

Кинетика и катализ, 2019, T. 60, № 2, стр. 152-168
Научное наследие Георгия Константиновича Борескова
Т. В. Андрушкевич 1, *, В. И. Бухтияров 1, **
1 ФГБУН Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: andrushk@catalysis.ru
** E-mail: vib@catalysis.ru
Поступила в редакцию 11.12.2018
После доработки 11.12.2018
Принята к публикации 11.12.2018
Аннотация
Обзор представляет собой краткую аннотацию огромного научного наследия академика Георгия Константиновича Борескова – известного русского ученого в области катализа, химической кинетики и химической технологии. Г.К. Боресков был убежденным сторонником химической теории катализа, основные положения которой в гетерогенном катализе он обосновал и последовательно развивал. К таким положениями прежде всего относятся: химическая природа промежуточного взаимодействия реактантов с катализатором; ключевая роль энергии связи промежуточных поверхностных соединений в скорости и направлении протекания реакций; динамическое единство системы “реактанты–катализатор”; приблизительное постоянство удельной каталитической активности веществ одинакового химического состава и структуры. Г.К. Боресков внес значительный вклад в теорию кинетики каталитических реакций и в создание и становление метода математического моделирования. Он показал решающее значение пористой структуры в приготовлении практических катализаторов. Технологии многих процессов были разработаны под руководством или непосредственном участии Георгия Константиновича. Положения теории катализа Г.К. Борескова являются основой современной науки о катализе и в существенной мере определяют ее развитие.
ВВЕДЕНИЕ
20 апреля 2017 г. исполнилось сто десять лет со дня рождения академика Георгия Константиновича Борескова – известного русского ученого в области катализа, химической кинетики и химической технологии. В честь этого события основанный им в 1958 г. и получивший в 1992 г. его имя Институт катализа им. Г.К. Борескова СО РАН организовал и провел ставшие уже традиционными “Боресковские Чтения”, в которых приняли участие около 200 специалистов из разных стран мира, в том числе из России, Казахстана, США, Польши, Испании, Германии, Италии, Японии, Нидерландов и Великобритании. Тематика конференции соответствовала основным научным интересам и достижениям Георгия Константиновича:
1) Формирование активного состояния катализатора под воздействием реакционной среды.
2) Структурная чувствительность, размерные эффекты, одноцентровый катализ.
3) Поверхностные формы активного кислорода: локальная структура, электронное строение, каталитические свойства.
4) Механизмы окислительных реакций.
5) От кинетики каталитических реакций до технологических процессов.
На конференции, которая состоялась 19–21 апреля 2017 г., было представлено 3 пленарных и 11 ключевых лекций, 36 устных и около 100 постерных презентаций. Конференцию принимали Дом ученых СО РАН и Институт катализа.
Конференция памяти академика Борескова стала поводом поговорить о Георгии Константиновиче, о влиянии его идей и разработок на развитие каталитической науки и каталитических технологий. Можно привести слова, сказанные профессором Кардиффского Университета (Великобритания) Грэмом Хатчингсом (G. Hutchings): “Многие статьи Борескова были опубликованы в одном русском журнале (прим. “Кинетика и катализ”), который был переведен на английский. В нем печатали очень короткие статьи, но в них было много интересных идей. Я смог их использовать в проектах, над которыми работал в промышленности. Мы осознавали значимость этих идей и работали с ними. Вот в чем проявилось для меня влияние Борескова.” Другой приглашенный участник конференции профессор Валерий Давидович Соколовский (Renovia Inc. США) указал на сохранение актуальности многих научных идей Борескова, подчеркнув важность того, что “Боресковские Чтения” позволяют новому поколению исследователей приобщиться к тем идеям, которые были заложены при организации Института катализа и которые не потеряли свою ценность спустя много лет.
Желая расширить круг молодых исследователей, которые, начиная свой творческий путь в области катализа, могли бы познакомиться с основополагающими идеями Георгия Константиновича Борескова и оценить вклад отечественных ученых в мировую науку, мы и решили подготовить этот обзор, в котором в кратком виде изложены основные теоретические представления Г.К. Борескова и показано, какое влияние они оказали на развитие технологических основ катализа.
ФУНДАМЕНТАЛЬНЫЕ ИССЛЕДОВАНИЯ
1. Удельная каталитическая активность. Правило Борескова
Исторический фон, на котором происходило становление представлений Г.К. Борескова, характеризовался интенсивным развитием и конкуренцией между собой разнообразных теоретических концепций. Теоретические и экспериментальные исследования Г.К. Борескова оказались исключительно актуальными и сыграли важную роль в формировании науки о катализе. В 1953 г. Георгий Константинович выступил на Всесоюзном совещании по гетерогенному катализу в химической промышленности с изложением своих взглядов на сущность каталитического действия [1]. В этом докладе он последовательно обосновал принципы химического подхода к катализу и сформулировал ряд важных положений в гетерогенном катализе: о химической природе поверхностных промежуточных соединениях и роли их энергии связи с катализатором, об удельной каталитической активности и принципе ее постоянства, о взаимосвязи катализатора и реакционной среды как единой системы.
Суть взглядов Георгия Константиновича, отраженных в докладе, выражена его словами: “Химическая природа промежуточного взаимодействия проявляется в соответствии химических свойств катализатора со свойствами участвующих в реакции веществ и в специфичности действия катализаторов. Cилы связи, обуславливающие образование поверхностных соединений, существенных для катализа, имеют ту же природу, что и силы связи в обычных химических соединениях. Построение теории катализа, способной объяснить и предвидеть каталитическое действие, возможно только на основе представлений о катализе как о химическом в своей сущности явлении, путем изучения особенностей промежуточного химического взаимодействия” [1]. Сейчас идею о химической сущности катализа разделяют все исследователи, но ее признание потребовало длительного времени и сопровождалось острыми дискуссиями.
Боресков предложил относить скорость каталитической реакции к единице поверхности, назвав полученную величину удельной каталитической активностью (УКА). В настоящее время УКА является общепринятой характеристикой катализаторов, без которой не мыслится количественное описание и сравнение их активности. Введение этой величины оказалось очень своевременным, так как именно тогда были разработаны надежные адсорбционные методы измерения внутренней поверхности пористых катализаторов и раздельного измерения поверхности отдельных компонентов некоторых сложных систем, прежде всего, в нанесенных металлических катализаторах.
Систематические исследования многих металлических и оксидных катализаторов в отношении различных реакций, проведенные под руководством Г.К. Борескова в 1945–1955 гг., показали, что УКА приблизительно постоянна при значительной вариации величины поверхности и условий приготовления катализатора одного и того же химического состава. Первое экспериментальное подтверждение этого правила было получено для реакции гидролиза хлорбензола на силикагеле [2]. При изменении в семь раз поверхности силикагеля, обусловленном температурой прокаливания образцов, его активность, отнесенная к квадратному метру площади поверхности, оставалась практически на одном уровне. При этом активность грамма катализатора возрастала пропорционально величине поверхности.
Постоянство УКА было выявлено и в дегидратации этилового спирта на γ-Al2O3, полученном из разных веществ и прокаленном при различных температурах [3]. При замене γ-Al2O3 на α-Al2O3 активность резко менялась, при этом изменялось и направление реакции – дегидратирующий катализатор становился дегидрирующим. Этот эксперимент продемонстрировал, что на свойства катализаторов влияет и их структура даже при неизменном химическом составе. Постоянство удельной активности в окислении диоксида серы было также показано для образцов Рt с поверхностью, различающейся на пять порядков [4].
Эти данные позволили сделать вывод, что удельная каталитическая активность мало зависит от величины поверхности и определяется, в основном, химическим составом и строением катализатора. Другими словами, при неизменном химическом составе и структуре катализатора его каталитическая активность, отнесенная к единице величины поверхности, приблизительно постоянна. Принцип постоянства УКА вошел в науку о катализе под названием “правило Борескова”.
Однако сам Георгий Константинович указывал, что этот принцип не универсален. На основе накопившегося экспериментального материала он детально проанализировал причины отклонений от УКА [5]. Так, в его работах [6] было продемонстрировано, что сокращение размера кристаллов нанесенного металла ниже 5 нм может приводить к увеличению или уменьшению активности в полном соответствии с введенным Мишелем Бударом (M. Boudart) понятием структурной чувствительности реакций для нанесенных металлических катализаторов [7]. Позднее концепция структурной чувствительности каталитических реакций, вызывающей соответствующие изменения УКА в зависимости от размера частиц нанесенного металла, нашла активное развитие в работах Мишеля Ше (M. Che) [8] и Рутгера Ван Сантена (R. Van Santen) [9, 10]. Активное участие в проведении подобных исследований принимают и российские каталитики, в том числе в Институте катализа имени Г.К. Борескова СО РАН [11–13].
Другой важной причиной отклонения от правила постоянства УКА Боресков указал трансформацию катализатора в условиях реакции под воздействием среды, которое может проявляться в изменении химического состава и структуры поверхности, размера кристаллов активного компонента, структурной чувствительности реакции, развитии различных граней кристаллов, разном распределении компонентов при одинаковом химическом составе в случае сложных катализаторов и т. д. [5].
