

Химия твердого топлива, 2021, № 5, стр. 3-14
ОСОБЕННОСТИ ТЕХНОЛОГИИ ПОЛУЧЕНИЯ АКТИВИРОВАННЫХ УГЛЕЙ НА ОСНОВЕ АНТРАЦИТА
К. Б. Хоанг 1, *, З. К. Ондаганова 1, **, С. М. Пестов 1, ***, В. Р. Флид 1, ****, О. Н. Темкин 1, *****
1 ФГБОУ ВО “МИРЭА – Российский технологический университет” (РТУ МИРЭА)
119571 Москва, Россия
* E-mail: vkb846@yandex.ru
** E-mail: azure_91_17@mail.ru
*** E-mail: pestovsm@yandex.ru
**** E-mail: vitaly-flid@yandex.ru
***** E-mail: olegtemkin@mail.ru
Поступила в редакцию 30.11.2020
После доработки 18.01.2021
Принята к публикации 06.04.2021
Аннотация
В результате исследования влияния химической модификации и режимов активации антрацита паровоздушной смесью на адсорбционные свойства и характеристики углеродных сорбентов установлено, что важнейшими факторами повышения адсорбционных характеристик активированных углей (АУ) в процессе активации являются переход к паровоздушной активации и выбор оптимальных значений температуры активации, расхода паровоздушной смеси и скорости подъема температуры в реакторе активации.
ВВЕДЕНИЕ
В настоящее время возрастает потребность в производстве активированных углей, которые используются в системах водоочистки, водоподготовки, обработки жидкофазных технологических растворов в медицине, пищевой промышленности, гидрометаллургии, при очистке сточных вод, отходящих газов и их разделении [1–3], а также в качестве носителей для приготовления катализаторов.
При получении углеродных сорбентов в настоящее время используются различные виды растительного сырья и ископаемых углей. Антрациты широко используются в качестве сырья для углеродных и графитовых материалов [4–6], для получения активированного угля различного функционального назначения [5], в качестве базисных компонентов угольных смесей [6, 7], бездымных видов топлива [2, 7–10] и восстановительного агента в доменных печах, поэтому антрацит наиболее пригоден в качестве сырья для получения углеродных сорбентов благодаря высокому содержанию углерода (95–98%), относительно низкому содержанию летучих органических веществ (5–7%), низкой зольности (3–5%), большим природным запасам и относительной дешевизне.
В настоящее время известен ряд технологических приемов, предшествующих парогазовой активации антрацита, способствующих созданию пористых углеродных сорбентов. Предлагается двухступенчатый процесс, включающий проведение предварительного пиролиза антрацита в токе N2 при температурах 850–900°С с последующей активацией диоксидом углерода [2, 4, 13–15].
Авторы работ [2, 9–18] считают, что пористость углеродных сорбентов в большой степени определяется их предварительной подготовкой, предшествующей активированию. В этих работах описано предварительное оксидирование антрацита воздухом до активации с целью образования транспортной структуры пор, которая облегчает процесс активации.
Известен метод модификации, включающий обработку антрацита химическими активаторами [5, 9–11, 16, 17, 20, 22, 26] с последующим активированием водяным паром при температурах 850–900°С до степени обгара 10–20%. После чего полученный уголь пропитывают водными растворами NaOH, Na2CO3 или Na2SO4, сушат и обжигают при таких же температурах. Обожженный остаток промывают водой и разбавленной серной кислотой. Оставшийся углеродный сорбент дополнительно обрабатывают водяным паром при 750°С и получают мезопористый активный уголь с удельной поверхностью около 900 м2 г–1. Известны работы, в которых применяют предварительную термообработку [2, 3] и химическую обработку растворами кислот HCl, NH4F(HF) [25] или сильных окислителей HClO4 [6, 11], HNO3 [8] и HNO3 + H2SO4 [3, 18, 22] с последующей активацией модифицированного антрацита водяным паром [1, 11, 16, 17, 20–22, 26–29], воздухом [15], водяным паром + CO2 [3, 12, 13, 19, 25] и паровоздушной смесью [18–20, 23].
Из анализа литературных данных по удельной поверхности, объему пор (V∑) и адсорбционной емкости по I2, бензолу и метиленовому голубому (МГ), приведенных в работах [3, 5, 7, 11, 12, 16–20, 23–25, 27–29], следует, что в результате сочетания “химической” обработки, активации водяным паром и смесью водяного пара с СО2 получается, как правило, углеродный адсорбент с высокими адсорбционными характеристиками и относительно широкими микропорами.
Наиболее полный набор характеристик приведен в работах [7, 12, 24], а именно: суммарный объем пор (АУ) по бензолу V∑ = 0.24 см3 г–1, Vмикро = = 0.12 см3 г–1, Vмезо = 0.03 см3 г–1; удельная поверхность Sуд = 570 м2 г–1 (расчет авторов – по приведенной в статье изотерме); адсорбционная емкость по йоду ≈60%, по бензолу 60 мг г–1 и по МГ 58 мг г–1. Заметим, что некоторые характеристики углеродных сорбентов из антрацита, полученные другими авторами [2, 7, 16–25], превосходят результаты работы [24], но в этих работах не приведены адсорбционные характеристики сорбентов. Так, например, сорбенты, полученные в работах [7, 12, 24, 28, 29], при помощи химической обработки, термообработки и активации окислителями и активаторами имеют высокую удельную поверхность и пористость с небольшими размерами пор, что приводит к низким адсорбционным характеристикам.
Ранее в лаборатории УНЦ “Адсорбция” (МИТХТ) был получен уголь АН-К1, который характеризуется относительно большим количеством микропор и высокой удельной поверхностью, однако адсорбционная емкость по С6Н6 и МГ при этом невысока, что связано, вероятно, с малым диаметром пор (d = 4.86 Å), меньшим диаметров молекул С6Н6 и МГ (6 и 16 Å соответственно). Для адсорбции больших молекул, в частности красителей, предпочтительными являются сорбенты, размеры пор которых соизмеримы с размерами молекул адсорбирующих веществ.
С учетом изложенного было предпринято изучение различных технологических вариантов получения АУ для сравнения методов активации и выяснения роли деминерализации сырья, окисления поверхности в производстве активированных сорбентов из антрацита.
Цель работы – исследование влияния химической модификации антрацита (деминерализация растворами НСl и NH4OH) и режимов его активации паровоздушной смесью на адсорбционные свойства и характеристики углеродных сорбентов. Такая информация необходима для усовершенствования технологии АУ на основе антрацитов.
