]. Одномерная стационарная модель совместной слоевой газификации угля и биомассы была предложена в [28].
В работе [29] с помощью CFD-модели (в трехмерной постановке) рассмотрены режимы совместной газификации угля и биомассы в реакторе типа Shell с долей биомассы до 20%. Наблюдалось снижение температурного уровня и повышение химического к.п.д. процесса газификации с ростом доли биомассы. В связи с большими вычислительными затратами для анализа был выбран очень ограниченный набор режимов.
В данной статье с помощью математического моделирования проведен анализ процесса совместной газификации углебиомассных смесей при различных составах топлива и дутья.
МАТЕМАТИЧЕСКАЯ МОДЕЛЬ И ИСХОДНЫЕ ДАННЫЕ
В работе использовалась пространственно одномерная модель процесса газификации угля в потоке, аналогичная моделям [30, 31]. При формулировке математической модели принимаются следующие допущения: скорость сушки лимитируется внешним массообменом с окружающим воздухом; скорость пиролиза пропорциональна содержанию летучих в частице и зависит от температуры по аррениусовскому закону; скорость газификации отдельных частиц описывается диффузионно-кинетической теорией горения углерода; теплопотерями реактора можно пренебречь; не учитывается влияние рециркуляции газов; а также влияние шлаковой пленки.
Уравнение теплового баланса для угольной частицы записывается следующим образом:
Скорость сушки вычисляли по формуле
Коэффициент конвективного тепло- и массообмена для частицы в потоке рассчитывали по формуле Сокольского:
где Nu – число Нуссельта; Sh – число Шервуда; Rep – число Рейнольдса для скорости движения частицы относительно несущего потока.Скорость пиролиза описывается кинетическим уравнением первого порядка:
Скорость реагирования топлива с газообразными окислителями записывается следующим образом:
где mc – масса топлива, кг; keff – эффективная константа скорости гетерогенной реакции, м/с; S – площадь поверхности топлива, м2; Cox – концентрация окислителя, кг/м3.Эффективная константа скорости выражается через кинетические и массообменные коэффициенты (в предположении, что кинетический порядок реакции по окислителю равен единице) следующим образом:
Здесь kc – кинетическая константа скорости гетерогенной реакции, м/с; kd – коэффициент массообмена частицы с потоком, м/с.Кинетическая константа скорости гетерогенной реакции зависит от температуры по экспоненциальному закону:
где $k_{c}^{0}$ – предэкспоненциальный коэффициент, м/с; ${{E}_{{\text{а }}}}$ – энергия активации, Дж/моль.Химическая кинетика реакций в газовой фазе не рассматривается: полагается, что выходящие в газовую фазу вещества мгновенно переходят в состояние термохимического равновесия. Таким образом, химические превращения описываются с помощью термодинамической модели с макрокинетическими ограничениями на скорость гетерогенных превращений [32, 33]. Такой подход применим для высокотемпературных процессов, в которых скорость газофазных процессов достаточно высока по сравнению со скоростью гетерофазных. Возможные эффекты взаимодействия органической и минеральной частей топлив [25, 34] не рассматриваются, поэтому неаддитивность может проявляться из-за изменения состава системы “топливо–дутье” и теплоты сгорания топлива. Эти два параметра в основном определяют эффективность процесса газификации [35].
Составы топлив приведены в табл. 1: состав 1 использовался при проверке адекватности модели (взят из опубликованных экспериментальных данных), состав 2 – для вариантных расчетов. Кинетические коэффициенты гетерогенных реакций для угля и биомассы приведены в табл. 2. Для других сортов топлива эти коэффициенты могут быть получены кинетическими методами термического анализа [36, 37].
Таблица 1.