Особенно Г.К. Боресков подчеркивал необходимость сопоставления УКА в стационарном состоянии катализаторов в условиях реакции. Когда вышеуказанное состояние, а вместе с ним и стационарная активность устанавливаются сравнительно медленно (при низких температурах), влияние предыстории катализатора на УКА может сказываться длительное время. При проведении реакции в мягких условиях создание определенного состояния катализатора открывает дополнительные возможности для его регулирования. Таким образом, установив правило постоянства УКА, Георгий Константинович четко определил и границы его применимости. Для подавляющего большинства промышленных каталитических реакций, реализуемых при повышенных температурах, стационарное состояние устанавливается быстро, так что правило приблизительного постоянства УКА может служить надежной основой подбора оптимальных катализаторов.
Г.К. Боресков отмечал, что УКА является усредненной характеристикой каталитического действия поверхности катализатора. В тех случаях, когда молекулярное описание механизма реакции требует оперирования каталитическими функциями структурных элементов поверхности (атомов или ионов), целесообразно перейти от УКА к атомной каталитической активности (АКА), отнеся величину каталитической активности к поверхностной концентрации тех атомов (ионов), которые обусловливают эту активность [6]. В настоящий момент именно АКА, получившая в англоязычной литературе широко используемое название turnover frequency (TOF), рассматривается в качестве универсальной характеристики, обеспечивающей количественное сравнение различных катализаторов.
2. Воздействие реакционной среды. Единство системы катализатор–реакция
Одним из главных принципов в представлениях Г.К. Борескова, вытекающих из химического подхода к катализу, является взаимосвязь и взаимозависимость катализатора и реагирующих веществ [14]. Рассматривая систему катализатор–реагенты как единую, нельзя характеризовать поверхность катализатора заранее заданными свойствами, не зависящими от состава реакционной среды. Боресковым было выделено три группы изменений химического состава катализаторов под действием реакционной среды в ходе реакции [14]. Это изменения:
1) химического состава, приводящие к фазовому превращению активного компонента катализатора;
2) объемного состава катализатора, не сопровождающиеся фазовыми превращениями;
3) состава поверхностного слоя катализатора.
Фазовые превращения каталитически активного компонента в результате воздействия реакционной смеси приводят обычно к резкому росту или падению каталитической активности. Благодаря этому их легко обнаружить и учитывать при исследовании катализаторов. Указанные превращения широко распространены и играют большую роль при подборе катализаторов и выборе оптимальных условий проведения реакции.
Более общее значение для оксидов переходных металлов имеют изменения состава без фазового превращения. Они практически всегда присутствуют, но влияние их на активность проявляется менее отчетливо и обычно не учитывается. Так, под воздействием окислительно-восстановительной среды оксиды переходных металлов могут частично восстанавливаться. Это проявляется в появлении кислородных вакансий не только на поверхности, но и в объеме твердого катализатора. Под влиянием реакционной системы может стать другим соотношение компонентов, входящих в состав катализатора, произойти растворение новых компонентов или удаление старых.
Трансформация поверхностного состава катализатора происходит под действием каталитических стадий в результате увеличения или уменьшения соотношения их скоростей при изменении состава реагирующей смеси. Как правило, варьировании состава поверхности распространяется на весь объем катализатора или, по крайней мере, на приповерхностные слои путем диффузии. Под воздействием реакционной среды состав катализатора может меняться, приближаясь с большей или меньшей скоростью к некоторой постоянной, стационарной величине, зависящей от состава реакционной смеси. Если реакционная система находится в равновесии, то стационарный состав катализатора однозначно определяется условиями равновесия с любым из компонентов реакционной смеси. В общем случае, когда равновесие катализируемой реакции не достигнуто, стационарный состав катализатора может варьироваться в определенных пределах в зависимости от соотношения скоростей взаимодействия катализатора с отдельными компонентами реакционной смеси [14].
В условиях реакции действует следующее правило: “Твердые катализаторы являются лабильными компонентами реакционной системы и под воздействием реакционной смеси меняют химический состав, структуру поверхности и каталитические свойства. Каждому составу реакционной смеси и температуре отвечает определенное состояние катализатора, независящее от его исходного состояния” [15].
Идея о воздействии реакционной среды на катализатор получила убедительное подтверждение в многочисленных экспериментальных исследованиях, выполненных под руководством Г.К. Борескова. Были обнаружены различные конкретные механизмы превращения катализатора в ходе реакции, сводящиеся как к изменению состава поверхностного слоя, так и к его структурной перестройке вплоть до другого фазового состава. Так, в реакции окислительного дегидрирования бутилена на железосурьмяном катализаторе вариация соотношения С4Н8/О2 сопровождается изменением поверхностного состава, активности и селективности катализатора [16]. В случае оксида железа уменьшение концентрации кислорода в реакционной смеси приводит к фазовому превращению.
Структурные трансформации катализаторов происходят и при хемосорбции некоторых газов. Под влиянием кислорода происходит реконструкция граней (100) и (110) никеля и грани (110) иридия с образованием в обоих случаях фасетированных граней (111) [17].
Изменение строения и состава поверхности при приближении катализатора к стационарному состоянию может происходить под действием стадий каталитической реакции, так и под влиянием побочных процессов. Эти механизмы иллюстрирует пример окисления акролеина на ванадий-молибденовом катализаторе [18, 19]. Установление стационарного состояния связано с восстановлением катализатора, которое является стадией каталитической реакции образования акриловой кислоты. Восстановление приводит к удалению поверхностного кислорода, увеличению его энергии связи и понижению степени окисления ванадия. Эти процессы сопровождаются резким повышением скорости и селективности образования основного продукта – акриловой кислоты.
Таким образом, по Борескову важным является “подход к катализатору и реагирующим веществам как к единой химической системе, в которой превращения испытывают не только реагенты под влиянием катализатора, но и катализатор в результате химического взаимодействия с реагентами” [15]. Многообразие явлений, происходящих с катализатором в реакционной среде, чрезвычайно велико. В cборнике научных трудов сотрудников Института катализа [20], посвященном тридцатилетию выхода первой статьи, в которой Г.К. Боресков сформулировал систему представлений об изменении катализатора под воздействием реакционной среды, приведены многие примеры изменения структурных, химических и каталитических свойств в условиях реакции, предложены теоретические модели и кинетика каталитических реакций, учитывающие воздействие реакционной среды.
Огромный вклад в развитие проблемы воздействия среды на катализатор внес А.Я. Розовский, который ввел понятия о специфических и неспецифических взаимодействиях компонентов реакции с катализатором. В его монографии [21] представлены многочисленные примеры и сделаны обобщения, подтверждающие действенность концепции влияния реакционной среды.
Концепция выражает одну из важнейших закономерностей гетерогенного катализа и время подтверждает ее теоретическое и практическое значение. Так, одним из генеральных направлений поиска катализаторов для реакций селективного окисления стал дизайн новых материалов, основанный на понимании работающего состояния твердого катализатора [22]. Понимание важности получаемых в данном направлении науки результатов обеспечило на рубеже XXI века бурное развитие физических методов для исследования катализаторов в режиме in situ, т.е. в присутствии реакционной смеси [23–26]. Это привело к появлению новых возможностей для установления сути явлений воздействия среды на катализатор и, как следствие, пониманию процессов формирования активных центров гетерогенных катализаторов. Исследователи из Института катализа им. Г.К. Борескова СО РАН занимают лидирующее положение среди отечественных ученых и сохраняют мировой уровень в данной области исследований [26, 27].
3. Значение энергии промежуточного взаимодействия реагентов и катализатора
Изменение скорости реакции в присутствии катализатора обусловлено промежуточным химическим взаимодействием реагирующих веществ с катализатором. Их энергия связи с катализатором определяет скорость и направление протекания реакции. Представление об оптимальной величине энергии промежуточного взаимодействия было сформулировано А.А. Баландиным в форме принципа энергетического соответствия мультиплетной теории [28–30].
Этот подход получил дальнейшее развитие в работах Makishima и сотр. [31], Sachtler и Farenfort [32], Темкина [33], Голодца и Ройтера [34], Tanaka и Tamaru [35]. Общим в вышеуказанном подходе является приближенная оценка величины оптимальной энергии промежуточного взаимодействия по известному уравнению Бренстеда–Поляни:
Знак плюс соответствует разрыву связи реактант–катализатор в лимитирующей стадии реакции, знак минус – образованию такой связи; α – постоянная для данной реакции величина, лежащая между нулем и единицей; q – теплота химической реакции. Для двухстадийной реакции это уравнение описывается хорошо известной вулканообразной кривой с ветвями, отражающими зависимости для разных лимитирующих стадии. Минимум на этой кривой соответствует оптимальной энергии связи и максимальной скорости реакции.
В основе подхода Г.К. Борескова [36] лежит предположение, что в пределах ограниченных групп катализаторов и реакций изменение энергии активного комплекса можно приближенно оценивать из величин энергии отдельных связей, разрывающихся или возникающих при образовании активного комплекса. Можно предвидеть линейную зависимость энергии активации реакции на различных катализаторах от изменения энергии связи реактант–катализатор. Строгое рассмотрение проблемы оптимальной энергии связи промежуточного взаимодействия реактантов с катализатором на основе уравнения Бренстеда–Поляни сделано Г.К. Боресковым в работах [37, 38].
