

Кинетика и катализ, 2019, T. 60, № 2, стр. 216-226
Влияние HF-модифицирования на каталитические свойства ферросфер в процессе окислительной конденсации метана
Н. П. Кирик 1, Н. Н. Аншиц 1, Е. В. Рабчевский 1, Л. А. Соловьев 1, А. Г. Аншиц 1, 2, *
1 Институт химии и химической технологии СО РАН – обособленное подразделение ФИЦ КНЦ СО РАН
660036 Академгородок, Красноярск, 50/24, Россия
2 Сибирский федеральный университет
660041 Красноярск, просп. Свободный, 79, Россия
* E-mail: anshits@icct.ru
Поступила в редакцию 19.06.2018
После доработки 27.11.2018
Принята к публикации 04.12.2018
Аннотация
Исследовано влияние HF-модифицирования ферросфер, выделенных из летучей золы от сжигания бурого угля, на их химический и фазовый составы, также каталитические свойства в процессе окислительной конденсации метана. Модифицирование приводит к изменению фазового состава, в котором по сравнению с исходными ферросферами появляется фаза CaF2, содержание фазы гематита возрастает, а феррошпинели падает. Выход С2-углеводородов при 750°C увеличивается в 1.5–2.0 раза, доля этилена в них возрастает до 30%, а при 850°C – до 65%. Предположено, что повышение эффективности HF-модифицированных ферросфер в образовании этана и его дегидрирования в этилен обусловлено формированием активных центров оксифторидного типа. Пирогидролиз фторсодержащих компонентов катализатора при 850°C за счет взаимодействия с парами воды реакционной среды приводит к формированию систем, активных в глубоком окислении, что проявляется в резком снижении селективности образования C2-углеводородов и повышении селективности по CO2.
Реакция окислительной конденсации метана (ОКМ) с образованием С2-углеводородов является привлекательным способом превращения метана, наиболее инертного в природном газе углеводорода, в этилен. В качестве катализаторов этого процесса детально исследованы индивидуальные и модифицированные оксиды щелочных, щелочноземельных, редкоземельных металлов, а также Mn-содержащие системы [1–6]. Изучено влияние введения других катионов в катионную подрешетку оксидов, а также замены кислородных ионов другими анионами, на активность и селективность образования продуктов реакции. В частности установлено, что модифицирование оксидных систем галогенидами щелочных, щелочноземельных и редкоземельных металлов приводит к повышению их эффективности в образовании С2-углеводородов за счет роста конверсии метана или значительного увеличения селективности по С2-продуктам, в которых часто отмечается увеличение доли этилена [7–22]. Аналогичные эффекты наблюдаются при введении хлорсодержащего компонента (например, дихлорметана) в газовую фазу при проведении процесса в присутствии оксида MgO в качестве катализатора [23].
Увеличение активности оксидов и их селективности в образовании этилена при модифицировании галогенидами связывают с подавлением центров глубокого окисления и формированием нового типа активных центров и/или фаз оксигалогенидного типа. Для Cl-промотированных катализаторов предполагается образование дефектов за счет замещения иона кислорода ионом хлора: в катализаторе Li/MgO – образование дефектных [Li+Cl–]-кластеров, в щелочноземельных и редкоземельных оксидах, а также в оксидах Mn – поверхностных оксихлоридных комплексов [7, 18–21]. Эффективность галогенсодержащих двухкомпонентных систем BaX2/Me2O3 (X = F, Cl, Br; Me = Y, La) зависит от природы оксидной составляющей и галогенида. В частности, показано, что в катализаторе BaF2/Y2O3 активные центры образуются в результате ионного обмена, что проявляется в дефектности решеток обоих компонентов [11]. Для систем BaX2/La2O3 (X = F, Br), из которых более эффективным является BaBr2/La2O3, установлено, что ион Br−, имеющий больший радиус, связывается с поверхностью La2O3, а ион F– диффундирует в объем с образованием LaF3 и LaOF. При этом на каталитических системах 10% BaF2/Me2O3 (Me = Y, La) и 10% BaBr2/La2O3 при 750°C отношение этилен : этан в продуктах составляет 1.03, 1.25 и 2.11 соответственно [11, 12].
Оксигалогенидные комплексы хлор- и броммодифицированных оксидов способны генерировать в условиях проведения процесса хлор- и бром-радикалы, которые участвуют в активации метана. Предполагается, что эти центры могут участвовать также в дегидрировании этана, а инициируемые галоген-радикалами реакции протекают в приповерхностном слое катализатора [8, 9, 14, 17–20].
Фтормодифицированные оксидные системы не способны генерировать фтор-радикал в условиях проведения процесса ОКМ. Улучшение каталитических свойств этих систем связывают с образованием оксифторидных центров/структур, эффективных в стабилизации электрофильных форм кислорода, необходимых для образования из метана метильных радикалов, рекомбинация которых приводит к образованию этана, и/или дегидрирования этана в этилен [11, 13, 24, 25].
Важно отметить, что монофазные хлориды щелочноземельных элементов (CaCl2, SrCl2 и BaCl2), а также фториды щелочноземельных и редкоземельных элементов (SrF2, BaF2 и LaF3), проявляют низкую активность в превращении метана и не селективны в образовании этилена [12, 21, 22].