Состав и свойства топлив
| Топливо | W r | Ad | V daf | C daf | H daf | N daf | S daf | Q r, МДж/кг | Плотность органической массы, кг/м3 | Средний размер частиц, мкм | Tf, K | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % | ||||||||||||
| Уголь | состав 1 | 9.5 | 10.4 | 37.1 | 77.98 | 5.4 | 1.09 | 2.7 | 24.7 | 1200 | 100 | 1773 |
| состав 2 | 2 | 14.2 | 27 | 85.33 | 4.77 | 1.98 | 0.93 | 27.8 | 1200 | 50 | 1773 | |
| Биомасса | состав 1 | 4.6 | 0.63 | 82.2 | 49.06 | 6.34 | 0.12 | 0 | 14.6 | 900 | 150 | 1573 |
| состав 2 | 2 | 1.1 | 83 | 50.05 | 8.19 | 0.08 | 0.02 | 17.5 | 900 | 100 | 1573 | |
Таблица 2.
Кинетические характеристики угля и биомассы
| Реакция | Уголь | Биомасса | ||
|---|---|---|---|---|
| $k_{c}^{0}$ | Eа, кДж/моль | $k_{c}^{0}$ | Eа, кДж/моль | |
| Пиролиз | 3.38 · 105 1/с | 113.3 | 5 · 104 1/с | 96 |
| C + O2 | 622.7 г/м2/атм | 101.8 | 2.4 · 104 г/м2/атм | 142 |
| C + CO2 | 3000 г/м2/атм | 210.6 | 1.32 · 105 г/м2/атм | 259 |
| C + H2O | 2470 г/м2/атм | 175.6 | 9.3 · 103 г/м2/атм | 175 |
Валидацию модели проводили с использованием экспериментальных данных, полученных на лабораторном реакторе [38] с высотой реакционной зоны 2 м и внутренним диаметром 25.5 см. Расход топлива (состав 1 в табл. 1) меняется от 10 до 14 кг/ч, доля древесины в рабочем топливе составляет 0, 20, 50 и 100%. Особенность экспериментов – подсветка факела метаном. Результаты расчетов и сравнение с экспериментальными данными [38] приведены в табл. 3. Сравнение показывает, что модель лучше предсказывает степень конверсии и химический к.п.д. для топлив с высокой долей угля, что может быть объяснено большей значимостью пиролитической стадии, которая в используемой математической модели описывается упрощенным выражением. Расчетные значения качественно согласуются с экспериментальными данными.
Таблица 3.
Сравнение результатов расчета с экспериментом [38] (верхнее значение – эксперимент, нижнее значение – расчет)
| $\frac{{{{G}_{{BM}}}}}{{{{G}_{{BM}}} + {{G}_{C}}}}$ | Конверсия углерода, % | Химический к.п.д. (CGE), % | ||||
|---|---|---|---|---|---|---|
| удельный расход окислителя | удельный расход окислителя | |||||
| 0.55 | 0.65 | 0.75 | 0.55 | 0.65 | 0.75 | |
| 0 | 71.56 | 80.65 | 85.2 | 33.33 | 33.16 | 26.60 |
| 71.91 | 98.26 | 99.99 | 31.87 | 39.97 | 28.94 | |
| 0.2 | 84.72 | 93.52 | 98.83 | 34.46 | 33.49 | 29.76 |
| 77.90 | 99.56 | 99.99 | 36.70 | 41.75 | 29.59 | |
| 0.5 | 80.36 | 89.16 | 95.35 | 39.81 | 40.94 | 31.46 |
| 80.71 | 99.42 | 99.99 | 39.67 | 42.42 | 29.96 | |
| 1 | 95.93 | 99.90 | 99.70 | 48.31 | 49.85 | 44.75 |