Вывод об оптимальной энергии промежуточного взаимодействия является действенным и для более сложных реакций. Если реакция протекает по нескольким направлениям, энергия промежуточного взаимодействия будет влиять на скорость превращения начальных веществ и селективность процесса. Анализ экспериментальных данных указывает на многочисленные случаи линейных корреляций между изменениями энергии активации и различными характеристиками энергии связи реактант–катализатор [39, 40]. Концепция определяющего значения энергии связи кислород–катализатор была показана для реакций каталитического окисления на твердых катализаторах [41]. Г.К. Боресков с сотрудниками на базе огромного экспериментального материала (десятки реакций и катализаторов) исследовал влияние прочности связи Ме–О катализаторов на каталитическую активность. Для определения прочности связи Ме–О были использованы независимые методы – изотопный обмен, измерение температурной зависимости парциального давления кислорода и адсорбционная микрокалориметрия. Развитие этих методов в существенной мере обязано Георгию Константиновичу и его ученикам. В результате этих исследований были установлены количественные закономерности между прочностью связи кислорода с поверхностью катализатора и его каталитической активностью в реакциях окисления водорода [42], метана [43], ацетилена [44], бензола [45], аммиака [46] и разложения оксидов азота [47]. Во всех случаях скорость реакций падала с увеличением энергии связи Ме–О, свидетельствуя о том, что лимитирующей стадией является отрыв кислорода от катализатора. Полученные количественные закономерности в дальнейшем были успешно применены при подборе катализаторов глубокого окисления вредных примесей в промышленных производствах.
В реакциях селективного окисления предсказание только величины энергии связи кислорода недостаточно. Более существенно для протекания реакции в селективном направлении “специфическое” взаимодействие окисляемого вещества (субстрата) [48–50]. Этот термин подразумевает способность катализатора генерировать поверхностное соединение определенной структуры, слабо связанное с активным центром. Так, в образовании карбоновых кислот при окислении функциональных углеводородов на оксидных ванадий-молибденовых и ванадий-титановых катализаторах непосредственными предшественниками являются соответствующие поверхностные карбоксилаты [51]. При окислении акролеина наблюдается корреляция прочности связи поверхностных акрилатов с селективностью: слабо связанные акрилаты превращаются в акриловую кислоту, прочно связанные – в продукты деструкции и глубокого окисления [52]. Обстоятельный обзор литературных данных по промежуточным соединениям различных гетерогенно-каталитических реакций содержится в монографии Крылова и Матышака [53]. Отметим, что авторы придерживаются химической теории катализа и разделяют концепцию Борескова воздействия реакционной среды.
Метод предсказания каталитической активности на базе изменения определенных связей между реактантом и катализатором, образующихся или разрывающихся при протекании реакции, имеет общее значение и успешно применим не только к реакциям глубокого окисления, но и к процессам гидрирования, гидрогенолиза, фиксации азота и другим. Позднее роль энергетического фактора в катализе была рассмотрена Георгием Константиновичем в более общей форме, позволившей соотнести оптимальные энергии связи реагентов с характеристиками стадийного механизма реакции [54].
4. Классификация механизмов
Проблема верификации механизмов окислительного катализа поставлена достаточно давно и требует выбора между двумя возможностями – происходит ли реакция каталитического окисления путем попеременного окисления и восстановления катализатора (раздельный или стадийный механизм) или через одновременное взаимодействие с катализатором участников реакции (слитный, концертный или ассоциативный механизм). Концепция стадийного механизма была сформулирована в работах Марса и Ван Кревелена [55], слитного – обоснована в общей форме Ройтером [56]. Справедливость того или иного механизма решается путем сопоставления скорости каталитической реакции со скоростями отдельных этапов – окисления и восстановления катализатора, находящегося в стационарном состоянии. Равенство скорости каталитической реакции скоростям этапов восстановления и окисления катализатора говорит о стадийном механизме протекания реакции, превышение скорости реакции над скоростями этапов – об ассоциативном механизме. Дополнительным подтверждением стадийного механизма является совпадение энергий активации каталитической реакции и ее предполагаемых стадий [54, 57]. Однако большинство попыток ответить на этот вопрос не было подкреплено обоснованными экспериментами: при раздельном проведении стадий не контролировалось изменение поверхности катализатора, а при использовании метода меченых атомов не учитывались побочные процессы изотопного обмена.
В цикле работ, выполненных в Институте катализа под руководством Г.К. Борескова, принципиально важным был строгий контроль состояния поверхности катализатора. Для реакций окисления водорода, оксида углерода и серы, парафинов, олефинов и функциональных углеводородов в присутствии большой серии катализаторов были сопоставлены скорости раздельно проводимых стадий (окисления и восстановления) со скоростью катализа на катализаторе в стационарном состоянии [54, 58].
Было показано, что при повышенных температурах некоторые реакции глубокого окисления протекают по стадийному механизму, а при понижении температуры преобладающим становится слитный (ассоциативный) механизм. В качестве примера это положение демонстрирует реакция окисления СО на окислах металлов четвертого периода. При высоких температурах скорости восстановления и реокисления катализаторов в стационарном состоянии совпадают со скоростью каталитической реакции. При снижении температуры скорость каталитической реакции падает медленнее, чем скорость ее отдельных этапов, и превышение скорости реакции тем больше, чем ниже температура. К такому же выводу привели проведенные Г.К. Боресковым исследования механизма изотопного обмена и переноса кислорода при каталитическом окислении [58].
Реакции парциального окисления преимущественно протекают по стадийному механизму. Экспериментально это было показано для окислительного дегидрирования бутилена на железосурьмяном катализаторе [59], окисления пропилена в акролеин на висмут- или молибденсодержащих катализаторах [60], окисления акролеина в акриловую кислоту на ванадий-молибденовом катализаторе [61]. При этом полное окисление осуществляется при значительном участии кислорода газовой фазы по ассоциативному механизму.
Полученные результаты имеют принципиальное значение при рассмотрении основных форм каталитического действия. Согласно Борескову, сущность каталитического действия заключается в том, что катализатор, входя в состав активного комплекса основных стадий реакции, способствует сохранению химической связанности в процессе превращения. Мерой такой связанности может служить степень компенсации энергии разрывающихся связей энергией образующихся связей. Основные формы каталитического действия определяются характером такой компенсации, которая может происходить двумя принципиально различными способами, обуславливающими механизм промежуточного химического взаимодействия реактантов с катализатором: стадийный или слитный. Этот подход, разрабатываемый Боресковым в последние годы, позволил ему связать воедино различные проявления химической природы катализа, рассмотренные выше.
По степени компенсации энергии он выделил три основные формы воздействия катализатора на химическую реакцию [54]:
1) реализация стадийного превращения в результате раздельного взаимодействия реактантов с катализатором;
2) увеличение степени компенсации при вхождении катализатора в состав активного комплекса при синхронном протекании реакции;
3) осуществление цепного механизма с межстадийной компенсацией благодаря участию катализатора.
При стадийном механизме, когда разрыв связей происходит поэтапно, снижение энергии активации итогового превращения и увеличение его скорости достигается только в тех случаях, когда разделение на стадии приводит к уменьшению молекулярности реакции; при этом существенную роль могут играть также степень компенсации на всех стадиях, простота строения активных промежуточных частиц. На стадиях механизма этого типа катализатор испытывает обратимые химические превращения, что приводит к быстрому установлению стационарного состояния.
Основным фактором ускоряющего действия катализатора при слитных механизмах является значительное повышение степени компенсации энергии за счет синхронного образования всего комплекса связей молекул продуктов реакции. В таких механизмах катализатор вступает в непосредственное химическое взаимодействие со всеми реагентами, входя в состав сложного активированного комплекса и высвобождаясь из него практически неизменным. В этих случаях реакционная среда влияет на катализатор сравнительно слабо и стационарный состав устанавливается медленно.
При цепном механизме синхронное взаимодействие заменяется стадийным, цепным, с образованием на каждой стадии частиц, аккумулирующих энергию химической реакции и вступающих в контакт с реактантами в последующих стадиях. Высокая скорость реакции достигается благодаря тому, что концентрация богатых энергией промежуточных частиц может стать много больше равновесной за счет свободной энергии реакции. На всех стадиях цепного механизма в реакции участвует лишняя по сравнению со стехиометрией частица, роль которой выполняет катализатор.
В подавляющем большинстве химических превращений в гомогенном, гетерогенном и ферментативном катализе наблюдается равновесное распределение энергии в промежуточных продуктах, и катализ осуществляется по первым двум механизмам. Если равновесное распределение энергии не устанавливается на каком-либо этапе реакции, то избыточная энергия может снизить энергию активации следующей стадии. Эта форма межстадийной компенсации (компенсация второго рода) не выходит за пределы одного реакционного цикла и тем существенно отличается от компенсации в цепных реакциях.
Более подробно условия реализации различных форм каталитического действия и их роль в природных и промышленных химических процессах была рассмотрена Г.К. Боресковым в работах [37, 54, 62, 63].