В задачи исследования входило системное изучение термохимических превращений и формирования пористой структуры в процессе одностадийной активации антрацита паровоздушной смесью при различных температурных режимах, скоростях подъема температуры нагревания исходных материалов в реакторе и расходах паровоздушной смеси. Проводилось также сравнение паровой и паровоздушной активации (для исходного и химически обработанного сырья). При использовании паровоздушной активации можно было ожидать, что наличие воздуха в процессе паровой активации приведет к повышению содержания активных кислородсодержащих центров на поверхности АУ, обычно возникающих в процессе окисления углеродных материалов [19] (фенольные и карбоксильные группы), активных при адсорбции полярных молекул и ионов металлов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исходного материала использовали антрацит марки А шахты ПО “Термоантрацит” Донецкого бассейна. После активации антрацита с характеристиками, описанными выше, и размером частиц антрацита 0.5–2.0 мм, при исходном объеме сорбирующих пор по бензолу менее 0.01 см3 г–1 и насыпной плотности ≈1.4 г см–3 насыпная плотность уменьшается в 4–5 раз (0.3–0.4 г см–1).
Технология состоит из двух этапов:
1. Предварительная обработка исходного антрацита последовательно разбавленными растворами HCl и NH4OH (0.7%) при соотношении объемов раствора и антрацита (ρ = 1.40 г см–3), равном 25, и комнатной температуре. Такая концентрация реагентов обеспечивает протекание процесса деминерализации антрацита в течение 5 ч с удалением микроэлементов и минеральных глинистых загрязнений в исходном сырье [19]. Использование более концентрированных растворов этих реагентов по результатам экспериментов нецелесообразно, так как дальнейшее повышение концентраций кислоты и NH4OH не приводит к заметному улучшению качества АУ. После обработки HCl и NH4OH антрацит промывают водой до значения рН = 7.0. Время обработки каждым раствором составляет 7 и 5 ч соответственно. Затем образец подвергают прокаливанию при Т = 500°С для удаления влаги и значительного количества летучих органических веществ. Замечено [19, 21, 29], что прокаливание при такой температуре не разрушает структуру антрацита вследствие повышенной стабильности этого типа каменных углей. Приведенный режим обработки был установлен экспериментально и является оптимальным.
2. Одностадийная активация антрацита (при температурах 750–900°С и расходе пара 3.5 нл мин–1 на 1500 г антрацита при выбранных объемных соотношениях водяной пар : азот (3.5–4.0) : 0.004 (для паровой активации) и водяной пар : воздух (3.5–4.0) : 3 (для паровоздушной активации)). Такой расход пара обеспечивает увеличение реакционной способности антрацитовой матрицы и степень равномерности обгара антрацита.
Активацию проводили на пилотной установке, состоящей из вращающегося реактора, изготовленного из нержавеющей стали, длиной 1.4 м и диаметром 0.07 м, помещенного в электрическую печь с наклоном реактора 15°. В реактор загружают 1.5 ± 0.1 кг антрацита и нагревают печь до температуры 800°С, со скоростью подъема температуры нагревания материала 17–47°С мин–1, временем активации 3 ч и вращением реактора 12 об/мин.
Свойства АУ оценивали по адсорбции паров бензола [1, 6, 7, 28] и метиленового голубого из водных растворов. В результате паровоздушной активации были получены две серии активных углей: АН-КПВ и АН-КПВА и две серии активных углей для паровой активации: АН-КП и АН-КПА (принятые авторами обозначения: АН-К–серия АУ из антрацита (использовано в работах [27–29]), нижние индексы – тип активации (п–паровая, пв–паровоздушная) при наличии химической модификации, т.е. предварительной обработки сырья, индекс А – отсутствие предварительной обработки сырья: пА, пвА). На всех стадиях исследования измеряли пористость полученных углей путем измерения общего объема сорбирующих пор по бензолу WS на приборе Pore-Sizer 9300-France (ГОСТ 8703–58). В процессе активации фиксировали выходы образца по массе (Rm) и насыпному объему (Rv), определяли насыпную плотность (ρ) и степень обгара (ω). Механическую прочность АУ определяли с помощью лабораторной шаровой мельницы путем истирания сорбента стальными шариками в металлическом барабане (ГОСТ 16188-70).
Адсорбционную емкость по метиленовому голубому (МГ) (с исходной концентрацией 1.5 г л–1) изучали в статических условиях (ГОСТ 4453-74). В связи с использованием красителя профлавин-ацетата (ПА) в фотокаталитическом методе очистки питьевой воды [27–29] адсорбцию ПА исследовали по той же методике. Определение сорбционной активности по йоду проводили методом, описанным в ГОСТ 6217-74 (п. 4). Адсорбцию паров бензола и азота измеряли весовым методом на высоковакуумной адсорбционной установке с использованием пружинных кварцевых весов Мак-Бена. По результатам изучения адсорбции при начальном давлении Р = 10–5 торр и температуре 25 °С рассчитывали величины удельных поверхностей по уравнению БЭТ, объемы и структурные константы пор активированных углей по уравнению Дубинина–Радушкевича [30, 31]. Для определения значений адсорбционных емкостей и построения изотерм адсорбции МГ, ПА, йода и ионов металлов использовали различные концентрации тестовых веществ, включая концентрации, указанные в ГОСТ 6217-74. Так, например, в каждую из набора колб загружали по 10 г АУ и по 1000 мл растворов с различной исходной концентрацией тестовых веществ. Концентрации МГ и ПА определяли фотоколориметрическим методом (фотоколориметр марки ФЭК-15) по времени до момента достижения равновесия. Измерения считали законченными, когда количество адсорбированного вещества практически переставало меняться при переходе к раствору с большей концентрацией. Концентрацию ионов металлов определяли по стандартной методике на атомно-абсорбционном спектрофотометре С-115М-1. Максимальную адсорбционную емкость при соответствующих равновесных концентрациях определяли по изотермам адсорбции.
Кинетические кривые адсорбции МГ, ПА строили следующим образом. Исследованные нами образцы углей АН-КПВ, АН-КП, АН-КПВА, АН-КПА предварительно сушили при 250 ± 5°С в течение 3 ч. Одновременно с этим готовилось определенное количество раствора (например, 3 л раствора МГ с концентрацией 20 или ПА с концентрацией 50 мг л–1). Затем угли АН-КПВ, АН-КП и АН-КПВА, АН-КПА каждый объемом 3 см3 (ρ = 0.317, 0.325 и 0.337, 0.342 г см–3) загружали в колбу емкостью 500 мл и наливали туда определенное количество исследуемого раствора заданной концентрации, например по 100 мл. Этот момент считается началом опыта. Время выдержки каждой порции раствора МГ – 10 мин, а ПА –30 мин. Осветленный раствор первой порции отфильтровали от угля, затем к углю добавляли новую порцию раствора красителя (по 100 мл). Такая процедура продолжалась до тех пор, пока сорбент переставал осветлять новую порцию, тогда адсорбционный процесс считался законченным. Адсорбционная емкость АУ определяется разностью концентрации исходного и осветленного раствора в заданное время и делится на массу АУ. Погрешность трех параллельных опытов составила 3–4%.