| 93.73 | 99.99 | 99.99 | 52.82 | 45.00 | 31.75 | |
РАСЧЕТЫ ДЛЯ РЕАКТОРА ПРОМЫШЛЕННОГО МАСШТАБА
Параметры реактора взяты из работ [29, 39]. Рассматривается цилиндрический реактор с производительностью по топливу GС + GBM = 70 т/ч (состав 2 в табл. 1), рабочее давление в реакторе 4.4 МПа. Геометрические размеры реактора: длина реакционной зоны 6.7 м; внутренний диаметр 3.7 м. Температура топлива, поступающего в реактор, составляет 25°С; температура пара 200°С; температура дутьевого кислорода 200°С. Расход пара (steam-to-fuel ratio, SFR) меняется от 0 до 0.2 моль/моль углерода. Коэффициент расхода окислителя α меняется в пределах от 0.1 до 0.7 (с шагом 0.05). Дутье состоит из технического кислорода (чистота 95%) и водяного пара, однако для подачи топлива используется сжатый азот, поэтому концентрация кислорода в дутье принята 85% [30]. Другим варьируемым параметром является доля биомассы в смеси с углем GBM/(GBM + GC): эта величина меняется от 0 до 1 (с шагом 0.1). В первом приближении температура плавления смесевой золы $T_{f}^{{mix}}$определяется как линейная функция состава зольного остатка:
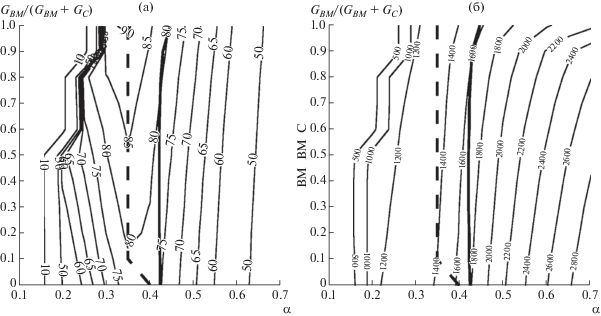
Рис. 1.
Зависимость (%) химического к.п.д. газификации (а) и температуры (K) выхода генераторного газа (б) от коэффициента избытка кислорода и состава угольно-биомассного топлива без подачи дутьевого пара.
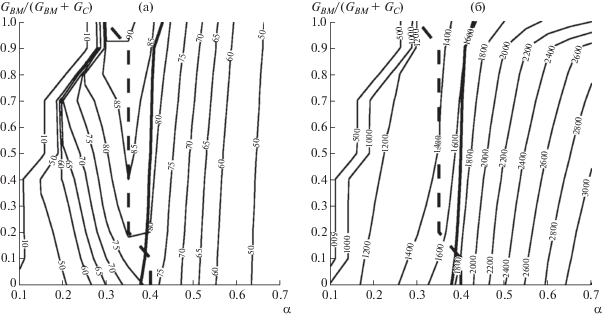
Рис. 2.
Зависимость (%) химического к.п.д. газификации (а) и температуры (K) выхода генераторного газа (б) от коэффициента избытка кислорода и состава угольно-биомассного топлива при фиксированном удельном расходе пара 0.1 моль/моль углерода.
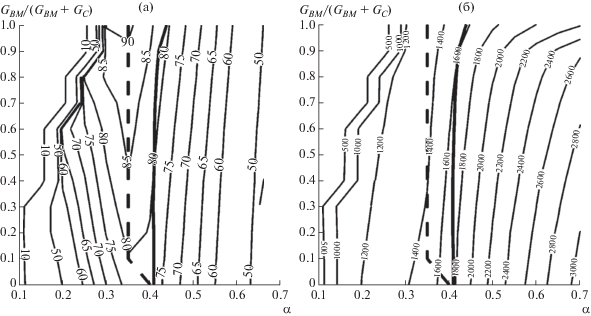
Рис. 3.
Зависимость (%) химического к.п.д. газификации (а) температуры (K) выхода генераторного газа (б) от коэффициента избытка кислорода и состава угольно-биомассного топлива при фиксированном удельном расходе пара 0.2 моль/моль углерода.
Основной энергетической характеристикой процесса газификации является химический к.п.д. газификации (cold gas efficiency, CGE), равный отношению теплоты сгорания газа к теплоте сгорания топлива, из которого он был получен:
Здесь Qg – удельная теплота сгорания газа, Дж/кг; Gg – расход генераторного газа, кг/с; Qf – удельная теплота сгорания топлива, Дж/кг; Gf – расход топлива на входе в газогенератор, кг/с.Одним из варьируемых параметров – это коэффициент расхода окислителя, рассчитанный для смесей угля и биомассы, поэтому массовый расход окислителя зависит не только от состава топлива.