Поиск недорогих и термостабильных катализаторов, селективных в образовании этилена, остается актуальным вопросом. Хлормодифицированные катализаторы, при более высокой эффективности, чем оксидные системы в процессе ОКМ имеют существенный недостаток, связанный со снижением их активности во времени за счет потери хлора [4, 21]. Оксифторидные системы по сравнению с хлоридными и бромидными более устойчивы в условиях проведения процесса ОКМ. В ранних работах нами было показано, что после обработки плавиковой кислотой ферросфер с содержанием Fe2O3 87.5 мас. %, выделенных из энергетических зол, конверсия метана при 850°C повышается с 9.5 до 16%, а селективность по С2-углеводородам – с 61 до 67% [26]. Исследование катализатора до и после проведения реакции методом РФЭС показало, что наряду с удалением стеклофазы происходит модифицирование ферросфер с образованием нового типа железооксидных центров, содержащих фтор.
Целью данной работы являлось детальное исследование влияния HF-модифицирования катализаторов на основе ферросфер с содержанием Fe2O3 84–98 мас. % на их фазовый состав и каталитические свойства в реакции ОКМ.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Получение и химический состав исследованных катализаторов
Исходные образцы ферросфер получали из двух фракций высококальциевых ферросфер (SMF –0.160 + 0.100 мм (90.4 мас. % Fe2O3) и SMF –0.100 + 0.063 мм (90.8 мас. % Fe2O3)) с помощью дополнительной магнитной классификации в градиентном магнитном поле с использованием магнитного сепаратора 138 Т (Россия). Исходные фракции ферросфер были получены из летучих зол от сжигания бурого угля Березовского месторождения (Красноярский край). Детальное описание способа получения и определения физико-химических характеристик фракций приведено в работах [27, 28]. Из каждой фракции ферросфер при магнитной сепарации было получено по 4 образца. В табл. 1 представлены значения удельной поверхности и химический состав (в расчете на оксиды) полученных образцов, обозначенных как SMF(n), где в скобках приведено массовое содержание Fe2O3. Результаты показывают, что исходные образцы ферросфер, за исключением SMF(84.0) и SMF(84.7), представляют собой оксидную железо-кальциевую систему с содержанием суммы Fe2O3 и CaO не менее 98 мас. %.
Таблица 1.
Химический состав и удельная поверхность исходных и HF-модифицированных ферросфер
| Образец | Sуд, м2/г |
Содержание, мас. % | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SiO2 | Al2O3 | Fe2O3 | CaO | MgO | Na2O | K2O | TiO2 | MnO | SO3 | ||
| Фракция −0.100 + 0.063, мм | |||||||||||
| SMF(84.7) | 0.12 | 4.08 | 1.62 | 84.69 | 8.92 | 0.78 | 0.18 | 0.05 | 0.20 | 0.15 | 0.96 |
| SMF(94.2) | 0.10 | 0.29 | <0.01 | 94.20 | 4.97 | 1.28 | 0.11 | 0.05 | <0.01 | 0.03 | 0.39 |
| SMFm(94.2) | 0.14 | 0.35 | <0.01 | 94.18 | 4.06 | 1.28 | 0.11 | 0.05 | <0.01 | 0.07 | 0.40 |
| SMF(95.2) | 0.16 | 0.10 | <0.01 | 95.25 | 4.06 | 0.64 | 0.10 | 0.03 | <0.01 | 0.05 | 0.52 |
| SMF HF(94.6) | 3.25 | 0.12 | <0.01 | 94.57 | 4.23 | 0.62 | 0.01 | 0.16 | <0.01 | 0.10 | 0.12 |
| SMF HF(95.9) | 3.38 | 0.10 | 0.29 | 95.90 | 3.32 | 0.70 | 0.05 | 0.74 | <0.01 | 0.08 | 0.20 |
| SMF HF(96.7) | 2.15 | 0.18 | <0.01 | 96.70 | 2.60 | 0.80 | 0.10 | 0.12 | <0.01 | 0.09 | 0.18 |
| SMF HF(97.8) | 2.78 | 0.16 | <0.01 | 97.80 | 1.60 | 0.97 | 0.01 | 0.11 | <0.01 | 0.11 | 0.05 |
| Фракция −0.160 + 0.100, мм | |||||||||||
| SMF(84.0) | 0.24 | 4.42 | 1.03 | 84.03 | 9.15 | 0.32 | 0.20 | 0.08 | 0.38 | 0.18 | 0.89 |
| SMF(97.0) | 0.20 | 0.70 | <0.01 | 97.05 | 3.22 | 0.30 | 0.08 | 0.05 | <0.01 | 0.07 | 0.40 |
| SMF(97.5) | 0.16 | 0.35 | <0.01 | 97.50 | 2.92 | 0.10 | 0.08 | 0.05 | <0.01 | 0.08 | 0.38 |
| SMF(98.1) | 0.32 | 0.10 | <0.01 | 98.10 | 2.70 | 0.32 | 0.10 | 0.03 | <0.01 | 0.04 | 0.42 |
| SMF HF(91.4) | 3.00 | 0.12 | 0.80 | 91.40 | 7.40 | 0.70 | 0.02 | 0.09 | <0.01 | 0.12 | 0.40 |
| SMF HF(95.4) | 3.58 | 0.22 | <0.01 | 95.40 | 4.20 | 0.16 | 0.04 | 0.15 | <0.01 | 0.09 | 0.50 |
| SMF HF(96.5) | 2.15 | 0.26 | <0.01 | 96.50 | 2.80 | 0.98 | 0.01 | 0.02 | <0.01 | 0.09 | 0.52 |
| SMF HFm(96.7) | 2.75 | 0.00 | <0.01 | 96.70 | 3.43 | 0.21 | 0.01 | 0.01 | <0.01 | 0.07 | 0.85 |
HF-модифицированные ферросферы получали обработкой исходных образцов ферросфер концентрированной плавиковой кислотой (“х. ч.”) в полипропиленовой емкости в следующих условиях: соотношение жидкая фаза (мл)/твердая фаза (г) = 5, длительность обработки 10 мин при периодическом перемешивании. Для удержания магнитной части образца при декантации травильного раствора пропиленовую емкость устанавливали на постоянный магнит. Продукт промывали дистиллированной водой до отрицательной качественной реакции на фтор-ионы с хлоридом бария (0.05 М) и сушили при температуре 110 ± 5°C до постоянной массы.