Адсорбционная емкость сорбента (АУ) при сорбции ионов металлов Cu2+, Cd2+, Pb2+ из растворов солей Cu(NO3)2 ⋅ 3H2O, Cd(NO3)2 ⋅ 4H2O и Pb(NO3)2 с исходными концентрациями ионов: Cu+2 = 200–500 мг л–1; Cd+2 = 600–1300 мг л–1; Pb+2 = 350–800 мг л–1, определяется в статических условиях.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
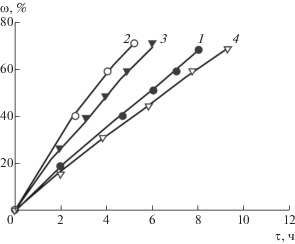
Закономерности активации. Для понимания закономерностей процесса паровоздушной активации антрацита большое значение имеет изучение кинетики самого процесса активации. На рис. 1 представлены кинетические кривые зависимости степени обгара (ω) антрацита от времени в разных температурных режимах активации водяным паром и паровоздушной смесью, с расходом водяного пара в паровоздушной смеси 3.5 нл мин–1 при объемном соотношении пар : : воздух (3.5 : 3); в смеси (пар + N2)–3.5 нл мин–1 при объемном соотношении пар: азот (3.5 : 0.004). Аналогичные зависимости наблюдали авторы работы [20]. Из приведенных данных следует, что использование в качестве активирующего агента смеси водяного пара и воздуха (кривая 2) ускоряет процесс активации по сравнению с использованием чистого водяного пара (кривая 1) при температуре 900°С. Так, например, степени обгара 40–65% в первом случае достигаются за время от 2 до 4 ч, в то время как при активировании чистым паром время составляет 5–7 ч соответственно.
Рис. 1.
Зависимость скорости активации для образцов на основе обработанного антрацита при активации водяным паром (АН-КП) при температуре: 1 – 900°С и активации паровоздушной смесью (АН-КПВ) при температурах: 2 – 900°С, 3 – 850°С, 4 – 800°С.
Значения объемов сорбирующих пор по бензолу возрастают до 0.3–0.35 см3 г–1 по мере увеличения степени обгара до 70%, но незначительно понижаются в случае применения комбинированного активатора. Влияние паровой и паровоздушной активации на характер пористой структуры антрацитовых сорбентов следует из зависимости индекса активации (Rm/Rv) от степени обгара [16, 17]. Значение индекса активации в случае процесса горения описывается прямой Г (индекс активации не зависит от степени обгара и равен единице, Rm/Rv = 1), а в случае процесса порообразования при активации изменение индекса активации в зависимости от степени обгара описывается прямой А, т.е. Rm/Rv = (1–10–2 ω). Из рис. 2 следует, что при активации паром процесс ближе находится к процессу порообразования (увеличивается объем микропор и удельная поверхность; табл. 2).
Рис. 2.
Изменение индекса активирования при активации антрацита водяном паром при температуре: 1 – 900°С и активатором пар–воздух при температурах: 2 – 800°С, 3 – 850°С, 4 – 900°С.
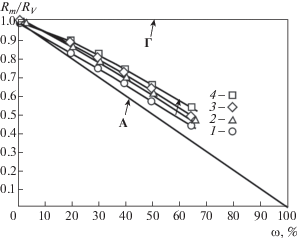
Таблица 1.
Сравнение характеристик АУ по литературным данным
| Образец АУ |
Технологический режим | Sуд, м2 г–1 |
Vмикро, см3 г–1 |
Vмезо, см3 г–1 |
V∑, см3 г–1 | R, Å | Адсорбционная емкость | ||
|---|---|---|---|---|---|---|---|---|---|
| I2, % | С6Н6, мг г–1 | МГ, мг г–1 | |||||||
| [16] АУа |
Антрацит Донецкого бассейна. Окисление влажным воздухом при 270–300°С. Химическая модификация раствором NaNO3. Активация водяным паром | 900 | – | – | 0.44 | – | 70 | – | – |
| [2] АУ–ОП |
Активация органопластиков при 850–900°С воздухом + Н2О. Скорость подъема температуры 10°С мин–1. Степень обгара 36% | 1740 | – | – | – | – | 27.1 | – | 576.8* |
| [2] АУ АЦБ–О |
Модификация углеродного целлолигнина H3PO4 под давлением 1000–1200 кг/м2. Карбонизация при 550–600°С со скоростью подъема температуры 20–60°С /мин, затем активация дробленного карбонизата при температуре 870–950°С | – | 0.56 | – | 1.12 | – | 110 | – | 445* |
| [3] АУ–№ 4 |
Антрацит Донецкого бассейна. Термическая обработка при 800°С. Деминерализация HCl и плавиковой кислотой, химическая модификация HNO3+H2SO4 (время ∑обр = 16 ч). Активация смесью СО2 + Н2О + О2 при 900°С (4.5 ч) | 653 | 0.27 | 0.047 | 0.32 | – | ≈30 | – | – |
| [18] АУО |
Антрацит Донецкого бассейна. Химическая модификация HClО4. Активация СО2 при 850°С (24 ч) | 1150 | 0.25 | – | – | 18.9 | – | – | – |
| [6] AC |
Антрацит La Mure-France. Химическая модификация HClО4. Активация СО2 при 850°С (τакт=24 ч при ω = 50%) | 1600 | 0.61 | 0.30 | – | – | – | – | – |
| [29] АН–К1 |
Антрацит Донецкого бассейна. Деминерализация 0.5%-ным раствором HCl при Т = 50–60°С. Активация водяным паром при 850°С (4 ч) | 1450 | 0.77 | – | – | 2.43 | 71 | 65 | 723* |
Таблица 2.