5. Кинетика реакций гетерогенного катализа
Кинетические исследования, проводимые Г.К. Боресковым, с одной стороны, всегда были направлены на выяснение детального механизма каталитической реакции, а с другой – служили надежной основой для расчета химических реакторов. Для создания кинетической модели он подчеркивал необходимость всестороннего изучения системы “катализатор–реакция” [64, 65]. Чем более обоснованы теоретические представлении о протекании реакции и чем более достоверны экспериментальные данные, полученные независимыми некинетическими методами, тем надежнее кинетические уравнения.
Классическим примером кинетических исследований является цикл работ по изучению кинетики окисления SO2 на ванадиевых катализаторах. Одновременно с анализом наблюдаемых кинетических закономерностей определяли состояние и состав катализатора. Было установлено, что в ванадиевом катализаторе при условиях реакции активный компонент находится в жидком состоянии и представляет собой раствор сульфованадатов калия в расплавленном пиросульфате калия [66]. Благодаря такому уникальному свойству ванадиевого катализатора реакция протекает не на поверхности, а во всем объеме расплава. Всестороннее изучение катализатора обусловило достоверность кинетического уравнения, которое применяли для расчета вплоть до 60-х гг. В настоящее время пользуются его уточненным вариантом, называемым именем Борескова–Иванова [67]:
В дальнейшем был изучен детальный механизм реакции. Немаловажную роль в кинетических исследованиях сыграл и метод измерения каталитической активности в проточно-циркуляционных установках, исключающих градиенты температур и концентраций по слою катализатора и позволяющих определять скорость реакции в широком интервале степени превращения реагентов. Уравнение, выведенное на основе детального механизма, совпадает с прежним уравнением в области промышленных условий осуществления процессах [68].
При рассмотрении кинетического описания каталитической реакции Боресков принимал в расчет тот факт, что катализатор – не просто место осуществления реакции, а участник химического взаимодействия, активность которого может существенно меняться при вариации состава реакционной смеси. Результатом такого подхода было предположение о том, что влияние компонентов реакционной смеси на скорость каталитической реакции должно проявляться двояко: во-первых, через их взаимодействие на поверхности катализатора в соотношениях, соответствующих механизму реакции; во-вторых, в результате воздействия реакционной смеси на свойства катализатора, приводя к изменению констант скорости реакций, в которых участвует катализатор. В результате величина, рассматриваемая как константа, не постоянна, а является функцией состава [65].
В 1959 г. в статье “Влияние взаимодействия реакционной системы и катализатора на кинетику каталитических реакций” Боресков предложил формулу, учитывающую эти обе составляющие [69]:
где w – скорость каталитической реакции; k – константа скорости реакции; Р – парциальные давления реагентов в смеси; θ – степень покрытия поверхности. Здесь и в [65] Георгий Константинович указал на необходимость раздельного определения обоих сомножителей этого уравнения. Раскрытие функциональной зависимости во втором множителе дает более строгую базу для выявления механизма реакции, а определение функциональной зависимости первого множителя позволяет подойти к объяснению природы действия катализатора и возможности регулирования его свойств.Учет воздействия реакционной среды не исключает влияния на кинетические закономерности неоднородности поверхности катализатора. Но нужно иметь в виду, что характер неоднородности не является постоянным и наперед заданным, а формируется под действием реакционной среды и в определенной степени представляет собой функцию ее состава.
Подходы, предложенные Георгием Константиновичем, были применены при изучении кинетики реакции окисления двуокиси серы [68], окислительного дегидрирования бутиленов [70], окисления пропилена [71].
Процессы влияния реакционной смеси на катализатор не обязательно являются стадиями каталитической реакции; изменение катализатора в условиях реакции возможно и из-за сторонних процессов. Их времена могут существенно различаться, в результате чего под действием реакционной среды на катализатор его стационарное состояние может устанавливаться крайне медленно. Вывод Борескова о том, что взаимодействие реакционной среды и катализатора приводит к зависимости кинетики реакций гетерогенного катализа от скорости достижения стационарного состава катализатора, имеет не только теоретическое, но и практическое значение.
Процессы с нестационарным состоянием катализатора все шире используются в промышленности. Это имеет место при быстрых изменениях свойств катализатора, требующих его непрерывной регенерации (каталитический крекинг, дегидрирование); в реакторах с кипящим слоем катализатора, где интенсивное продольное перемешивание катализатора приводит к отклонениям от стационарности в отношении реакционной смеси; когда стационарный состав катализатора отличается от оптимального и требуется специальная обработка катализатора перед поступлением в реактор и т.д. Медленное установление стационарного состояния в ряде случаев обуславливает выгодность ведения процесса с нестационарным составом катализатора в искусственно создаваемых условиях. Примером является известный “реверс-процесс” – переключение направления подачи реакционной смеси в слой катализатора, что приводит к ускорению реакции и регенерации тепла [72]. Технологическая перспективность нестационарного катализа делает необходимым широкое изучение реакций с учетом изменения состава не только реакционной смеси, но и твердого катализатора.
Являясь одним из крупнейших специалистов в области теории и технологии разделения и применения изотопов, Г.К. Боресков эффективно использовал в каталитических исследованиях метод меченых атомов. Большой цикл работ Георгий Константинович с сотрудниками посвятили выяснению механизма реакций и реакционной способности поверхности катализаторов с применением изотопов водорода [73], кислорода [74], азота [75]. Метод меченых атомов получил дальнейшее развитие в трудах самого Георгия Константиновича и его учеников. В частности, были получены строгие кинетические уравнения для одновременного протекания двух процессов обмена – гомомолекулярного и гетеромолекулярного – на основе введенных представлений о типах механизмов обмена [76]. Эти уравнения позволяют выявить существенные молекулярные черты механизмов процессов на поверхности катализаторов.
Особое место в научном творчестве Г.К. Борескова занимает статья “Cоотношение между молекулярностью и энергиями активации реакции в прямом и обратном направлениях” [77]. Это небольшая работа, однако без нее невозможно представить себе современную кинетику сложных реакций. В статье есть ответ на общий вопрос, как правильно вывести кинетическое уравнение сложной обратимой реакции. Дж. Хориути исследовал эту задачу в 1939 г. для частного случая – реакции, протекающей на водородном электроде, и ввел известное понятие “стехиометрического числа” [78]. Боресков, который из-за условий военного времени не был знаком с работой Хориути, независимо от него дал общее решение (Хориути впоследствии не раз указывал на оригинальность результата Георгия Константиновича).
Г.К. Боресков установил следующие соотношения для обратимых реакций:
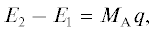
В заключение данного раздела хочется еще раз подчеркнуть, что, характеризуя ситуацию в кинетике гетерогенного катализа, Боресков постоянно подчеркивал необходимость дальнейшего развития теории в направлении учета воздействия реакционной среды на катализатор.
6. Научные основы приготовления катализаторов
Работами Г.К. Борескова были заложены научные основы приготовления катализаторов [80]. Выдвинув правило приблизительного постоянства удельной каталитической активности (УКА), Георгий Константинович указывал на необходимость принятия во внимание факторов, отражающих пористую структуру катализатора [81]. При одном и том же УКА производительность объема катализатора определяется формулой:
где А – величина УКА; S – величина активной поверхности в единице объема зерна катализатора; η – степень использования внутренней поверхности. Для количественных расчeтов надо исходить из модели пористой структуры катализатора.Степень использования внутренней поверхности определяется параметром Тиле ψ:
характеризующим отношение скорости реакции к скорости диффузионного переноса внутри зерна. Здесь L – размер зерна, D – коэффициент диффузии, ρ – истинная плотность катализатора, Сн – концентрация реагирующего вещества у наружной поверхности зерна. При малых значениях ψ (<0.5) внутренний перенос не оказывает заметного влияния на скорость реакции (область химической кинетики). При ψ > 2.5 концентрация реагирующего вещества в середине зерна становится настолько малой, что реакция практически осуществляется в слое определенной толщины, примыкающем к внешней поверхности зерна (область внутренней диффузии).
На основе этих представлений Георгий Константинович пришел к идее выбора оптимальной пористой структуры катализатора [82]. Так, для однокомпонентного катализатора поверхность единицы объема возрастает с уменьшением размера корпускул, из которых слагается зерно катализатора, а степень использования внутренней поверхности падает. С целью дальнейшего увеличения каталитической активности Г.К. Боресков предложил применять бидисперсную пористую структуру [82]. Она образуется крупными порами, гидравлический радиус которых превышает длину свободного пробега, и короткими тонкими порами, создающими большую внутреннюю поверхность. Преимущество бидисперсной структуры состоит в том, что степень использования внутренней поверхности малых частиц близка к единице. Что касается степени использования внутренней поверхности всего зерна катализатора, то она возрастает благодаря молекулярному характеру диффузии в крупных каналах между малыми частицами. Переход к бидисперсной структуре позволяет увеличить активность катализатора в 5–8 раз, а также в ряде случаев повысить и селективность сложных реакций.
Оптимальными, по Борескову, должны быть и форма, и размер зерна катализатора. Они определяются компромиссом между стремлением ослабить влияние внутридиффузионного торможения (оно падает с понижением отношения объема зерна к его поверхности) и ограничением по допустимому гидравлическому сопротивлению, которое возрастает с уменьшением размера зерен и свободного объема между ними. Георгий Константинович показал, что оптимальный размер зерен отвечает протеканию каталитической реакции в области, промежуточной между кинетической и внутридиффузионной. По Борескову, для процессов при атмосферном давлении целесообразнее применять катализаторы в виде зерен со значительно увеличенным отношением наружной поверхности к объему и большим свободным объемом между зернами. Этим условиям соответствуют кольца с тонкими стенками, особой формы лепестки и т. п. При высоких давлениях оптимальной является однородная тонкопористая структура.