При обработке ферросфер HF происходит частичное разрушение глобул, а удельная поверхность образцов увеличивается более чем на порядок (табл. 1). После стадии сушки проводили контрольный рассев образца на ситах с ячейками, соответствующими размеру исходной фракции. Потери после завершения всех стадий составляли 45–74 мас. % от исходной фракции. Химический состав модифицированных образцов (в пересчете на оксиды), обозначенных как SMF HF(n), где в скобках указано массовое содержание Fe2O3, приведен в табл. 1. В качестве примера на рис. 1 показан микроснимок образца SMF HFm(96.7). Из анализа составов HF-модифицированных образцов ферросфер следует, что все образцы представляют собой двухкомпонентную Fe2O3–CaO систему с суммой оксидов не менее 98 мас. %. При монотонном снижении содержания Fe2O3 в интервале 98–91 мас. % наблюдается рост содержания CaO в интервале 2–7 мас. %.

Каталитические измерения
Каталитические свойства полученных образцов в реакции ОКМ изучали в проточной установке со стационарным слоем катализатора в кварцевых трубчатых микрореакторах с внутренним диаметром 4–8 мм при температурах 750–850°C, в составе реакционной смеси, об. %: СН4 – 80, O2 – 10, Не – 10 и атмосферном давлении. Навеска образцов составляла 0.3–1.3 г, время контакта – 1.2–1.3 гкат с/мл. Состав исходной смеси и продуктов реакции анализировали на хроматографе Agilent 7890A GC (США), оснащенном набивными колонками HP Plot Al2O3, Molecular Sieve 5A и капиллярной колонкой DB–1. Относительная ошибка определения концентраций газовых компонентов составляла ±5%. Каталитические свойства образцов определяли через 4 ч после начала эксперимента.
Физико-химические методы исследования катализаторов
Удельную поверхность образцов оценивали методом многоточечной БЭТ по низкотемпературной адсорбции азота на сорбционном анализаторе NOVA 3200e (“Quantachrome Instruments”, CША).
Химический состав полученных образцов определяли по ГОСТ 5382–91.
Съемку рентгенограмм образцов производили на порошковом рентгеновском дифрактометре X’Pert Pro MPD (“PANalytical”, Нидерланды) с твердотельным детектором PIXcel и вторичным графитовым монохроматором для CoKα-излучения аналогично описанным в работах [27, 29].
Количественный фазовый состав образцов в исходном состоянии и после проведения катализа, а также микроструктурные характеристики фаз определяли полнопрофильным методом Ритвельда и методом минимизации производной разности, как описано в работах [27, 29].
Мессбауэровские спектры HF-модифицированных ферросфер после катализа получали на гамма-резонансном спектрометре МС-1104Ем (“НИИ физики ЮФУ”, Россия) с источником Со57(Cr) при комнатной температуре, расшифровку спектров проводили, как описано ранее [29].
Состав, строение поверхности глобул и их полированных срезов исследовали методом сканирующей электронной микроскопии (СЭМ) на микроскопе ТМ–3000 (“Hitachi”, Япония), оборудованном системой микроанализа Quantax70 с энергодисперсионным спектрометром (ЭДС) Bruker XFlash 430H (Германия). Время накопления определяли по качеству сборки спектра, позволяющему проводить количественную обработку, что составляло не менее 10 мин.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Каталитические свойства HF-модифицированных ферросфер
При проведении процесса ОКМ при 750°C в присутствии как исходных, так и HF-модифицированных образцов ферросфер, основными продуктами являются С2-углеводороды – этан и этилен, а также оксиды углерода CO и CO2. На рис. 2 представлены каталитические характеристики исходных и HF-модифицированных ферросфер в виде зависимостей селективности образования целевых продуктов – суммы С2-углеводородов – от конверсии метана. Для двух групп ферросфер наблюдается снижение селективности образования С2-углеводородов с ростом конверсии метана, что согласуется с общим характером изменения селективности по С2-продуктам от конверсии метана, установленной для катализаторов ОКМ [2, 4–6]. Исходные ферросферы в исследованных условиях характеризуются конверсией метана 1–2% и С2-селективностью 51–61%. Селективность по CO на исследованных образцах варьирует в интервале 9–14%.
Рис. 2.
Зависимость селективности образования С2-продуктов от конверсии метана при 750°C в присутствии немодифицированных (1) и HF-модифицированных ферросфер (2). Для каждой точки в скобках приведено содержание железа в образце (табл. 1).
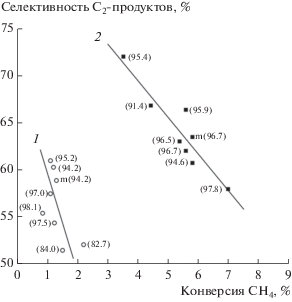
HF-модифицирование ферросфер приводит к существенному изменению каталитических свойств. Для этих систем наблюдается увеличение конверсии метана до 3.5–7.0% и С2-селективности до 58–72% (рис. 2). Селективность образования CO на всех модифицированных образцах снижается до 3–4%, что согласуется с литературными данными для двухкомпонентных систем фториды стронция и бария–редкоземельные оксиды [12, 22].