Параметры АУ при температурах активации Т = 850°С и 900°С на основе обработанного (АН-КП, АН-КПВ) и необработанного (АН-КПВА) антрацита
| Образец АУ | $W_{0}^{1}$, см3 г–1 | $W_{0}^{2}$, см3 г–1 | Sуд по N2, м2 г–1 | ρнасып.плотн., г см–3 | R, Ǻ | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 850 | 900 | 850 | 900 | 850 | 900 | 850 | 900 | 850 | 900 | |
| АН-КП (на обработанный антрацит) | 0.393 | 0.305 | 0.310 | 0.377 | 1200 | 930 | 0.412 | 0.498 | 7–9 | 11–13 |
| АН-КПВА (на необработанный антрацит) | 0.310 | 0.232 | 0.371 | 0.443 | 517 | 406 | 0.487 | 0.574 | 19–21 | 22–25 |
| АН-КПВ (на обработанный антрацит) | 0.357 | 0.271 | 0.325 | 0.388 | 900 | 688 | 0.435 | 0.530 | 8–11 | 12–15 |
При паровоздушной активации большой вклад вносит процесс горения и удаления с окисленной поверхности молекул СО2 и СО. Микропоры расширяются с образованием супермикропор, сопровождаемым уменьшением доли микропор и, соответственно, удельной поверхности (табл. 5).
Характеристики АУ. В пользу более заметного изменения пористой структуры угля в случае паровоздушной активации свидетельствует сравнение удельной поверхности (Sуд) образцов, подвергнутых активации водяным паром при температуре 850°С и активации смесью пар–воздух при той же температуре при степени обгара 35–40%. В первом случае эта величина составляет 1200 м2 г–1, во втором – 900 м2 г–1 (табл. 2). Понижение удельной поверхности образцов, проактивированных до одинаковой степени обгара, связано с преобладанием супермикропористой структуры над микропористой [10, 21, 23].
Установлено, что зависимость значений удельной поверхности (Sуд) от расхода паровоздушной смеси носит экстремальный характер для предварительно обработанного (АН-КПВ) и необработанного (АН-КПВА) образцов сорбента (рис. 3). Максимальное значение Sуд достигалось в интервале 3.5–4.0 нл мин–1. Характеристики АУ АН-КП, АН‑КПВ и АН-КПВА приведены в табл. 2. Из табл. 2 следует, что при увеличении температуры активации от 850 до 900°С объем микропор уменьшается на 20–22%, а объем супермикропор увеличивается на 21–25%. При этом наблюдается уменьшение удельной поверхности на ≈23%, увеличение насыпной плотности на ≈21% и уменьшение радиуса микропор и супермикропор на 40–46% (табл. 2). При выбранных с использованием опыта работы с АУ серии АН-К значениях скорости подачи активирующей смеси пар–воздух и скорости подъема температуры активации наблюдается экстремальная зависимость объемов микропор и супермикропор АН-КПВ от температуры активации (рис. 4), а также экстремальная зависимость удельной поверхности и активности по йоду с максимумом при 850°С (табл. 3). Важная роль скорости подъема температуры активации также подтверждается экспериментально.
Рис. 3.
Зависимость удельной поверхности активированных углей АН-КПВ и АН-КПВА от скорости подачи паровоздушной смеси при температуре активации 850°С и скорости подъема температуры 27°С мин–1.
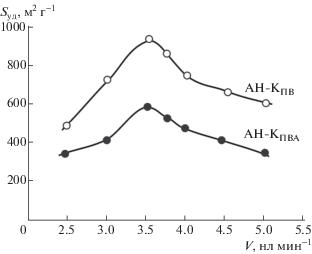
Рис. 4.
Зависимость предельных объемов адсорбционного пространства микропор ($W_{0}^{1}$) и супермикропор ($W_{0}^{2}$) АУ АН-КПВ от температуры активации.
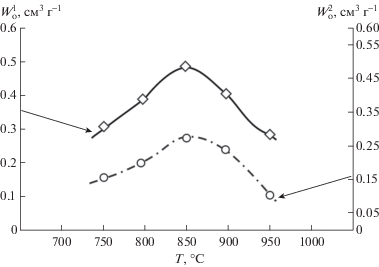
Таблица 3.
Зависимости значений удельной поверх-ности и адсорбционной емкости а по I2 активирован-ных углей, полученных на основе обработанного ан-трацита, от скорости подъема температуры активации
| Образец АУ АН-КПВ |
Тактив, °С | Vпод. т, °С мин–1 | ${{v}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$, л мин–1 | Sуд, м2 г–1 | а, по I2,% |
|---|---|---|---|---|---|
| 1 | 850 | 7 | 3.5 | 480 | 49 |
| 2 | 17 | 620 | 60 | ||
| 3 | 27 | 895 | 88 | ||
| 4 | 37 | 690 | 65 | ||
| 5 | 47 | 630 | 61 |
Из табл. 4 видно, что с ростом температуры активации до 850°С степень обгара увеличивается до 40–45%. Это приводит к образованию микропор и расширению их объема до максимального значения, в то же время структура углеродной матрицы не успевает разрушиться из-за медленного подъема температуры и отсутствия резкого термоудара. При скорости подъема температуры активации до 27 (°С/мин) формирование микропор завершается в результате дальнейшей 3-часовой активации при оптимальной температуре 850°С, соответствующей высокому значению удельной поверхности. Дальнейшее увеличение скорости подъема температуры активации приводит к повышению степени обгара, к заметному разрушению структуры углеродной матрицы еще до достижения заданной температуры активации и началу процесса образования супермикропор, вызывающему уменьшение удельной поверхности. Увеличение расхода водяного пара или паровоздушной смеси в реакторе активации антрацита и соответствующее уменьшение времени контакта активатора с углем приводят к уменьшению степени превращения пара и кислорода, т.е. к увеличению парциальных давлений активаторов, а следовательно, к увеличению скорости окисления поверхности и степени обгара (рис. 3).
Таблица 4.
Зависимости значений удельной поверхности и адсорбционной емкости по I2 активированных углей, полученных на основе обработанного антрацита, от температуры активации
| Образец АУ АН–КПВ |
Тактив, °С | Vпод. т, °С мин–1 | ${{v}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$, л мин–1 | Sуд, м2 г–1 | а, по I2,% |
|---|---|---|---|---|---|
| 1 | 750 | 27 | 35 | 495 | 51 |
| 2 | 800 | 680 | 63 | ||
| 3 | 850 | 900 | 90 | ||
| 4 | 900 | 789 | 71 | ||
| 5 | 950 | 550 | 55 |
Таблица 5.