Пористая структура катализатора оказывает значительное влияние и на избирательность катализаторов [83]. Переход из кинетической области во внутридиффузионную всегда сопровождается падением селективности при наличии последовательных стадий превращения основного продукта; при паралельном образовании побочных продуктов изменение селективности определяется кинетическими зависимостями основной и побочной реакций.
Поставив задачу определения оптимальных характеристик катализатора – пористой структуры, формы и размера зерна, Г.К. Боресков положил начало целому направлению исследований. Выбор носителя, обеспечивающего необходимую термическую стойкость катализатора, генезис катализатора и изменение его характеристик, в первую очередь удельной поверхности, в зависимости от условий приготовления – этим вопросам посвящен цикл исследований Г.К. Борескова и сотрудников (прежде всего, с В.А. Дзисько), начатый еще в 1930-х годах. В результате этих работ для многих катализаторов определены закономерности и найдены приемы достижения оптимальной пористой структуры, величины внутренней поверхности, распределения активного компонента по поверхности носителя и др. характеристик катализаторов. Вследствие многочисленности катализаторов и разнообразия методов приготовления обобщение этих приемов затруднительно, но основные подходы могут быть использованы достаточно широко.
7. Математическое моделирование и оптимизация каталитических процессов
Каталитический процесс включает в себя, с одной стороны, совокупность физико-химических элементарных актов, протекающих в системе “реакционная смесь–катализатор”, а с другой стороны, процессы массо- и теплообмена. Комплексное рассмотрение каталитического процесса было свойственно Г.К. Борескову еще в 1930-х гг.: эта точка зрения нашла выражение в курсах, которые он читал тогда студентам. Однако сама постановка вопроса была в ясной и сжатой форме дана Георгием Константиновичем в статье “Физико-химический расчет контактных аппаратов” еще в 1935 г. [84]. На примере окисления SO2 был впервые найден оптимальный температурный режим обратимой экзотермической реакции – падение температуры с ростом степени превращения. Именно из концепции совокупности физико-химических процессов в системе “реакционная смесь–катализатор” и процессов массо- и теплообмена в реакторе и выросло новое направление – математическое моделирование каталитических (и вообще химических) процессов, основные положения которого были сформулированы Г.К. Боресковым и М. Г. Слинько в конце 1940-х–начале 1950-х годов [85, 86].
Применение метода математического моделирования нуждается в знании закономерностей собственно химического превращения, выражаемых в виде кинетических уравнений. Последние характеризуют зависимость скорости химического превращения от состава реакционной смеси, температуры, давления и свойств катализатора. Уравнения, описывающие химические превращения, в сочетании с известными зависимостями для процессов массо- и теплообмена образуют математическое описание процесса. Метод математического моделирования получил большое распространение при расчете аппаратов с неподвижным слоем катализатора в стационарном режиме. Он широко используется при описании жидкофазных и многофазных каталитических процессов, при анализе нестационарно протекающих процессов, для расчета процессов в псевдоожиженном слое катализатора. Применение метода математического моделирования оказалось существенно важным для понимания условий устойчивой работы каталитических реакторов. Особое внимание Георгий Константинович обращал на выбор условий, обеспечивающих наибольшую скорость процесса, максимальное использование объема контакта, т. е. на оптимизацию процесса в целом [87].
Следует отметить, что становление метода математического моделирования происходило в обстановке оживленной дискуссии. Г.К. Боресков и М.Г. Слинько убедили научную общественность в плодотворности этого направления, показав невозможность применения традиционных методов теории подобия для описания химических процессов [86, 87]. На основе математического моделирования произошло формирование специальной дисциплины – “Теоретические основы химической технологии”. Отечественная наука находится здесь на мировом уровне, и это в немалой степени связано с “заделом”, созданным Г.К. Боресковым.
РАЗВИТИЕ КАТАЛИТИЧЕСКОЙ ТЕХНОЛОГИИ
Отличительной чертой деятельности Борескова являлась тесная связь теоретических работ с практическими задачами. Он участвовал в создании многих новых каталитических технологий, оказывал консультативную помощь производствам в процессе их становления, черпая, в свою очередь, из промышленной практики темы новых исследований. Конечным итогом каталитических исследований Боресков всегда считал усовершенствование известных промышленных процессов и создание принципиально новых.
Первый этап трудовой деятельности Георгия Константиновича приходится на 30-е гг. прошлого века. В химической промышленности Советского Союза того времени самым крупнотоннажным каталитическим производством было сернокислотное. Составляя основу для многих других химических производств, именно оно требовало первоочередного подъема. Боресков занялся решением проблем сернокислого катализа.
При изыскании нового сернокислотного катализатора за основу им была выбрана пятиокись ванадия. Как катализатор окисления SO2 пятиокись ванадия была известна с 1895 г. В патентной литературе было описано много составов и способов приготовления ванадиевых катализаторов. Их промышленное использование началось c 1927 г. фирмой ”Monsanto”. В Советском Союзе в контактном производстве серной кислоты для окисления сернистого ангидрида на тот момент применялся платиновый катализатор, имеющий высокую стоимость и низкую стойкость к контактным ядам. Эти недостатки существенно ограничивали возможности роста производства. Таким образом, прогресс сернокислотной промышленности прежде всего требовал разработки нового катализатора. С 1929 г. многими исследователями Советского Союза начали разрабатываться ванадиевые катализаторы.
Г.К. Боресков в результате систематических исследований различных способов приготовления, целенаправленно варьируя носители и промоторы, смог добиться высокой каталитической активности и устойчивости и за сравнительно короткое время был создан новый высокоэффективный катализатор сложного состава, получивший название БАВ (барий-алюмо-ванадиевый). Ключевым в подборе катализаторов оказалось понимание, что активный компонент в условиях реакции находится в расплавленном состоянии. Создание катализатора явилось результатом глубокого изучения физико-химических основ каталитических процессов, детального исследования кинетики и механизма реакций. Достаточно сказать, что еще в 1937 г. Георгий Константинович получил кинетическое уравнение окисления сернистого ангидрида на ванадиевом катализаторе [88], которое десятки лет использовалось во всем мире для расчета контактных аппаратов и лишь сравнительно недавно уступило место более точному уравнению Борескова–Иванова [67, 68].
Превзойдя по эксплуатационным качествам все известные ранее, этот катализатор совершил переворот в отечественном сернокислотном производстве – уже в конце 30-х гг. на катализатор БАВ перешли заводы Советского Союза, вырабатывающие серную кислоту контактным способом. Применением этого катализатора удалось увеличить производственные мощности, и в тяжелое военное время промышленность была обеспечена важным сырьем. Разработанный Г.К. Боресковым сернокислотный катализатор дал начало целому поколению эффективных катализаторов, которые широко используются в современных промышленных производствах.
Г.К. Боресков занимался всеми этапами интенсификации серно-кислотного производства – от лабораторной разработки катализатора до внедрения его в промышленность. Под его руководством были спроектированы, созданы и введены в строй новые мощные контактные аппараты. Увеличение их производительности стало возможным не только благодаря новому катализатору, но и тому, что Георгием Константиновичем были рассчитаны оптимальные условия проведения процесса, а затем в соответствии с этими условиями усовершенствованы конструкции аппаратов и технологические режимы. Много раз он принимал непосредственное участие в заводских испытаниях катализаторов, руководил пуском контактных аппаратов, сернокислотных цехов и катализаторных производств.
Важным итогом этого периода деятельности Г.К. Борескова является создание научных основ комплексного подхода к разработке и промышленному освоению катализаторов. Широкий круг возникающих при этом научных и инженерно-технических проблем сформулирован в заключение его классической монографии “Катализ в производстве серной кислоты” (1954) [89]: “Изыскание катализатора, изучение на нем кинетики контактной реакции, выяснение роли процессов переноса реагирующих веществ к поверхности катализатора, определение оптимальной внутренней структуры и размеров зерен катализатора, вычисление оптимальных температур и оптимального состава газовой смеси, расчет перепада температур внутри зерен, между их поверхностью и газовым потоком, вычисление необходимого теплоотвода на разных стадиях контактирования, создание конструкции, обеспечивающей осуществление найденного теплоотвода, и, наконец, проверка с помощью моделей равномерности распределения газа по сечению выбранной конструкции – таков неполный перечень различных, но тесно связанных между собой задач, возникающих перед исследователем каждого контактного процесса” (с. 340). В монографии представлен опыт исследований каталитического окисления двуокиси серы, отразившей на примере этой реакции различные аспекты катализа: кинетику и механизм процесса, свойства и структуру катализатора, конструирование и расчет промышленного аппарата. Такое уникальное сочетание сделало эту книгу настольной для русскоязычных специалистов в области гетерогенного катализа.
К процессу окисления двуокиси серы Г.К. Боресков обращался неоднократно в различные периоды своей научной деятельности.