Значения активности и селективности (S) HF-модифицированных ферросфер также превышают соответствующие характеристики немодифицированных образцов с содержанием Fe2O3 ≥ 89 мас. %, для которых было показано, что активным центром образования С2-углеводородов является дефектный центр феррошпинели [FeVV3+–O2−–vac] [29]. Этот центр включает Са2+-катион в тетраэдрической позиции и октаэдрическую катионную вакансию среди ближайших соседей иона Fe3+ и способен стабилизировать электрофильный кислород, необходимый для образования СН3-радикала из метана. Результаты статьи [29] позволяют сравнить селективности образования С2-углеводородов при одинаковой конверсии метана. При конверсии метана 3.5–4.5% на исходных ферросферах с содержанием Fe2O3 91.4 и 89.1 мас. % ${{S}_{{{{{\text{C}}}_{2}}}}}$ составляет 53.6 и 49.7% соответственно, тогда как ${{S}_{{{{{\text{C}}}_{2}}}}}$ фторированных ферросфер с содержанием Fe2O3 95.4 и 91.4 мас. % значительно выше и составляет 72 и 67% соответственно (рис. 2). Приведенное сравнение свидетельствует о разной природе центров ОКМ на исходных и модифицированных ферросферах.
Кроме того, исследование методом мессбауэровской спектроскопии состояния и распределения железа по фазам и кристаллографическим позициям в HF-модифицированных ферросферах в стационарном состоянии, аналогично работе [29], показало, что доля позиций железа в феррошпинели, отнесенных к дефектным центрам [FeVV3+–O2−–vac], во всех образцах составляет ∼0.02 и находится в пределах ошибки определения, что свидетельствует об отсутствии такого типа центров в HF-модифицированных ферросферах. На основании вышеизложенного можно сделать вывод, что каталитические свойства HF-модифицированных ферросфер по сравнению с исходными определяются активными центрами другого типа.
Ранее нами с использованием метода РФЭС было показано, что обработка плавиковой кислотой ферросфер, содержащих 87.5% Fe2O3, приводит к образованию дефектных состояний железа. В спектре фторированного катализатора наблюдается сдвиг линии Fe2p3/2 в область больших энергий с 710.3 до 711.0 эВ, на основании чего было сделан вывод о замещении части ионов кислорода на ионы фтора [26].
Совокупность литературных и экспериментальных данных позволяет предположить, что каталитические свойства HF-модифицированных ферросфер определяются составом и строением сложного оксифторидного комплекса на основе железа. Для проверки этого предположения мы исследовали фазовый состав модифицированных ферросфер в исходном состоянии и после проведения каталитической реакции, а также химический состав их поверхности.
Фазовый состав ферросфер в исходном состоянии и после проведения каталитической реакции
Данные рентгенофазового исследования исходных и HF-модифицированных образцов до и после катализа приведены в табл. 2. Основными железосодержащими фазами исходных ферросфер являются феррошпинель и гематит. С увеличением содержания Fe2O3 в образце количество фазы шпинели возрастает с 42 до 62 мас. %, а фазы гематита снижается с 30 до 17 мас. %. Параметр решетки шпинели во всех исследованных в работе исходных образцах (как и в исследованных ранее ферросферах более мелких фракций [29, 30]) лежит в интервале 8.398–8.399 Å и превышает значение для стехиометрического магнетита (8.396 Å). Более высокое по сравнению со стехиометрическим магнетитом значение параметра феррошпинельной фазы в образцах ферросфер свидетельствует о формировании решетки с изоморфным замещением железа на кальций [31, 32]. Содержание аморфного остатка в исходных ферросферах до и после катализа практически не меняется и составляет от 18 до 29 мас. %. В образцах после катализа содержание гематита увеличивается до 46–50 мас. %, а феррошпинели снижается до 22–33 мас. %, что хорошо согласуется с ранее полученными данными для мелких фракций ферросфер с высоким содержанием железа [29].
Таблица 2.
Фазовый состав немодифицированных и HF-модифицированных ферросфер до и после катализа при 750°C
| Образец | Исходное состояние, мас. % | После катализа, мас. % | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| шпинель | гематит | кварц | аморфный остаток |
CaSO4 | FeO | шпинель | гематит | кварц | CaF2 | Ca2Fe2O5 | аморфный остаток |
|
| Фракция −0.100 + 0.063, мм | ||||||||||||
| SMF(84.7) | 42.3 | 30.5 | 1 | 25.6 | 0.4 | 0.3 | н.о. | н.о. | н.о. | н.о. | н.о. | н.о. |
| SMF(94.2) | 50.5 | 25.5 | 0.8 | 22.8 | 0.2 | 0.2 | 26.2 | 48.1 | 0.7 | 0 | 0.4 | 23.9 |