Сравнение характеристик АУ АН-К1 и АН-КПВ с другими марками АУ
| АУ | Sуд, м2 г–1 |
$W_{0}^{1}$, см3 г–1 | $W_{0}^{2}$, см3 г–1 | R, Å | η, % | Адсорбционная емкость | Технологический режим | |||
|---|---|---|---|---|---|---|---|---|---|---|
| I2, % | С6Н6, мг г–1 |
МГ, мг г–1 |
ПА, мг г–1 |
|||||||
| АУ–ОП [2] |
1740 | – | – | – | – | 27.1 | – | 576.8* | – | Активация органопластиков при 850–900°С воздухом + + Н2О. Скорость подъема температуры 10°С мин–1. Степень обгара 36% |
| АГ–3И [2] |
900–920 | 0.24–0.28 | 0.12 | – | – | 85–90 | – | 200–300* | – | На основе каменных углей и АУ АГ–3 и АГ–5. Активация парогазовой смесью при 850–950°С |
| АГ–3–О(б) [2] |
800 | 0.319** | 0.067** | – | – | 85 | – | 232.5* | – | На основе АГ–3 (по ГОСТу 20464-75). Измельченный АГ–3. Обработка 10% HCl c промывкой водой |
| БАУ–А [24] | 400–500 | 0.21–0.24 | – | – | – | 73.6 | 104 | 211* | – | На основе березового угля-сырца. Активация парогазовой смесью при 850–900°С |
| ДАУ–А [24] | – | 0.28–0.32 | – | – | – | 62 | 48 | 86* | – | На основе каменного угля СС Кузнецкого бассейна. Активация водяным паром при 850–980°С |
| CКТ | 990 | 0.414 | 0.130 | 12 | 63.5 | 63 | 56 | 225* | 397* | Данные получены в УНЦ “Адсорбция” МИТХТ. Адсор-бционная емкость по МГ и ПА определена в статическом ре-жиме |
| ГИД–5 | 895 | 0.423 | 0.152 | 13 | 58.6 | 65 | 57 | 285* | 412* | |
| Сarbsorb | 530 | 0.259 | 0.272 | 25 | 65.4 | 62 | 51 | 174* | 200* | |
| АН–К1 | 1450 | 0.55 | 0.22 | 2.4 | 71.6 | 71 | 65 | 723* | 916* | |
| АН–КП | 1200 | 0.393 | 0.310 | 7–9 | 73.7 | 75 | 72 | 873* | 1130* | |
| АН–КПВ | 900 | 0.357 | 0.325 | 8–11 | 74.3 | 92 | 82 | 1076* | 1904* | |
Для выявления эффекта предварительной обработки антрацита (образец АН-КПВ) при активации паровоздушной смесью проводили активацию исходного необработанного антрацита (образец АН-КПВА) при тех же температурах и расходе пара. Было установлено, что значение удельной поверхности (по адсорбции азота) угля АН-КПВ в 1.75 раза больше, чем угля АН-КПВА–900 м2 г–1 и 517 м2 г–1 соответственно. При этом адсорбционная емкость АУ АН-КПВ по метиленовому голубому в 2.5 раза больше, чем у АН-КПВА, и в 2.8 раза больше, чем у АН-КПА (рис. 5, 6). Заметное повышение количества адсорбированного метиленового голубого вызвано, вероятно, тем, что образец АН-КПВ на основе обработанного антрацита имеет большее количество микропор с более широким размером пор, соизмеримых с размером молекул МГ, по сравнению с необработанным углем вследствие эффективной деминерализации сырья.
Рис. 5.
Кинетические кривые адсорбции метиленового голубого (МГ) (С0 = 0.02 г л–1) углеродными сорбентами при 25°С: АН-КПВА, АН-КПА (необработанный антрацит); АН-КПВ, АН-КП (обработанный антрацит).
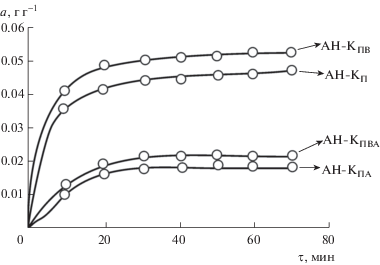
Рис. 6.
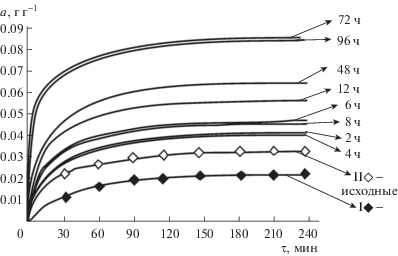
Кинетические кривые адсорбции профлавинацетата (ПА) (С0 = 0.05 г л–1) на АУ АН-КПВ при 25°С. Необработанный (кривая I) и обработанный (кривая II) образцы антрацита при разных временах выдержки, ч.
Сравнение предварительно обработанных образцов АУ при различных вариантах активации показало (см. табл. 2), что переход от процесса активации только водяным паром к паровоздушной активации при 850°С сопровождается заметным понижением удельной поверхности (на 25%) при незначительном снижении суммарного объема микро- и супермикропор (всего на 4%). Это может быть связано с заметным расширением супермикропор при паровоздушной активации и, соответственно, с повышением адсорбционной емкости по большим молекулам красителей метиленового голубого и профлавинацетата.
Адсорбционная емкость АУ. В связи с фотокаталитическим методом очистки питьевой воды [27–29] адсорбционную емкость по метиленовому голубому (МГ) изучали по ГОСТ 4453-74 в статических условиях; адсорбцию профлавинацетата (ПА) исследовали по той же методике; сорбционную активность по йоду определяли с помощью метода, описанного в ГОСТ 6217-74 (п. 4).
Для определения значений адсорбционных емкостей и построения изотерм адсорбции МГ, ПА, йода и ионов металлов использовали различные концентрации тестовых веществ, включая концентрации, указанные в ГОСТ. Так, например, в каждую из набора колб загружали по 1 г АУ и по 100 мл растворов с различной исходной концентрацией тестовых веществ. Концентрации МГ и ПА определяли фотоколориметрическим методом (фотоколориметр марки ФЭК–15) по времени до момента достижения равновесия. Измерения считали законченными, когда количество адсорбированного вещества практически переставало меняться при переходе к раствору с большей концентрацией. Концентрацию ионов металлов определяли по стандартной методике на атомно-абсорбционном спектрофотометре С-115М-1. Максимальную адсорбционную емкость при соответствующих равновесных концентрациях определяли по изотермам адсорбции (рис. 7, 8).
Рис. 7.
Изотермы адсорбции ПА на АУ АН-КПВ, насыщенном ПА при температуре 25°С: I – время сушки 72 ч; II – время сушки 96 ч; III – изотерма адсорбции на исходном АН-${\text{К}}_{{{\text{ПВ}}}}^{*}$ (без промежуточной сушки) для трех параллельных опытов (кривые I и II); погрешность измерения для трех опытов не превышает 3.7%.