Второй этап истории разработки сернокислотных катализаторов начался в шестидесятые годы в связи с интенсификацией процессов за счет внедрения агрегатов большой мощности (1000–1500 т/сут), переработки высококонцентрированного сернистого газа, внедрения систем двойного контактирования с промежуточной адсорбцией, которые обеспечивали высокие степени переработки сернистого газа и уменьшали выбросы сернистого газа в атмосферу. Реализация новых технологий была возможной только при разработке катализаторов, активных при низкой температуре и в условиях новых технологических решений. Эта проблема стала одной из ведущих в создаваемом Институте катализа. Ее второму дыханию способствовали новые возможности, полученные благодаря появлению новейших приборов для физико-химических исследований природы катализаторов, всех стадий их синтеза, генезиса, а также современные методики исследования катализаторов. Они позволили впервые обнаружить, что реакция окисления SO2 протекает в пленке расплава активного компонента [90]. Это открытие коренным образом изменило представление о природе катализатора и дало новую ориентацию для дальнейшей работы.
Под руководством Борескова, а впоследствии – его учениками, была развита научная концепция регулирования свойств сернокислотных ванадиевых катализаторов, эффективных в широком диапазоне условий, реализуемых в действующих и перспективных контактных системах, в различных слоях систем с двойным и тройным контактированием, в процессах под давлением, в нестационарных режимах и в кипящем слое [91]. Были разработаны теоретические основы оптимизации химического состава катализатора и его пористой структуры, позволяющие для любых составов реакционных смесей и температур предсказать качественный и количественный состав промотирующих добавок, обеспечивающих при заданных условиях максимальную скорость реакции. Основываясь на этих представлениях, был предложен широкий ассортимент катализаторов для различных условий ведения процесса, в том числе, новые типы катализаторов для разбавленных газов, супернизкотемпературный аналог Cs-содержащих катализаторов для повышенного давления, создана технология их производства. Под научным руководством Борескова на Воскресенском химкомбинате в 1970-е гг. построен крупнейший в Европе цех для получения сернокислотных катализаторов. В 1976 г. была пущена технологическая линия первой очереди цеха, производящая 1000 т низкотемпературного гранулированного катализатора в год. При непосредственном участии Георгия Константиновича осуществился стремительный рост мощности реакторов окисления двуокиси серы: по сравнению с довоенным временем их мощность была увеличена в десятки раз и теперь составляет 1000–1500 т серной кислоты в сутки.
Технологическая интуиция, присущая Георгию Константиновичу, проявлялась в том, что выбранные им направления исследований приводили к важным результатам, за которыми следовали и другие, причем не только в первоначально выбранной конкретной области. Успешно решая проблемы в сернокислотном катализе, Г.К. Боресков в то же время формулировал направления исследований в других практически важных областях катализа. Под его руководством была создана серия окcидных катализаторов для различных реакций окисления – парциального и полного. Эффективные катализаторы на основе ванадия были разработаны для парциального окисления о-ксилола во фталевый ангидрид, формальдегида в муравьиную кислоту, 3-пиколина в никотиновую кислоту. Серьезным достижением явился разработанный Г.К. Боресковым (совместно с Г.Д. Коловертновым, Б.И. Поповым и др.) оксидный железомолибденовый катализатор для получения формальдегида из метанола [92]. Этот катализатор обладает теми преимуществами, что образующийся на нем формальдегид содержит крайне мало метанола (безметанольный формальдегид), а процесс характеризуется гораздо меньшими расходными коэффициентами.
С начала 1950-х годов в Научно-исследовательском физико-химическом институте им. Л.Я. Карпова и в Государственном институте азотной промышленности Г.К. Боресковым были развернуты работы по изысканию катализаторов очистки в различных химических системах. Были найдены новые катализаторы для процессов тонкой очистки газов от примесей кислорода, для очистки выхлопных газов, для очистки азотно-водородной смеси, применяемой в синтезе аммиака. Накопленный опыт был в дальнейшем использован в Институте катализа, где эти работы были продолжены в направлении поиска катализаторов для экологических целей – очистки от вредных примесей промышленных выбросов и обезвреживания сточных вод. Были предложены эффективные катализаторы, прежде всего медноокисные, которые нашли применение в очистительных устройствах для различных процессов – эмалирования проводов, производства окиси этилена, производства фталевого ангидрида, производства формалина и др.
Для Борескова как для технолога был характерен интерес к “новым возможностям” катализа, т. е. к расширению применения каталитических методов и в химической технологии, и в энергетике, а также к нетривиальным технологическим решениям. Знаменательно, что именно Г.К. Боресков в 1931 г. впервые в мире предложил способ осуществления каталитических процессов в псевдоожиженном (“кипящем”) слое [93]. Псевдоожиженный слой является сейчас распространенным и весьма перспективным способом осуществления крупнотоннажных каталитических процессов. Развитию этого направления в Институте катализа Георгий Константинович придавал большое значение.
Под руководством Борескова были начаты и интенсивно продолжаются работы по применению каталитических методов для получения жидкого топлива из угля и природного газа через промежуточный синтез Фишера–Тропша [94]. Разработан метод прямого синтеза жидкого топлива на эффективных полифункциональных катализаторах [95].
Георгий Константинович руководил исследованиями по применению катализаторов в процессах сжигания топлива – новой перспективной области катализа. Некаталитическое сжигание топлив в большом избытке воздуха приводит к значительным потерям тепла с отходящими газами. Применение катализатора позволяет снизить расход воздуха и устранить этот недостаток. В Институте катализа под руководством Г.К. Борескова и Э.А. Левицкого разработаны каталитические генераторы тепла, в которых горение происходит на катализаторах в условиях псевдоожижения. К.п.д. такого генератора может составлять 80–90% [96].
Интересные возможности привнесла “нестационарная технология”, идеи которой предложены Г.К. Боресковым и Ю.Ш. Матросом [97]. Возможность осуществления каталитического процесса при нестационарном состоянии катализатора появляется, если времена релаксации велики по сравнению с длительностью работы катализатора. Это открывает широкие горизонты для повышения интенсивности и избирательности каталитических реакций. Действительно, стационарное состояние катализатора, отвечающее заданному составу реакционной смеси и температуре, лишь случайно может совпасть с оптимальным в отношении его каталитических свойств. Катализатор предварительно можно приводить в оптимальное состояние, периодически возвращаясь к нему после достижения стационарного состояния в условиях реакции. Одно из решений этой технологии состоит в том, что катализатор выполняет не только свою основную функцию ускорителя реакции, но также является и регенератором тепла. Периодическая смена мест ввода и вывода реакционной смеси приводит к перемещению зоны реакции по слою катализатора. Здесь отпадает необходимость во внутренних теплообменниках, что существенно упрощает конструкцию реактора и уменьшает расход металла. Этот метод успешно используется на нескольких российских и зарубежных производствах для обезвреживания промышленных выбросов от органических примесей, окисления SO2 в SO3, восстановления оксидов азота.
ЗАКЛЮЧЕНИЕ
Боресков жил и работал во времена интенсивного развития катализа как в научном, так и в практическом направлениях. Мы в этой статье не обсуждаем неоспоримый вклад огромного научного сообщества в развитие этих направлений. Боресков в своих публикациях всегда отдавал должное работам современников и предшественников, о чем свидетельствует впечатляющий список цитирований, сопровождающий его обзорные работы.
В этом обзоре мы представили краткую аннотацию огромного научного наследия Г.К. Борескова. Концепция катализа Борескова полно и детально изложена в монографии “Гетерогенный катализ” [64]. В этой статье мы хотели напомнить о наиболее важных положениях теории катализа, которые он обосновал и последовательно развивал. Такими положениями в гетерогенном катализе, прежде всего, являются:
• химическая природа промежуточного взаимодействия;
• ключевая роль энергии связи промежуточных поверхностных соединений в скорости и направлении протекания реакций;
• динамическое единство системы реактанты–катализатор;
• приблизительное постоянство удельной каталитической активности веществ одинакового химического состава и структуры.
Г.К. Боресков обосновал подход к приготовлению практических катализаторов, поставив в центр внимания процессы переноса реагирующих веществ и тепла внутри зерна и определив решающее значение пористой структуры при постоянном химическом составе катализатора.
Важный вклад Г.К. Боресков внес в химическую кинетику, установив соотношение между энергиями активации прямой и обратной реакций, дав общее выражение для скорости обратимых реакций и введя понятие молекулярности. Он предложил подход к кинетике гетерогенных реакций, учитывающий изменение свойств поверхности катализаторов под воздействием реакционной среды.
Боресков первым стал широко применять химическую кинетику в инженерных расчетах. Из его ранних работ по сернокислотному производству возникло новое направление – математическое моделирование химических процессов, которое сейчас является теоретической основой химической технологии.
Многостороння деятельность Г.К. Борескова была исключительно плодотворна. Постоянно выдвигая свои оригинальные идеи, воспринимая чужие, способствуя реализации всего разумного и возможного, он генерировал вокруг себя атмосферу научного творчества. Г.К. Боресков создал научную школу, которая продолжает и развивает его идеи в разных областях катализа.