| SMFm(94.2) | 56.9 | 22.3 | 0.5 | 20 | 0.2 | 0.2 | 28.2 | 48.7 | 0.9 | 0 | 0.2 | 21.5 |
| SMF(95.2) | 60.2 | 20.9 | 0.6 | 17.9 | 0.2 | 0.2 | 33.2 | 46.4 | 0.8 | 0 | 0.2 | 18.8 |
| SMF HF(94.6) | 21.3 | 45.6 | 0.2 | 32.9 | 0 | 0 | 9 | 70.5 | 0.6 | 1.3 | 1.5 | 17.1 |
| SMF HF(95.9) | 39.1 | 43.5 | 0.2 | 17.2 | 0.1 | 0 | 9.7 | 73.9 | 0.5 | 1.7 | 0.5 | 13.7 |
| SMF HF(96.7) | 42.5 | 42.1 | 0.2 | 15.2 | 0 | 0 | 9.3 | 79 | 0.7 | 1.1 | 0.3 | 9.7 |
| SMF HF(97.8) | 42.5 | 37.6 | 0.2 | 19.7 | 0 | 0 | 9.9 | 76.5 | 0.7 | 0.2 | 0.2 | 12.5 |
| SMF HF(97.8)* | 20 | 58.1 | 0.6 | 0.1 | 0.9 | 20.3 | ||||||
| Фракция −0.160 + 0.100, мм | ||||||||||||
| SMF(84.0) | 45 | 24.3 | 1.3 | 28.7 | 0.6 | 0.2 | н.о. | н.о. | н.о. | н.о. | н.о. | н.о. |
| SMF(97.0) | 50.3 | 20.8 | 0.8 | 27.6 | 0.3 | 0.3 | 21.6 | 48.5 | 1 | 0 | 0.5 | 28.1 |
| SMF(97.5) | 62.5 | 16.8 | 0.7 | 19.6 | 0.3 | 0.2 | 26.8 | 49.7 | 0.8 | 0 | 0.1 | 21.7 |
| SMF(98.1) | 60.8 | 19 | 0.7 | 18.7 | 0.2 | 0.6 | 25.8 | 48.9 | 0.9 | 0 | 0.1 | 23.4 |
| SMF HF(91.4) | 44.6 | 34.4 | 0.2 | 20.6 | 0.1 | 0.2 | 12.3 | 68.5 | 0.3 | 2.9 | 0.3 | 15.7 |
| SMF HF(95.4) | 35.7 | 33.9 | 0.4 | 29.8 | 0.2 | 0.1 | 10.2 | 61.1 | 0.6 | 3.8 | 0.8 | 23.5 |
| SMF HF(95.4)* | 4.8 | 59.4 | 0.6 | 0.3 | 2.6 | 32.3 | ||||||
| SMF HF(96.5) | 51.5 | 29.5 | 0.1 | 18.8 | 0 | 0.1 | 15.1 | 70.2 | 0.5 | 1.5 | 0 | 12.7 |
| SMF HFm(96.7) | 50.2 | 33 | 0.1 | 16.5 | 0.1 | 0.1 | 13.5 | 72.9 | 0.5 | 1.4 | 0 | 11.6 |
Фазовый состав HF-модифицированных ферросфер в состоянии до катализа качественно не изменяется, но образцы содержат больше фазы гематита (30–46 мас. %) и меньше фазы шпинели (21–52 мас. %) (табл. 2). С увеличением содержания феррошпинельной фазы наблюдается снижение параметра решетки (8.401–8.397 Å), который также превышает значение для стехиометрического магнетита (8.396 Å).
СЭМ–ЭДС-исследование поверхности глобул и приповерхностных участков на полированных срезах HF-модифицированных ферросфер (SMF HF(97.8) и SMF HF(95.4)) с минимальной и максимальной селективностью образования С2-углеводородов показало, что в большинстве случаев концентрации Fe2O3, CaO и F изменяются в пределах 63–98, 0.3–19 и 0.1–6 мас. % соответственно. На поверхности отдельных глобул в образце SMF HF(97.8) концентрации достигают 39, 28 и 20 мас. %, в образце SMF HF(95.4) – 13, 26 и 44 мас. % соответственно. Полученные результаты свидетельствует об обогащении поверхности модифицированных ферросфер фтором. Отсутствие в этих ферросферах контролируемого количества фторсодержащих фаз объясняется формированием рентгеноаморфных соединений или поверхностных фторжелезооксидных и/или кальцийфторжелезооксидных комплексов.
После катализа в образцах HF-модифицированных ферросфер содержание гематита возрастает до 61–79 мас. %, содержание шпинели снижается до 9–15 мас. %, а значение параметра ее решетки (8.396–8.398 Å) по сравнению с образцами до катализа приближается к значению для магнетита. Содержание аморфного остатка снижается и изменяется в пределах 10–23 мас. % по сравнению с образцами в исходном состоянии, в которых остаток составляет 15–33 мас. %. Снижение содержания аморфного остатка, очевидно, связано с его кристаллизацией в условиях проведения реакции. Подтверждением этому служит появление в образцах после катализа 0.2–3.8 мас. % фазы CaF2 и 0.2–1.5 мас. % Ca2Fe2O5 (табл. 2).
Полученные результаты показывают, что псевдостационарное состояние модифицированных ферросфер при 750°C включает фазы гематита, феррошпинели, характерных для исходных ферросфер, и CaF2. Исследованные ферросферы можно представить в виде катализаторов общей системы CaF2–Fe2O3, в которой каждая фракция содержит разное количество CaF2. Подтверждением этому служит зависимость выходов основных продуктов ОКМ при 750°C от содержания фазы CaF2 в стационарном состоянии (рис. 3). С ростом содержания CaF2 в образцах наблюдается монотонное снижение выходов CO2 и С2-углеводородов (рис. 3), что, по-видимому, обусловлено частичным блокированием активной поверхности фторидом кальция. Для ферросфер фракции SMF HF(97.8) с минимальным содержанием CaF2 (0.2 мас. %) степень превращения CH4 (${{X}_{{{\text{C}}{{{\text{H}}}_{4}}}}}$) и селективность образования С2-углеводородов (${{S}_{{{{{\text{C}}}_{2}}}}}$) соответствуют значениям 7.0 и 58 соответственно. Для образца SMF HF (95.4) с максимальным содержанием CaF2 (3.8 мас. %) параметры процесса ${{X}_{{{\text{C}}{{{\text{H}}}_{4}}}}}$ и ${{S}_{{{{{\text{C}}}_{2}}}}}$ равны величинам 3.5 и 72 соответственно (рис. 2, табл. 2).
Рис. 3.