Как видно из рис. 1–3, при фиксированном составе топлива и при изменении коэффициента расхода окислителя химический к.п.д. имеет единственный максимум, обычно совпадающий с условиями полной конверсии твердого топлива [41]. С ростом доли биомассы химический к.п.д. возрастает, что согласуется с результатами работы [29]. Максимальный химический к.п.д. (91%) достигается при газификации биомассы без добавок угля. Область стационарных режимов газификации биомассы начинается при α ≈ 0.25. При добавлении угля эта область расширяется за счет увеличения теплоты сгорания топлива. Оптимальный коэффициент расхода окислителя для всех случаев лежит в диапазоне α = 0.3–0.4.
На рис. 4 показаны пределы достижимого химического к.п.д. с учетом ограничения температуры шлакования (т.е. значения химического к.п.д. на жирной сплошной линии на рис. 1–3). Несмотря на увеличение химического к.п.д. процесса газификации с ростом доли биомассы в смеси топлива, условия жидкого шлакования сдвигают область технологически осуществимых режимов в сторону больших избытков окислителя и меньшей эффективности [42, 43]. Добавление пара к дутью улучшает характеристики газификации угля, однако приводит к снижению температуры в реакционной зоне при газификации биомассы. В результате при уменьшении теплотворной способности топливной смеси условия жидкого шлакования достигаются при еще больших значениях коэффициента избытка окислителя.
Рис. 4.
Зависимость химического к.п.д. совместной газификации угля и биомассы от состава топлива и удельного расхода пара на линии температуры плавления золы.
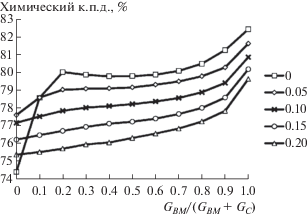
Как видно из рис. 4, при доле биомассы выше 10% добавление пара не эффективно. Водяной пар образуется в достаточном количестве при сгорании частиц биомассы (которая более богата водородом, чем уголь), и дополнительная его подача перестает быть целесообразной. Тем не менее во всех случаях добавление биомассы позволяет повысить эффективность конверсии топлива в горючий газ (по сравнению с чистым углем).
С увеличением доли биомассы растет разность между максимальным химическим к.п.д. и химическим к.п.д. в точке шлакования. Для увеличения эффективности совместной конверсии биомасса должна иметь более легкоплавкую золу, либо взаимодействовать с угольной золой с образованием таковой [18]. Для этого можно воздействовать на температуру плавления золы, добавляя к топливу минеральные компоненты [44], которые образуют более легкоплавкие или тугоплавкие составы.
По сравнению с результатами работы [29] расчетный химический к.п.д. совместной газификации биомассы и угля оказался на несколько процентных пунктов ниже, что может быть связано с разными условиями теплообмена: если в данной работе рассматривали адиабатический процесс (без подвода теплоты извне), то авторы работы [29] задавали постоянную температуру внутренних стенок.
ВЫВОДЫ
С помощью математического моделирования рассмотрена реакционная зона газогенератора типа Shell при подаче угольно-биомассного топлива. Расчеты показали, что добавление биомассы способствует повышению эффективности конверсии твердого топлива за счет высокой реакционной способности, однако области максимального химического к.п.д. могут быть недостижимы по условиям жидкого шлакования. Добавление биомассы позволяет снизить потребности процесса в дутьевом водяном паре без потерь эффективности. Термодинамически достижимый химический к.п.д. газификации биомассы достигал 90%, но при наложении ограничения на достижение температуры плавления золы предельная эффективность уменьшалась до 80%.