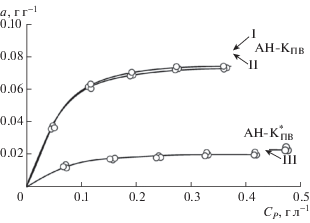
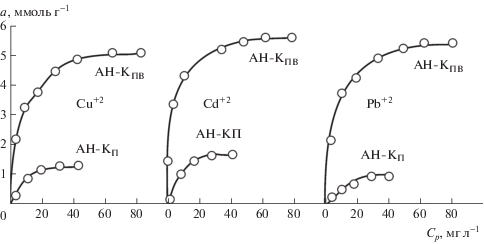
Рис. 8.
Изотермы сорбции ионов металлов из раствора активированными углями АН‑КП и АН‑КПВ при объемном отношении сорбент : раствор = 1 : 100. Исходные концентрации ионов, мг л–1 : Cu+2 = 200–500; Cd+2 = 600–1300; Pb+2 = 350–800.
Кинетические кривые адсорбции МГ, ПА (рис. 5, 6) строили следующим образом. Исследованные образцы углей АН-КПВ, АН-КП, АН-КПВА, АН-КПА предварительно сушили при 250 ± 5°С в течение 3 ч. Одновременно с этим готовилось определенное количество раствора (например, 3 л раствора МГ с концентрацией 20 или ПА с концентрацией 50 мг л–1). Угли АН-КПВ, АН-КП и АН-КПВА, АН-КПА объемом каждый в 3 см3 (ρ = = 0.317, 0.325 и 0.337, 0.342 г см–3) загружали в колбу емкостью 500 мл и наливали туда определенное количество исследуемого раствора заданной концентрации, например по 100 мл. Этот момент – начало опыта. Время выдержки каждой порции раствора МГ – 10 мин, а ПА – 30 мин. Осветленный раствор первой порции отфильтровали от угля, затем к углю добавляли новую порцию раствора красителя (по 100 мл). Такая процедура продолжалась до тех пор, пока сорбент переставал осветлять новую порцию, тогда адсорбционный процесс считался законченным. Адсорбционная емкость АУ определяется значением разности концентраций исходного и осветленного раствора в определенное время и делится на массу АУ. Погрешность трех параллельных опытов составила 3–4%.
Максимальную адсорбционную емкость, равновесную исходным концентрациям красителей 0.2; 0.5 или 1.5 г л–1, определяли также статическим методом. Для этого образцы АУ загружали в определенном количестве в колбу емкостью 500 мл и наливали туда требуемый для достижения объемного соотношения 100 : 1 объем раствора красителя. После достижения равновесия адсорбции в первой порции раствора сорбент отфильтровывали и добавляли новую порцию раствора той же исходной концентрации. Такая процедура продолжалась до момента, когда концентрация раствора переставала отличаться от исходной, например 1.5 г л–1 (см. табл. 1 и 5).
Определение адсорбционной емкости АУ при одинаковой, но низкой концентрации красителей показало, что наблюдаемая адсорбционная емкость по метиленовому голубому образцов АН-КПВ и АН-КП, полученных на основе обработанного антрацита (0.05–0.042 г г–1), более чем в 2 раза превышает емкость АУ (0.022–0.018 г г–1), полученных на основе необработанного антрацита при концентрации красителя 0.02 г л–1.
Адсорбционная емкость активированных углей АН-К1, АН-КП, АН-КПВ по МГ (с исходной концентрацией 1.5 г л–1 по ГОСТ 4453-74) составляет 723, 873 и 1076 мг г1 соответственно, а по ПА при такой же концентрации – 916, 1130 и 1904 мг г–1 соответственно (табл. 1 и 5).
Отметим, что адсорбционная емкость промышленных активированных углей АУ-ОП, АГ-3И, АГ-3-О, АЦБ-О, БАУ-А, ДАУ-А по МГ составляет 576.8; 200–300; 232.5; 445; 211 и 86 мг г–1 соответственно, по данным работ [2 и 24]. Таким образом, адсорбционная емкость по МГ образцов АН-КПВ и АН-КП, полученных на основе обработанного антрацита, более чем в 2–3 и даже 6 раз (по сравнению с АУ ДАУ-А) превышает емкости промышленных АУ (табл. 5).
Установлено, что максимальная сорбционная емкость по I2 по мере увеличения температуры активации имеет место при температуре 850°С (АН-КПВ, табл. 2–4). Это также свидетельствует об уменьшении объема микропор при высоких температурах (выше 850°С) активации с расширением супермикропор и, может быть, о начале образования мезопор с ростом температуры (табл. 5), что согласуется и с ростом сорбционной способности по МГ.
При изучении адсорбции профлавинацетата (ПА) наблюдался интересный эффект, не обнаруженный в случае МГ. При повторном измерении адсорбции ПА на образце, хранившемся в эксикаторе после первичного насыщения поверхности ПА, было установлено повышение адсорбционной емкости сорбента. В связи с этим наблюдением исследовали адсорбцию ПА на АН-КПВ подробнее, тем более, что активированные угли серии АН-К1 (см. табл. 5) в течение ряда лет применяли на пилотной установке Рублевской станции Мосводоканала для удаления фотокатализатора очистки воды – ПА. После достижения равновесия при первой адсорбции, удаления раствора и выдерживания отфильтрованного сорбента в эксикаторе над прокаленным хлоридом кальция для удаления влаги в течение 2, 4, 6, 8, 12, 48, 72 и 96 ч проводилась повторная адсорбция растворов ПА той же концентрации до достижения постоянных величин адсорбционной емкости. Было установлено, что адсорбционная емкость угля, уже насыщенного ПА, увеличивается по мере увеличения времени выдерживания сорбента после адсорбции первой порции красителя (рис. 5). Увеличение адсорбционной емкости прекращается после 72 ч выдерживания сорбента в эксикаторе и превышает первоначальное значение в 4.6 раза. Величина a = 0.083 г г–1 является максимальной величиной адсорбционной емкости и при построении изотермы адсорбции ПА с промежуточной сушкой и выдерживанием насыщенных ПА образцов в течение 72–96 ч при концентрации 0.05 г л–1 (рис. 6). На рис. 7 приведены изотермы адсорбции ПА, полученные при различных режимах адсорбции: кривая I, АН-КПВ (время сушки 72 ч, равновесие адсорбции достигается за 15 мин при всех концентрациях С0 = 0.20; 0.30; 0.40 и 0.50 г л–1). Кривая II, АН-КПВ (время сушки 96 ч, равновесие адсорбции достигается примерно за 20 мин при тех же концентрациях ПА). Кривая III, исходный АН-КПВ (без промежуточной сушки, равновесие адсорбции достигается за 20 мин для трех параллельных опытов с четырьмя различными концентрациями на каждую новую порцию угля). Погрешность из трех опытов не выше 3–4%.