Зависимость выходов образования этана (1), этилена (2) и диоксида углерода (3) от содержания CaF2 в модифицированных катализаторах после катализа при 750°C.
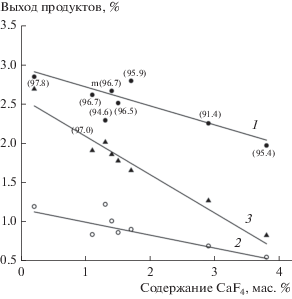
По аналогии с другими оксифторидными системами, можно полагать, что высокий выход С2-углеводородов на ферросферах системы CaF2–Fe2O3 обеспечивается оксифторидными комплексами, которые образуются при частичном замещении ионов кислорода на ионы фтора в решетке феррошпинели, а CaF2, как неактивный в ОКМ компонент, частично блокирует поверхность катализатора. Так как фтор имеет большее значение электроотрицательности по сравнению с кислородом, замещение в оксиде кислородных позиций на фтор приводит, очевидно, к образованию центров стабилизации электрофильной формы кислорода. Можно предположить, что в качестве активных центров, подобно установленным для оксида магния с примесным содержанием фтора и переходных металлов [33], выступают катионные вакансии, имеющие в ближайшем окружении ион фтора и ион кислорода с недостатком электронной плотности (дырку).
Проведение каталитических экспериментов в присутствии модифицированных ферросфер системы CaF2–Fe2O3 сопровождается помутнением стенки кварцевого реактора, что свидетельствует о присутствии HF в газовой фазе. Фторид водорода образуется в реакционных условиях за счет пирогидролиза фторсодержащих компонентов катализатора (оксифторидных комплексов, CaF2) парами воды реакционной среды. За счет пирогидролиза CaF2 и повторного взаимодействия HF с оксидами при 750°C поддерживаются определенная концентрация активных центров (предположительно оксифторидного типа) и стабильность работы катализатора. Представляло интерес изучить характер трансформации каталитических систем под действием реакционной среды при высоких температурах.
Трансформация каталитических систем под действием паров воды реакционной среды
Процесс пирогидролиза CaF2 и фторсодержащих комплексов под действием паров воды протекает при высоких температурах и должен привести к изменению каталитических свойств ферросфер системы CaF2–Fe2O3. Процесс трансформации каталитических систем изучали с использованием двух фракций ферросфер с минимальным (SMF HF(97.8)) и максимальным (SMF HF(95.4)) содержанием CaF2. Каталитические свойства в зависимости от времени протекания реакции определяли при температурах 750°C с последующим повышением до 850°C и далее с понижением до исходной температуры 750°C (рис. 4). При 750°C в присутствии наименее селективного в образовании С2-продуктов образца SMF HF(97.8) значения селективностей ${{S}_{{{{{\text{C}}}_{2}}}}},$ ${{S}_{{{{{\text{C}}}_{{2 = }}}}}}$ и ${{S}_{{{\text{C}}{{{\text{O}}}_{2}}}}}$ со временем мало меняются, и через 4 ч составляют 41, 17 и 38% соответственно (рис. 4а, табл. 2). Повышение температуры до 850°C приводит к изменению селективностей по всем продуктам. В начальный момент селективности имеют следующие значения: ${{S}_{{{{{\text{C}}}_{2}}}}}$ = 23%, ${{S}_{{{{{\text{C}}}_{{2 = }}}}}}$ = 46%, ${{S}_{{{\text{C}}{{{\text{O}}}_{2}}}}}$ = 19%. Кроме того, в продуктах появляются С3-углеводороды пропан и пропилен с селективностями ∼0.5 и 6% соответственно. Примерно через 40 мин в ходе процесса селективность по этилену резко снижается с 46 до 36%, а селективность по CO2 возрастает с 19 до 30%. В течение последующих 3 ч селективность по CO2 монотонно растет, а по C2H4 падает, достигая близких значений ~33%. Селективность образования этана при 850°C практически не меняется и составляет 23–24%. Следует отметить, что при повышении температуры реакции (825°C) селективности образования всех продуктов в присутствии немодифицированных ферросфер не изменяются в ходе эксперимента [29].
Рис. 4.
Изменение селективностей образования продуктов в присутствии модифицированных ферросфер SMF HF(97.8) (а) и SMF HF(95.4) (б) от времени проведения эксперимента при изменении температуры процесса (750 → 850 → 750°C): 1 – C2H6, 2 – C2H4, 3 – CO2.
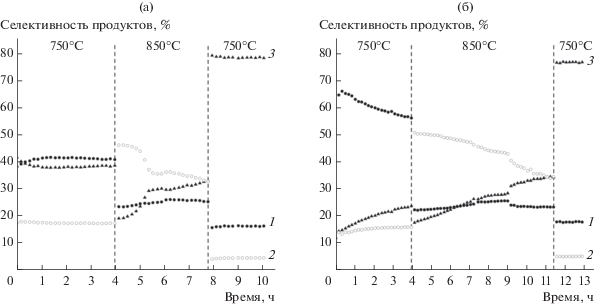
Из полученных результатов следует, что при высокой температуре протекания процесса происходит трансформация катализатора, активного в ОКМ, в катализатор глубокого окисления. Эффект трансформации еще более четко проявляется при возращении к исходной температуре 750°C. Значение ${{S}_{{{\text{C}}{{{\text{O}}}_{2}}}}}$ возрастает до 78%, а ${{S}_{{{{{\text{C}}}_{2}}{{{\text{H}}}_{6}}}}}$ и ${{S}_{{{{{\text{C}}}_{2}}{{{\text{H}}}_{4}}}}}$ снижаются до 16 и 5% соответственно (рис. 4а) при конверсии метана 6.8%.