Статья имела целью представить портрет Георгия Константиновича Борескова как ученого. В основу ее положен материал прижизненного издания книги “Георгий Константинович Боресков” (1982), создание которой состоялось благодаря работе одного из авторов этой статьи. Позднее изданные книги “Академик Георгий Константинович Боресков” (1997) и “Георгий Константинович Боресков” (2007) воссоздают образ Георгия Константиновича не только как выдающегося ученого, но и, благодаря воспоминаниям его учеников, как мудрого учителя и замечательного человека.
Список литературы
Боресков Г.К. // Механизм действия твердых катализаторов, в кн. Гетерогенный катализ в химической промышленности: Материалы Всесоюзного совещания. 1953. М., 1955. С. 5.
Боресков Г.К., Дзисько В.А. // Журн. физ. химии. 1950. Т. 24. № 9. С. 1135; Дзисько В.А., Вишневская А.А., Чесалова В.С. // Журн. физ. химии. 1950. Т. 24. № 9. С. 1416.
Боресков Г.К., Дзисько В.А., Борисова М.С., Краснопольская В.Н. // Журн. физ. химии. 1952. Т. 26. № 4. С. 492.
Чесалова В.С., Боресков Г.К. // Докл. АН СССР. 1952. Т. 85. № 2. С. 377.
Боресков Г.К. // Кинетика и катализ. 1977. Т. 18. № 5. С. 1111.
Боресков Г.К. // Теоретические проблемы катализа. Сб. научных трудов, Новосибирск: Изд. ИК СО АН СССР, 1977. С. 113.
Boudart M. // Adv. Catal. 1969. V. 20. P. 153.
Che M., Bennett C.O. // Adv. Catal. 1989. V. 36. P. 55.
van Santen R.A. // Acc. Chem. Res. 2010. V. 42. P. 57.
van Santen R.A., Neurlock M., Shetty S.G. // Chem. Rev. 2010. V. 110. P. 2005.
Bekk I.E., Bukhtiyarov V.I., Pakharukov I.Yu., Zaikovsky V.I., Kriventsov V.V., Parmon V.N. // J. Catal. 2009. V. 268. P. 60.
Гололобов А.М., Бекк И.Е., Брагина Г.О., Зайковский В.И., Аюпов А.Б., Телегина Н.С., Бухтияров В.И., Стахеев А.Ю. // Кинетика и катализ. 2009. Т. 50. № 6. С. 864.
Stakheev A.Yu., Batkin A.M., Telegina N.S., Bragina G.O., Khudorozhkov A.K., Bukhtiyarov V.I. // Topics Catal. 2013. V. 56. P. 306.
Боресков Г.К. // Журн. физ. химии. 1958. Т. 32. С. 1969; Боресков Г.К. // Журн. физ. химии. 1958. Т. 32. С. 2739.
Боресков Г.К. // Кинетика и катализ. 1980. Т. 21. С. 5.
Боресков Г.К., Веньяминов С.А., Сазонова Н.Н. // Докл. АН СССР. 1978. Т. 240. С. 619.
Дадаян К.А., Боресков Г.К., Савченко В.И. // Докл. АН СССР. 1976. Т. 230. С. 635.
Boreskov G.K. // 8th Intern. Congress on Catalysis, Berlin, 2–6 July, 1984. V. 3. P. 231.
Andrushkevich T.V. // Cat. Rev. Sci. Eng. 1993. V. 35. P. 213.
Взаимодействие катализатора и реакционной системы. Сб. науч. труд. под ред. Замараева К.И. Новосибирск: Ин-т катализа, 1988. 176 с.
Розовский A.Я. Катализатор и реакционная среда. М.: Наука, 1988. 304 с.
Centi G., Cavani F., Trifiro F. Selective Oxidation by heterogeneous Catalysis, Kluwer Academic Plenum Publishers, New York, Boston, Dordrecht, London, Moscow, 494c.
Hansen P.L., Helveg S., Datye A.K. // Adv. Catal. 2006. V. 50. P. 77.
Lamberti C., Groppo E., Spoto G., Bordiga S., Zecchina A. // Adv. Catal. 2007. V. 51. P. 1.
Bare S.R., Ressler T. // Adv. Catal. 2009. V. 52. P. 339.
Knop-Gericke A., Kleimenov E., Haevecker M., Blume R., Teschner D., Zafeiratos S., Schloegl R., Bukhtiyarov V., Kaichev V., Prosvirin I., Nizovskii A., Bluhm H., Barinov A., Dudin P., Kiskinova M. // Adv. Catal. 2009. V. 52. P. 213.
Бухтияров В.И. // Успехи химии. 2007. Т. 76. Т. 596.
Баландин А.А. // Журн. физ. химии. 1940. Т. 14. С. 1160.
Баландин А.А. // Журн. органич. химии. 1942. Т. 12. С. 337.
Баландин А.А. Мультиплетная теория катализа. Часть II. Энергетические факторы в катализе, М.: Из-во МГУ, 1964г. 244 с. (Для англ. версии. Balandin A.A. // The Multiplet Theory of Catalysis. Ch. 2. Energetic Factors in Catalysis. Izd. MGU, Moscow, 1964, 243 p. (in Russian).
Makishima S., Yonrda Y., Saito Y. // Actes 11 Congress Intern. de catalyse, Paris, 1961. V. 1. P. 617
Sachtler W., Farenfort J. // Actes 11 Congress Intern. de catalyse, Paris, 1961. V. 1. P. 831.
Темкин М.И. // Журн. физ. химии. 1957. Т. 31. C. 3.
Голодец Г.И., Ройтер В.А. // Укр. хим. журн. 1963. Т. 29. С. 667.
Tanaka K., Tamaru K. // J. Catal. V. 2. № 5. P. 366.
Boreskov G.K. / In Fundamentals of Forecasting the Catalytic Action. Proc. IV Intern. Congr. Catal. Nauka, Moscow, 1970. V. 2. P. 437. (in Russian); Боресков Г.К. // Кинетика и катализ. 1967. Т. 8. С. 1020; Боресков Г.К. // Кинетика и катализ. 1969. Т. 10. С. 5.
Боресков Г.К. Катализ. Новосибирск: Наука, 1971. 125 с.
Боресков Г.К. // Докл. АН СССР. 1971. Т. 201. С. 126.
Yoneda Y. // In: Fundamentals of Forecasting the Catalytic Action. Proc. IV Intern. Congr. Catal., Nauka, Moscow, 1970. V. 2. P. 35. (in Russian).
Boreskov G.K., Popovskii V.V., Sazonov V.A. // In: Fundamentals of Forecasting the Catalytic Action. Proc. IV Intern. Congr. Catal., Nauka, Moscow, 1970. V. 1. P. 343 (in Russian).
Boreskov G.K. // Discuss. Faraday Soc. 1966. № 41. P. 263.
Popovskii V.V., Boreskov G.K. // In: Physics and Physico-Chemistry of Catalysis. Izd. Akad. Nauk SSSR, Moscow, 1960. P. 67 (Problems of Kinetics and Catalysis, Vol. 10, in Russian).
Андрушкевич Т.В., Поповский В.В., Боресков Г.К. // Кинетика и Катализ. 1965. Т. 6. С. 860.
Поповский В.В. // Кинетика и катализ. 1969. Т. 10. С. 1068.
Кимхай О.Н., Поповский В.В., Боресков Г.К., Андрушкевич Т.В., Днепровская Т.Б. // Кинетика и катализ. 1971. Т. 12. С. 371.
Il’chenko N.I., Golodets G.I. // J. Catal. 1975. V. 39. P. 73.
Юрьева T.M., Поповский В.В., Боресков Г.К. // Кинетика и катализ. 1965. Т. 6. С. 1041.
Боресков Г.К., Кулиев А.Р., Соколовский В.Д. // Докл. АН СССР. 1973. Т. 212. С. 121.
Боресков Г.К., Веньяминов С.А., Сазонова Н.Н., Панкратьев Ю.Д., Питаева А.Н. // Кинетика и катализ. 1975. Т. 16. С. 1442.
Боресков Г.К. // Кинетика и катализ. 1973. Т. 14. С. 7.
Попова Г.Я., Андрушкевич Т.В., Захаров И.И., Чесалов Ю.А. // Кинетика и катализ. 2005. Т. 46. С. 233.
Андрушкевич Т.В. // Кинетика и катализ. 1997. Т. 38. С. 289; Андрушкевич Т.В., Чесалов Ю.А. // Успехи химии. 2018. Т. 87. № 6. С. 586.
Крылов О.В., Матышак В.А. Промежуточные соединения в гетерогенном катализе, М.: Наука, 1996. 316 с.
Боресков Г.К. Всесоюзная конференция по механизму гетерогенно-каталитических реакций, 9–13 сентября 1974 г., М., Сборник пленарных докладов, Черноголовка, 1977. С. 3.
Mars P., van Krevelen D.W. // Chem. Eng. Sci. Suppl. 1954. V. 3. P. 41.
Roiter V.A. // Actes du 11 Congress de Catalyse, Paris, 1961. V. 1. P. 759.
Боресков Г.К., Поповский В.В., Мамедов Э.А. // Докл. АН СССР. 1971. Т. 197. С. 373.
Boreskov G.K. // Catal. Sci. Technol. 1982. V. 3. P. 40.
Щукин В.П., Веньяминов С.А., Боресков Г.К. // Кинетика и катализ. 1970. Т. 11. С. 1236.