Список литературы
Roni M.S., Chowdhury S., Mamun S., Marufuzzaman M., Lein W., Johnson S. // Ren. and Sustain. Energy Rev. 2017. V. 78. P. 1089. doi https://doi.org/10.1016/j.rser.2017.05.023
Bhuiyan A.A., Blicblau A.S., Sadrul Islam A.K.M., Naser J. // J. Energy Inst. 2018. V. 91. № 1. P. 1. doi https://doi.org/10.1016/j.joei.2016.10.006
Sofia D., Llano P.C., Giuliano A., Hernandez M.I., Pena F.G., Barletta D. // Chem. Eng. Res. and Des. 2014. V. 92. P. 1428. doi https://doi.org/10.1016/j.cherd.2013.11.019
Thattai A.T., Oldenboek V., Schoenmakers L., Woudstra T., Aravind P.V. // Appl. Energy. 2016. V. 168. P. 381. doi https://doi.org/10.1016/j.apenergy.2016.01.131
Cormos A.-M., Dinca C., Cormos C.-C. // Appl. Thermal Eng. 2015. V. 74. P. 20. doi https://doi.org/10.1016/j.applthermaleng.2013.12.080
Long III H.A., Wang T. // Int. J. Energy Res. 2016. V. 40. № 4. P. 473. doi https://doi.org/10.1002/er.3452
Kamble A.D., Saxena V.K., Chavan P.D., Mendhe V.A. // Int. J. Mining Sci. and Technol. 2019. V. 29. P. 151. doi https://doi.org/10.1016/j.ijmst.2018.03.011
Frau C., Ferrara F., Orsini A., Pettinau A. // Fuel. 2015. V. 152. P. 138. doi https://doi.org/10.1016/j.fuel.2014.09.054
Haykiri-Acma H., Yaman S. // Ren. Energy. 2010. V. 35. P. 288. doi https://doi.org/10.1016/j.renene.2009.08.001
Idris S.S., Rahman N.A., Ismail K., Alias A.B., Rashid Z.A., Aris M.J. // Biores. Technol. 2010. V. 101. P. 4584. doi https://doi.org/10.1016/j.biortech.2010.01.059
Jones J.M., Kubacki M., Kubica K., Ross A.B., Williams A. // J. Anal. and Appl. Pyrolys. 2005. V. 74. P. 502. doi https://doi.org/10.1016/j.jaap.2004.11.018
Haykiri-Acma H., Yaman S. // Fuel Proc. Technol. 2008. V. 89. P. 176. doi https://doi.org/10.1016/j.fuproc.2007.09.001
Zhou C., Liu G., Wang X., Qi C. // Biores. Technol. 2016. V. 218. P. 418. doi https://doi.org/10.1016/j.biortech.2016.06.134
Mallick D., Mahanta P., Moholkar V.S. // Fuel. 2017. V. 204. P. 106. doi https://doi.org/10.1016/j.fuel.2017.05.006
Meng H., Wang S., Chen L., Wu Z., Zhao J. // Fuel. 2015. V. 158. P. 602. doi https://doi.org/10.1016/j.fuel.2015.06.023
Farid M.M., Hwang J. // Fuel. 2017. V. 207. P. 198. doi https://doi.org/10.1016/j.fuel.2017.06.074
Brar J.S., Singh K., Wang J., Kumar S. // Int. J. Forestry Res. 2012. Article ID 363058. doi https://doi.org/10.1155/2012/363058
Fang X., Jia L. // Biores. Technol. 2012. V. 104. P. 769. doi https://doi.org/10.1016/j.biortech.2011.11.055
Zhang G., Reinmoller M., Klinger M., Meyer B. // Fuel. 2015. V. 139. P. 457. doi https://doi.org/10.1016/j.fuel.2014.09.029
Muresan M., Cormos C.-C., Agachi P.-S. // Chem. Eng. Res. and Des. 2013. V. 91. P. 1527. doi https://doi.org/10.1016/j.cherd.2013.02.018
Kuo P.-C., Wu W. // Chem. Eng. Sci. 2016. V. 142. P. 201. doi https://doi.org/10.1016/j.ces.2015.11.030
Ali D.A., Gadalla M.A., Abdelaziz O.Y., Hulteberg C.P., Ashour F.H. // J. Natur. Gas Sci. and Eng. 2017. V. 37. P. 126. doi https://doi.org/10.1016/j.jngse.2016.11.044