Изменение селективности образования продуктов в присутствии наиболее селективного образца SMF HF(95.4) в аналогичном эксперименте представлено на рис. 4б. При 750°C на этом образце изменение селективности по продуктам происходит в большей степени, и через 4 ч значения ${{S}_{{{{{\text{C}}}_{2}}}}},$ ${{S}_{{{{{\text{C}}}_{{2 = }}}}}}$ и ${{S}_{{{\text{C}}{{{\text{O}}}_{2}}}}}$ составляют 56, 16 и 23% соответственно. При повышении температуры до 850°C характер изменения селективностей аналогичен первому образцу. В течение 7.5 ч наблюдается монотонный рост селективности образования CO2 и падение селективности C2H4. Их значения достигают одинаковой величины 33%. Селективность образования C2H6 в течение этого времени сохраняется на уровне 23–25%. При последующем снижении температуры до 750°C значения селективности по всем продуктам практически совпадают с образцом SMF HF(97.8) (рис. 4, табл. 2). При конверсии метана 7.6% селективность по CO2 возрастает до 77%, а селективности по этану и этилену снижаются до 17 и 5% соответственно.
Полученные результаты показывают (рис. 4), что для превращения катализаторов с разным содержанием CaF2 в системы с одинаковыми каталитическими характеристиками необходимо разное время воздействия реакционной среды при 850°C. Для ферросфер с исходным содержанием фазы CaF2 0.2 мас. % требуется 3.5 ч, а для катализатора с концентрацией CaF2 3.8 мас. % – 7.5 ч.
Исследование фазового состава образцов после катализа при 850°C показало снижение содержания фазы CaF2 и увеличение – Ca2Fe2O5 по сравнению с образцами в псевдостационарном состоянии при 750°C, что хорошо видно на примере образца SMF HF(95.4), в котором содержание CaF2 снижается с 3.8 до 0.3 мас. %, а содержание Ca2Fe2O5 увеличивается с 0.8 до 2.6 мас. % (табл. 2). Повышенное содержание фазы Ca2Fe2O5 в некоторых образцах, по сравнению с исходными, может быть обусловлено участием гематита в процессе пирогидролиза CaF2. Известно, что присутствие оксидов SiO2 и U3O8 снижает температуру пирогидролиза флюорита в результате образования силикатов кальция и CaU2O7. В качестве добавок рекомендуются также другие оксиды – PbO, Cr2O3 и ZrO2, включая Fe2O3 [34].
Образующийся феррит кальция Ca2Fe2O5 со структурой браунмиллерита (анион-дефицитная структура перовскита) и гематит Fe2O3 являются активными катализаторами глубокого окисления метана, пропилена и CO [35–37]. Их формированием можно объяснить возрастающую селективность в образовании СО2 при 850°C. Низкая селективность этих образцов по CO (∼1–2%) косвенно подтверждает, что в результате трансформации каталитических систем не происходит образования CaO, который в ОКМ проявляет высокую (до 33%) селективность по монооксиду углерода [38].
Таким образом, проведенное исследование показало, что повышение эффективности HF-модифицированных ферросфер системы CaF2–Fe2O3 обусловлено протеканием процесса ОКМ на активных центрах предположительно оксифторидного типа. Модифицированные ферросферы проявляют достаточно стабильную активность и селективность при температуре 750°C. При повышении температуры процесса до 850°C в результате пирогидролиза фторсодержащих компонентов катализатора происходит трансформация каталитических систем, активных в ОКМ, в катализаторы глубокого окисления.
ЗАКЛЮЧЕНИЕ
Проведенное исследование показало, что травление ферросфер летучих зол от сжигания бурого угля с содержанием Fe2O3 84–98 мас. % концентрированной плавиковой кислотой приводит к получению более эффективных катализаторов процесса ОКМ. При температуре процесса 750°C конверсия метана возрастает в 3.5–5 раз, а селективность по С2-продуктам – на 7–11% по сравнению с исходными ферросферами. На всех модифицированных образцах наблюдается увеличение доли этилена в С2-продуктах до 30%, а при 850°C – до 65%. Модифицирование приводит к изменению фазового состава ферросфер, в котором по сравнению с исходными ферросферами появляется фаза CaF2, содержание фазы гематита возрастает, а феррошпинели падает. Предположено, что повышение эффективности ферросфер системы CaF2–Fe2O3 в процессе ОКМ обусловлено формированием активных центров оксифторидного типа. Пирогидролиз фторсодержащих компонентов катализатора при 850°C за счет взаимодействия с парами воды реакционной среды приводит к формированию систем, активных в глубоком окислении, что проявляется в резком снижении селективности образования C2-углеводородов и увеличении селективности по CO2.
БЛАГОДАРНОСТИ
Авторы благодарны д-ру физ.-мат. наук Баюкову О.А. (Институт физики им. Л.В. Киренского ФИЦ КНЦ СО РАН) за исследование HF-модифицированных ферросфер методом ядерного гамма-резонанса и расшифровку мессбауэровских спектров.
ФИНАНСИРОВАНИЕ
Исследование выполнено за счет средств проекта фундаментальных исследований СО РАН V.45.3.3 в Институте химии и химической технологии СО РАН – обособленном подразделении Федерального исследовательского центра “Красноярский научный центр СО РАН”.
Список литературы
Миначев Х.М., Усачев Н.Я., Удут В.Н., Ходаков Ю.С. // Успехи химии. 1988. Т. 57. № 3. С. 385.
Maitra A.M. // Appl. Catal. A. General. 1993. V. 104. P. 11.
Voskresenskaya E.V., Roguleva V.G., Anshits A.G. // Catal. Rev. Sci. Eng. 1995. V. 37. P. 101.