Оказалось, что фактор времени выдерживания образцов в случае ПА решающий и установлено, что адсорбционная емкость полученных АУ существенно повышается, если насыщенный профлавинацетатом после адсорбции первой порции красителя и осушенный сорбент выдерживали в эксикаторе еще 72 ч.
Так, построение изотерм адсорбции ПА на образце АН-КПВ по аналогии с адсорбцией МГ, т.е. без промежуточной сушки выдерживанием образцов в эксикаторе, приводит к совершенно другим показателям максимальной адсорбции (рис. 6, кривая III).
Можно предположить, что повышение адсорбционной емкости после определенного времени выдерживания насыщенного адсорбатом активного угля в эксикаторе связано с освобождением закупоренных устьев микро- и супермикропор молекулами ПА, вызванным последующим медленным процессом диффузии ПА с поверхности внутрь супермикропор и микропор, сопровождаемых частичным освобождением активных центров адсорбции. Нельзя исключить и возможность окислительной деструкции ПА, катализируемой поверхностью активного угля, полученного паровоздушной активацией, с участием активных форм кислорода на поверхности. Такая деструкция также может привести к освобождению центров адсорбции или к появлению новых центров, содержащих фрагменты молекулы ПА. Не ясна и причина различного поведения МГ и ПА на одном и том же АУ. Для выяснения природы обнаруженного явления необходимы дополнительные исследования.
Адсорбция ионов переходных металлов. Сделанный ранее вывод о близости свойств АУ, полученных в результате паровоздушной активации антрацита, к свойствам катионообменников [10, 11], подтверждается исследованием адсорбционной емкости сорбента при сорбции ионов металлов Cu2+, Cd2+, Pb2+ из растворов солей Cu(NO3)2 · · 3H2O, Cd(NO3)2 · 4H2O и Pb(NO3)2 в статических условиях. Установлено, что уголь АН-КПВ на основе антрацита обладает большей способностью сорбировать ионы тяжелых металлов, чем АУ АН-КП (рис. 8).
Наблюдается крутой рост изотерм адсорбции ионов металлов на АН-КПВ при повышении концентрации ионов (рис. 8), что объясняется, вероятнее всего, обогащением поверхности сорбента в процессе окислительной паровоздушной активации активными в адсорбции ионов металлов карбоксильными группами. Процесс сорбции для всех изучаемых ионов протекает достаточно быстро – в течение примерно 5 мин достигается равновесие адсорбции. Контрольные анализы через 10 и 15 мин выдержки не приводили к изменению количества адсорбированных ионов. Максимальная адсорбционная емкость АУ АН-КПВ по ионам Cu2+, Cd2+, Pb2+ относится к равновесным концентрациям раствора по ионам металла 0.060 и 0.080 г л–1. Сорбционная емкость АУ АН-КПВ по Cd2+, Pb2+ и Cu2+ равна 5.85, 5.75 и 5.25 ммоль г–1 соответственно, а для АН-КП – 1.8; 1.75 и 1.28 ммоль г–1 соответственно. Все изотермы адсорбции ионов Cu2+, Cd2+, Pb2+ относятся к лэнгмюровскому типу и описываются уравнением
где a – удельная адсорбция, ммоль г–1; a∞ – величина максимальной сорбции, ммоль г–1; b – константа Ленгмюра, см3 ммоль–1. Значения b для серии ионов Cu2+, Pb2+ , Cd2+ на образце АН-КПВ равны 0.16; 0.085 и 0.075 л г–1 соответственно, т.е. константы равновесия сорбции b можно расположить по величине в ряду Сu2+ > Pb2+ > Cd2+. В том же ряду располагаются значения констант устойчивости моноацетатных комплексов М (ОСОСН3)+ [32] с большей константой для иона меди и с близкими константами для ионов свинца и кадмия. Это обстоятельство может свидетельствовать, вероятно, в пользу образования карбоксилатных групп на поверхности АУ в ходе активации и их важной роли в процессе сорбции ионов металлов [33]. Отметим, что отношение величин сорбционных емкостей для образцов АН-КПВ и АН-КП, приведенных на рис. 8, располагается аналогично рядам b и констант устойчивости ацетатных комплексов: Сu2+ > Pb2+ > Cd2+ (4.10 > 3.28 > > 3.25).В результате проведенной работы было установлено, что адсорбционные характеристики новой серии сорбентов (АН-КПВ), полученных в результате паровоздушной активации при температуре 850°С, скорости подъема температуры 27°С мин–1 и скорости подачи водяного пара 3.5 нл мин–1 и воздуха 3 нл мин–1, превосходят по адсорбционным характеристикам активированный уголь АН-К1, полученный при активации только водяным паром и много лучше сорбентов [3], взятых в качестве образца сравнения (табл. 1). Найдено, что объем микропор АН-КПВ (0.36 см3 г–1) в 1.5 раза меньше объема микропор АН-К1, а объем супермикропор АН-КПВ (0.32 см3 г–1) примерно во столько же раз больше, чем у АН-К1.
Было установлено, что адсорбционная емкость АН-КПВ по I2, C6H6 и МГ, примерно в 1.3 раза больше адсорбционной емкости АН-К1, из чего следует, что размер пор новой серии сорбентов соизмерим с размерами больших молекул адсорбатов. Варьирование технологических режимов процесса активации и использование оригинальной комбинации химических модификаторов для процедуры деминерализации антрацита показали важную роль скорости подъема температуры и значений скоростей расхода паровоздушной смеси в процессе активации антрацита.
ВЫВОДЫ
Проведено исследование закономерностей активации антрацита паровоздушной смесью до разных степеней обгара в интервале температур 750–900°С. Показано, что оптимальная комбинация температуры (850°С), скорости подъема температуры (25–30°С мин–1), продолжительности активации (3–4 ч) расхода водяного пара Vпар = = 3.5–4.0 нл мин–1 вместе с эффективной деминерализацией антрацита растворами HCl и NH4OH приводит к существенному повышению объема сорбирующих пор по бензолу (до 0.32 см3 г–1), удельной поверхности сорбента (до 900 м2 г–1) при достаточно высоком выходе готового продукта (60–65%) и сорбционной активности активированного угля по тестовым веществам (I2, C6H6, красители метиленовый голубой и профлавин-ацетат).
Показано, что в процессе активации смесью водяного пара и воздуха увеличивается реакционная способность антрацита, заключающаяся в повышении суммарного объема супермикропор до 0.32 см3 г–1 по сравнению с другими методами активации, а также в увеличении сорбционной активности по ионам Cd2+, Pb2+ и Cu2+ до 5.85, 5.75 и 5.25 ммоль г–1 соответственно.