Andrushkevich T.V., Pankratiev Yu.D., Popova G.Ya. // React. Kinet. Catal. Lett. 1985. V. 29. P. 457.
Popova G.Ya., Andrushkevich T.V., Metakova G.A. // React. Kinet. Catal. Lett. 1979. V. 12. P. 469.
Боресков Г.К. / Сущность каталитического действия на примере реакций окисления. Механизм катализа, Ч. 1. Природа каталитического действия, Новосибирск: Наука, 1984. 218 с.
Boreskov G.K. Heterogeneous Catalysis, eds. Zamaraev K.I., Khasin A.V., Nova Science Publishers, Inc., Hauppauge, 2003. 288 p.
Боресков Г.К. Гетерогенный катализ. М.: Наука, 1986 (1988). 304 с.
Боресков Г.К. // Кинетика и катализ. 1972. Т. 13. С. 543; Боресков Г.К. // Всесоюзная конференция по химическим реакторам: Теория, моделирование, расчет. Новосибирск, 1966. Т. 4. С. 607.
Frazer J.H., Kirkpatrick W.J. // J. Am. Chem. Soс. 1940. V. 62. P. 1659; Боресков Г.К. Дис. … докт. хим. наук. М., 1946.
Боресков Г.К., Буянов Р.А., Иванов А.А. // Кинетика и катализ. 1967. Т. 8. С. 153; Иванов А.А., Боресков Г.К., Буянов Р.А., Полякова Г.М., Давыдова Л.П., Кочкина Л.Д. // Кинетика и катализ. 1968. Т. 9. С. 560.
Ivanov A.A., Balzhinimaev B.S. // React. Kinet. Catal. Lett. 1987. V. 35. P. 413.
Boreskov G.K. Katalyse: Bericht von der Hauptjahrestagung, 1958, Berlin, 1959. P. 29. Abb. (Chemische Gesellschaft in der DDR. Mitteilundsblatt. Sonderheft, 1959).
Веньяминов С.А. / Механизм гетерогенно-каталических реакций окисления. Под ред. Музыкантова В.С., Новосибирск: Ин-т Катализа СО РАН, 1993. С. 73.
Boreskov G.K., Erenburg E.M., Andrushkevich T.V. // React. Kinet. Catal. Lett. 1981. V. 17. P. 341.
Боресков Г.К., Матрос Ю.Ш., Киселев О.В., Бунимович Г.А. // Докл. АН СССР. 1977. Т. 237. С. 160.
Боресков Г.К., Кучаев В.Л. // Докл. АН СССР. 1958. Т. 119. С. 302; Кучаев В.Л., Боресков Г.К. // Кинетика и катализ. 1960. Т. 1. С. 356; Боресков Г.К., Василевич О.В. // Докл. АН СССР. 1959. Т. 127. С. 1033; Боресков Г.К., Василевич О.В. // Кинетика и катализ. 1960. С. 69.
Boreskov G.K. // Adv. Catal. 1964. V. 15. P. 285.
Боресков Г.К., Горбунов А.И., Масанов О.Л. // Докл. АН СССР. 1958. Т. 123. С. 90; Боресков Г.К., Колчанова В.М., Рачковский Э.Э., Филимонова С.Н., Хасин А.В. // Кинетика и катализ. 1975. Т. 16. С. 1218; Панов Г.И., Боресков Г.К., Харитонов А.С. // Кинетика и катализ. 1984. Т. 25. С. 123; Kharitonov A.S., Boreskov G.K., Panov G.I., Pankratiev Yu.D. // React. Kinet. Catal. Lett. 1983. V. 22. P. 309; Panov G.I., Boreskov G.K., Kharitonov A.S., Moroz E.M. // React. Kinet. Catal. Lett. 1981. V. 16. P. 247; Панов Г.И., Боресков Г.K., Харитонов A.С. // Докл. AН СССР. 1980. Т. 252. С. 646.
Музыкантов В.С., Поповский В.В., Боресков Г.К. // Кинетика и катализ. 1964. Т. 5. С. 624.
Боресков Г.К. // Журн. физ. химии. 1945. Т. 19. С. 92.
Horiuti J., Ikishima M. // Proc. Imp. Acad. 1939. V. 15. P. 39; Horiuti J. // Proc. Japan. Acad. 1953. V. 29. P. 160.
Боресков Г.К. // Докл. АН СССР. 1959. Т. 129. С. 607.
Boreskov G.K. // 3-rd Intern. Congr. Catalysis, Amsterdam, 1964. Proccedings, Amsterdam, 1965. V. 1. P. 163; Боресков Г.К. Гетерогенный катализ. М.: Наука, 1988. С. 255.
Боресков Г.К. // Журн. физ. химии. 1935. Т. 6. С. 255; Боресков Г.К. // Пористая структура катализаторов и процессы переноса в гетерогенном катализе (IV Международный Конгресс по катализу, симпозиум 3), Новосибирск, 1970. С. 5; Боресков Г.К. // Катализаторы и каталитические процессы. Сб. научных трудов, Новосибирск, 1977. С. 29.
Боресков Г.К. Влияние процессов переноса тепла и вещества на скорость контактных реакций. / В кн.: Проблемы кинетики и катализа. Т. 6. Гетерогенный катализ: Тр. Всесоюз. конф. по катализу. М., Л., 1949. С. 404.
Боресков Г.К., Дзисько В.А., Борисова М.С. // Журн. физ. химии. 1954. Т. 28. С. 1055.
Боресков Г.К. Изыскание активных ванадиевых катализаторов для производства серной кислоты. / В кн.: Сборник. трудов Украинского НИИ треста "Укрхим". Вып. 1. Технология серной кислоты. Одесса: 1935. С. 88.
Боресков Г.К., Слинько М.Г. // Журн. прикл. Химии. 1943. Т. 16. С. 377; Боресков Г.К., Слинько М.Г. // Хим. пром. 1960. № 3. С. 17.
Боресков Г.К., Слинько М.Г. // Вестн. АН СССР. 1961. № 10. С. 25; Боресков Г.К., Слинько М.Г. // Хим. пром. 1962. № 6. С. 32; Боресков Г.К., Слинько М.Г. // Химия и технол. топлив и масел. 1965. № 8. С. 30; Боресков Г.К. Слинько М.Г. // Теор. основы химич. технол. 1967. Т. 1. С. 5.
Боресков Г.К. // Хим. пром. 1947. № 9. С. 5; Боресков Г.К. // Проблемы кинетики и катализа, М., Л., 1949. С. 404; Боресков Г.К. // Вестн. АН СССР. 1964. № 5. С. 47.
Боресков Г.К., Соколова Т.И. // Журн. хим. пром. 1937. Т. 14. С. 1241.
Боресков Г.К. Катализ в производстве серной кислоты, М.-Л.: Госхимиздат, 1954. 348 с.
Боресков Г.К., Дзисько В.А., Тарасова Д.В., Балаганская Г.П. // Кинетика и катализ. 1970. Т. 11. С. 181.
Симонова Л.Г., Дзисько В.А., Фенелонов В.Б., Боресков Г.К. // Сернокислотный катализ. Новосибирск. 1982. Ч.1, С. 3; Mastikhin V.M., Lapina O.B., Simonova L.G. // React. Kinet. Catal. Lett. 1984. V. 26. С. 431; Mastikhin V.M., Lapina O.B., Balzhinimaev B.S., Simonova L.G., Karnatovskaya L.M., Ivanov A.A. // J. Catal. 1987. V. 103. P. 160. Бальжинимаев Б.С., Мастихин В.М., Иванов А.А. // Расплавы. 1987. Т. 1. С. 100; Бальжинимаев Б.С., Беляева Н.П., Симонова Л.Г., Иванов А.А. // Кинетика и катализ. 1989. Т. 30. С. 885; Bal’zhinimaev B.S., Ivanov A.A., Lapina O.B., Mastikhin V.M., Zamaraev K.I. // Faraday Discuss. Chem. Soс. 1989. V. 87. P. 227; Lapina O.B., Bal’zhinimaev B.S., Boghosian S., Eriksen K.M., Fehrmann R. // Catal. Today. 1999. V. 51. P. 469.
АС 168649. Способ получения железомолибденового катализатора 01.01.65.
АС 28207. Способ ведения газовых каталитических реакций, 1932.
Хасин А.А. Автореферат дис. … докт. хим. наук, Новосибирск, 2006.
АС 1213583. Способ получения катализатора для синтеза бензина из окиси углерода и водорода, 10.10.99.
Боресков Г.К. // Вестн. АН СССР. 1980. № 12. С. 46; Боресков Г.К., Левицкий, Э.А., Исмагилов З.Р. // 6-й Советско-французский семинар по катализу. Сб. Докл. М., 1983. С. 133.
Боресков Г.К. // Вестн. АН СССР. 1983. С. 22; Боресков Г.К., Матрос Ю.Ш. // Советско-французский семинар по катализу. Сб. Докл. М., 1983. С. 29; Boreskov G.K., Matros Yu.Sh. // Appl. Catal. 1983. V. 5. P. 337; Boreskov G.K, Matros Ju.Sh. // Catal. Rev. Sci. Eng. 1983. V. 25. P. 551.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