Арутюнов В.С., Крылов О.В. Окислительные превращения метана. М.: Наука, 1998. 361 с.
Langfeld K., Frank B., Strempel V.E., Berger–Karin C., Weinberg G., Kondratenko E.V., Schomäcker R. // Appl. Catal. A. General. 2012. V. 417–418. P. 145.
Zavyalova Ul., Holena M., Schlögl R., Baerns M. // Chem. Cat. Chem. 2011. V. 3. P. 1935.
Otsuka K., Liu Q., Hatano M., Morikawa A. // Chem. Lett. 1986. V. 15. № 6. P. 903.
Shigapov A.N., Novozhilova M.A., Vereshchagin S.N., Anshits A.G., Sokolovskii V.D. // Reac. Kinet. Catal. Lett. 1988. V. 37. № 2. P. 397.
Anshits A.G., Kirik N.P., Roguleva V.G., Shigapov A.N., Selyutin G.E. // Catal. Today. 1989. V. 4. № 3–4. P. 399.
Dai H.X., Liu Y.W., Ng C.F., Au C.T. // J. Catal. 1999. V. 187. P. 59.
Au C.T., Zhou X.P., Liu Y.W., Ji W.J., Ng C.F. // J. Catal. 1998. V. 174. P. 153.
Au C.T., He H., Lai S.Y., Ng C.F. // J. Catal. 1996. V. 159. P. 280.
Au C.T., Zhang Y.Q., Ng C.F., Wan H.L. // Catal. Lett. 1994. V. 23. P. 377.
Burch R., Squire G.D., Tsang S.C. // Catal. Today. 1990. V. 6. P. 503.
Hiyoshi N., Ikeda T. // Fuel Proc. Technol. 2015. V. 133. P. 29.
Burch R., Squire G.D., Tsang S.C. // Appl. Catal. 1989. V. 46. P. 69.
Burch R., Chalker S., Loader P., Thomas J.M., Ueda W. // Appl. Catal. A. General. 1992. V. 82. P. 77.
Arndt S., Laugel G., Levchenko S., Horn R., Baerns M., Scheffler M., Schlögl R., Schomäcker R. // Catal. Rev. Sci. Eng. 2011. V. 53. P. 424.
Burch R., Chalker S., Loader P., Rice D.A., Webb G. // Appl. Catal. A. General. 1991. V. 79. P. 265.
Мамедов А.Х., Гулиев И.А., Ширяев П.А., Шашкин Д.П. // Кинетика и катализ. 1990. Т. 31. № 5. С. 1214.
Baldwin T.R., Burch R., Crabb E.M., Squire G.D., Tsang S.C. // Appl. Catal. 1989. V. 56. P. 219.
Long R., Zhou S., Huang Y., Weng W., Wan H., Tsai K. // Appl. Catal. A. General. 1995. V. 133. P. 269.
Burch R., Chalker S., Hibble S.J. // Appl. Catal. A. General. 1993. V. 96. P. 289.
Long R., Luo J., Chen M., Wan H. // Appl. Catal. A. General. 1997. V. 159. P. 171.
Wan H., Zhou X., Weng W., Long R., Chao Z., Zhang W., Chen M., Luo J., Zhou S. // Catal. Today. 1999. V. 51. P. 161.
Fomenko E.V., Kondratenko E.V., Sharonova O.M., Plekhanov V.P., Koshcheev S.V., Boronin A.I., Salanov A.N., Bajukov O.A., Anshits A.G. // Catal. Today. 1998. V. 42. № 2. P. 273.
Sharonova O.M., Anshits N.N., Solovyov L.A., Salanov A.N., Anshits A.G. // Fuel. 2013. V. 111. P. 332.
Sharonova O.M., Anshits N.N., Anshits A.G. // Inorg. Mater. 2013. V. 49. № 6. P. 586.
Anshits A.G., Bayukov O.A., Kondratenko E.V., Anshits N.N., Pletnev O.N., Rabchevskii E.V., Solovyov L.A. // Appl. Catal. A. General. 2016. V. 524. P. 192.
Верещагин С.Н., Кондратенко Е.В., Рабчевский Е.В., Аншиц Н.Н., Соловьев Л.А., Аншиц А.Г. // Кинетика и катализ. 2012. Т. 53. № 4. С. 470.
Orewczyk J. // J. Therm. Anal. Calorim. 2000. V. 60. P. 265.
De Sitter J., Govaert A., De Grave E., Chambaere D., Robbrecht G. // Phys. Stat. Sol. A. 1977. V. 43. P. 619.
O'Mara W.C., Davies J.J., Wertz J.E. // Phys. Rev. 1969. V. 179. P. 816.
Раков Э.Г., Тесленко В.В. Пирогидролиз неорганических фторидов. М.: Энергоатомиздат, 1987. 152 с.
Боресков Г.К., Поповский В.В., Лебедева Н.Е., Сазонов В.А., Андрушкевич Т.В. // Кинетика и катализ. 1970. Т. 11. № 5. С. 1253.
Исупова Л.А., Цыбуля С.В., Крюкова Г.Н., Буднева А.А., Паукштис Е.А., Литвак Г.С., Иванов В.П., Коломийчук В.Н., Павлюхин Ю.Т., Садыков В.А. // Кинетика и катализ. 2002. Т. 43. № 1. С. 132.
Hirabayashi D., Yoshikawa T., Mochizuki K., Suzuki K., Sakai Y. // Catal. Lett. 2006. V. 110. № 1–2. P. 155.
Roguleva V.G., Kondratenko E.V., Maksimov N.G., Selyutin G.E., Anshits A.G. // Catal. Lett. 1992. V. 16. P. 165.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