Cali G., Deiana P., Bassano C., Maggio E. // Fuel. 2017. V. 207. P. 671. doi https://doi.org/10.1016/j.fuel.2017.04.061
Mallick D., Buragohain B., Mahanta P., Moholkar V.S. Gasification of mixed biomass: analysis using equilibrium, semi-equilibrium, and kinetic models // Coal and biomass gasification. Recent advances and future challenges. Singapore: Springer Nature, 2018. P. 223. doi https://doi.org/10.1007/978-981-10-7335-9_9
Tchapda A.H., Pisupari S.B.V. // Energies. 2014. V. 7. P. 1098. doi https://doi.org/10.3390/en7031098
Xu Q., Pang S., Levi T. // Chem. Eng. Sci. 2011. V. 66. P. 2232. doi https://doi.org/10.1016/j.ces.2011.02.054
Xu Q., Pang S., Levi T. // AIChE J. 2015. V. 61. № 5. P. 1639. doi https://doi.org/10.1002/aic.14773
Donskoy I.G. // MATEC Web of Conferences. 2016. V. 72. Paper No. 01026. doi https://doi.org/10.1051/matecconf/20167201026
Jeong H.J., Hwang I.S., Park S.S., Hwang J. // Fuel. 2017. V. 196. P. 371. doi https://doi.org/10.1016/j.fuel.2017.01.103
Донской И.Г. // ХТТ. 2016. № 3. С. 54. [Solid Fuel Chemistry. 2016. V. 50. № 3. P. 191. 10.3103/S0361521916030034]
Donskoy I.G., Shamansky V.A., Kozlov A.N., Svishchev D.A. // Combustion Theory and Modelling. 2017. V. 21. № 3. P. 529. doi https://doi.org/10.1080/13647830.2016.1259505
Каганович Б.М., Филиппов С.П., Кейко А.В., Шаманский В.А. // Теплоэнергетика. 2011. № 2. С. 51. [Thermal Engineering. 2011. V. 58. No. 2. P. 143. doi 10.1134/S0040601511020054]
Kangas P., Koukkari P., Hupa M. // Energy Fuels. 2014. V. 28. № 10. P. 6361. doi https://doi.org/10.1021/ef501343d
Ding L., Zhang Y., Wang Z., Huang J., Fang Y. // Biores. Technol. 2014. V. 173, P. 11. doi https://doi.org/10.1016/j.biortech.2014.09.007
Svishchev D.A., Kozlov A.N., Donskoy I.G., Ryzhkov A.F. // Fuel. 2016. V. 168. P. 91. doi https://doi.org/10.1016/j.fuel.2015.11.066
Козлов А.Н., Свищев Д.А., Худякова Г.И., Рыжков А.Ф. // ХТТ. 2017. № 4. С. 12. [Solid Fuel Chemistry. 2017. V. 51 P. 205. doi 10.3103/S0361521917040061]
Дектерев А.А., Осипов П.В., Чернецкий М.Ю., Рыжков А.Ф. // ХТТ. 2017. № 1. С. 21. [Solid Fuel Chemistry. 2017. V. 51. № 1. P. 17. doi 10.3103/S0361521917010037]
Kobayashi N., Suami A., Itaya Y. // J. Chem. Eng. Japan. 2017. V. 50. № 11. P. 862. doi https://doi.org/10.1252/jcej.16we266
Gazzani M., Manzolini G., Macchi E., Ghoniem A.F. // Fuel. 2013. V. 104. P. 822. doi https://doi.org/10.1016/j.fuel.2012.06.117
Алехнович А.Н. Зола и шлакование в пылеугольных котлах. Челябинск: Абрис-принт, 2016. 798 с.
Biagini E. // Energy Conv. and Manag. 2016. V. 128. P. 120. doi https://doi.org/10.1016/j.enconman.2016.09.068
Yun Y., Yoo Y.D., Chung S.W. // Fuel Proc. Technol. 2007. V. 88. P. 107. doi https://doi.org/10.1016/j.fuproc.2004.08.009
Roberts D.G., Harris D.J., Tremel A., Ilyushechkin A.Y. // Fuel Proc. Technol. 2012. V. 94. № 1. P. 26. doi https://doi.org/10.1016/j.fuproc.2011.10.011
Liu X., Yu G., Xu J., Liang Q., Liu H. // Fuel Proc. Technol. 2017. V. 158. P. 115. doi https://doi.org/10.1016/j.fuproc.2016.12.013
Дополнительные материалы отсутствуют.
Инструменты
Химия твердого топлива