Активированный уголь, полученный в результате использования паровоздушной смеси для активации антрацита и выбора оптимальных технологических параметров (температуры активации, скорости подъема температуры активации и расхода пара и воздуха), обладает высокой адсорбционной емкостью по МГ (≈ в 2.5 раза), по ПА (в 4 раза) и по ионам тяжелых металлов (в 4–5 раз), превышающей адсорбционную емкость (при одинаковых концентрациях тестовых веществ) активированных углей, полученных при активации только водяным паром. АУ АН-КПВ может быть использован в качестве углеродного катионита в процессах водоочистки.
Список литературы
Хоанг Ким Бонг, Александров В.В. // Изв. АН УССР, Институт угля, Харьков. 1998. Вып. 2. С. 35.
Мухин В.М., Тарасов А.В., Клушин В.Н. Активные угли России. М.: Металлургия, 2000. Гл. 2, Гл. 3. 352 с.
Кузнецов Б.Н., Щипко М.Л., Чесноков Н.В., Милошенко Т.П., Сафонова Л.В. и др. // Химия в интересах устойчивого развития. 2005. № 13. С. 521.
Parra J.B., Pis J.J., de Sousa J.C., Pajares J.A., Bansal R.C. // Carbon. 1996. V. 34. № 6. P. 783. https://doi.org/10.1016/0008-6223 (96)00030-9
Lyubchik S.B., Benaddi H., Shapranov V.V., Beguin F. // Carbon. 1997. V. 35. P. 162.https://doi.org/10.1016/j.jpcs.2003.10.006
Daulan C., Lyubchik S.B., Rouzaud J.-N., Beguin F. // Fuel. 1998. V. 77. № 6. P. 495. https://doi.org/10.1016/S0016-2361(97) 00273-1
Bodoev N.V., Gruber R., Kucherenko V.A., Guet J.-M., Khabarova T.V., Cohaut N., Heintz O., Rokosova N.N. // Fuel. 1998. V. 77. № 6. P. 473. https://doi.org/10.1016/S0016-2361(97)00273-1
Тамаркина Ю.В., Хабарова В.А., Кучеренко В.А., Шендрик Т.Г. // ХТТ. 2005. № 3. С. 44.
Tamarkina Y.V., Shendrik T.G., Kurcherenko V.A. // Journal of Siberian Federal University. Chemistry. 2015. V. 1. № 8. P. 99.
Hoang Kim Bong, Temkin O.N., Hoang Huu Binh, Yamandiy D.I. // J. Chem. Chem. Eng. (USA). 2011. V. 5. P. 473.
Митракова Т.Н., Лозинская Е.Ф., Хоанг Ким Бонг, Темкин О.Н. // Вода: химия и экология. 2015. № 12. С. 56.
Lyubchik S.B., Benoit R., Beguin F. // Carbon. 2002. V. 40. P. 1287. https://doi.org/10.1016/S0008-6223(01)00288-3
Bratek K., Bratek W., Gerus-Piasecka I., Jasienko S., Wilk P. // Fuel. 2002. V. 81. P. 97. https://doi.org/10.1016/S0016-2361(01)00120-X
Кураков Ю.И., Самофалов В.С. // Изв. вузов. Сев.-Кавк. регион. Естеств. науки. Приложение. 2003. № 7. С. 96.
Бервено А.В., Бервено В.П. // Ползуновский вестник. 2009. № 3. С. 189.
Стрелко В.В., Герасименко Н.В., Картель Н.Т. // ЖПХ. 2001. Т. 74. Вып. 6. С. 931.
Картель Н.Т., Сыч Н.В., Стрелко В.В. // ЖПХ. 2002. Т .75. Вып. 9. С. 1443.
Kuznetsov B.N., Chesnokov N.V., Mikova N.M. et al. // Journal of Siberian Federal University, Chemistry. 2008. № 1. P. 3.
Стрелко В.В., Герасименко (Сыч) Н.В., Картель Н.Т., Миронюк Т.И. // Химия твердого топлива. 2003. № 1. С. 77.
Сыч Н.В., Картель Н.Т., Ковтун М.Ф. // ЖПХ. 2003. Т. 76. Вып. 9. С. 1463.
Hoang Kim Bong, Pestov S.M., Flid V.R., Karaeva A.R., Peshnev B.V. // Open J. Applied Sciences. 2017. V. 7. P. 720. https://doi.org/10.4236/OJAPPS 2017.712051
Волоскова Е.В., Полубояров В.А., Жданок А.А., Милошенко Т.П., Фетисова О.Ю., Меленевский В.Н. // ХТТ. 2010. Т. 4. С. 19.
Тамамьян А.Н., Мухин В.М., Зубова И.Н., Макеева В.А., Полякова В.А., Яковлева Е.Н., Таратун М.Н. // Патент РФ № 2228293. 2004.
Мухин В.М., Учанов П.В., Сотникова Н.И. // Сорбционные и хроматографические процессы. 2013. Т. 13. Вып. 1. С. 83.
Мухин В.М., Учанов П.В. // Успехи в химии и химической технологии. 2013. Т. XXVII. № 9. С. 35.
Гонцов А.А., Коссинский В.А., Черников А.Б. // Патент РФ № 2177906.
Хоанг Ким Бонг, Тимофеев В.С., Темкин О.Н., Тимошенко А.В., Калия О.Л., Ворожцов Г.Н., Кузнецова Н.А. // Патент РФ № 2303569. Опубликован 27.07.2007.
Хоанг Ким Бонг, Тимофеев В.С., Темкин О.Н., Валитова В.Э., Грешняева И.М., Черкасова О.А., Калия О.Л., Ворожцов Г.Н., Кузнецова Н.А. // Патент РФ № 2394760. Опубликован 27.07.2007.
Хоанг Ким Бонг, Темкин О.Н., Кузнецова Н.А., Калия О.Л. // Водоочистка. Водоподготовка. Водоснабжение. 2012. Т. 7. № 67. С. 20.
Дубинин М.М., Поляков Н.С., Устинов Е.А. // Изв. АН СССР, Серия химическая. 1985. № 12. С. 2680.
Карнаухов А.П. Адсорбция, Текстура дисперсных и пористых материалов, Новосибирск: Наука, Сибирское предприятие РАН, 1999. Гл. 2-3. 470 с.
Яцимирский К.Б., Васильев В.П. Константы нестойкости комплексных соединений. М.: Изд. АН СССР, 1959. Гл. 3. 210 с.
Русьянова Н.Д. Углехимия. М.: Наука, 2000. Гл. 4. 320 с.
Дополнительные материалы отсутствуют.
Инструменты
Химия твердого топлива