

Кинетика и катализ, 2019, T. 60, № 2, стр. 206-215
Простой и экологически безопасный каталитический синтез производных 2,4,6-триарилпиридина. Использование модифицированного мезопористого органического полимера на основе каликс[4]резорцинарена в качестве гетерогенного кислотного эффективного и стабильного катализатора
A. Mouradzadegun 1, 2, *, M. A. Mostafavi 1, **, M. R. Ganjali 2, 3, ***
1 Department of Chemistry, Faculty of Science, Shahid Chamran University of Ahvaz
Ahvaz, Iran
2 Center of Excellence in Electrochemistry, Faculty of Chemistry, University of Tehran
Tehran, Iran
3 Biosensor Research Center, Endocrinology & Metabolism Molecular-Cellular Sciences Institute,
Tehran University of Medical Sciences
Tehran, Iran
* E-mail: arash_m@scu.ac.ir
** E-mail: mostafavi.mahsa1366@yahoo.com
*** E-mail: ganjali@ut.ac.ir
Поступила в редакцию 06.03.2018
После доработки 18.08.2018
Принята к публикации 19.10.2018
Аннотация
Работа посвящена эффективному однореакторному синтезу производных 2,4,6-трехзамещенного пиридина в трехкомпонентной системе. В качестве катализатора использован мезопористый органический полимер на основе каликс[4]резорцинарена, функционализированный N-пропилсульфаминовой кислотой. Наиболее важными достоинствами предложенной методики являются высокий выход, непродолжительное время реакции, возможность повторного использования катализатора, а также простота операций, проводимых в отсутствие растворителя и при использовании слабой кислоты. Кислотный катализатор был охарактеризован такими физико-химическими методами, как ИК-Фурье-спектроскопия, полевая эмиссионная сканирующая электронная микроскопия, просвечивающая электронная микроскопия и элементный CHNS-анализ.
ВВЕДЕНИЕ
Реакции в многокомпонентной системе все шире внедряются в практику органического синтеза. При проведении этих реакций нет необходимости в стадиях выделения и очистки продуктов, методики отличаются простотой, не требуется также разделять и идентифицировать промежуточные соединения. Если к этому добавить невысокие экспериментальные затраты [1], то станет понятно, почему эти реакции получили распространение при синтезе различных гетероциклических соединений. К этим соединениям относятся производные пиридина [2–8], интерес к которым связан с их важной ролью в качестве ключевых соединений для фармацевтической и агрохимической промышленности [9–11].
В последние годы в практику повседневной жизни и в технологические процессы вошли синтетические пористые металлорганические каркасные полимеры, которые обладают системой микро- (<2 нм) и мезопор (2–50 нм). Они стали играть важную роль в гетерогенном катализе, органическом синтезе, в производстве сенсорных химических устройств, регулировании освещения, хранении и разделении газовых смесей [12–17]. В этой области хорошо зарекомендовал себя ряд разновидностей аморфных или кристаллических пористых материалов, таких как гиперсетчатые полимеры, микропористые полимеры, ковалентные органические каркасы, пористые органические полимеры [18–23]. В области мезопористых материалов значительное внимание уделяется вопросам разработки и синтеза мезопористых органических полимеров, которые образованы легкими неметаллическими элементами, такими как C, H, O и N. Эти полимеры обладают свойствами, характерными для мезопористых материалов, т.е. большой поверхностью, низкой плотностью и регулируемой структурой. При этом они проявляют также свойства, типичные для полимерных органических материалов: содержат ароматические группировки, стабильны к нагреванию и действию кислот и очень гидрофобны. Эти пористые структуры поддаются функционализации до или после синтеза, и прививка самых разных функциональных групп придает им свойства, которые делают их перспективными гетерогенными катализаторами для разнообразных химических реакций [24–26].
Твердые кислоты зарекомендовали себя в качестве активных каталитических систем в органическом синтезе, поскольку они выгодно отличаются от гомогенных катализаторов. К достоинствам гетерогенных кислотных катализаторов относятся нетоксичность, длительное время работы, соответствие экологическим требованиям, дешевизна, легкость выделения из реакционной смеси и возможность многократного использования [27–29]. Таким образом, прививка органических фрагментов к полимеру и последующая добавка кислотного агента к этим активным центрам приведет к формированию твердого кислотного катализатора [30, 31]. Соответственно, введение кислотных функциональных групп, таких как фрагменты сульфаминовой кислоты, на твердый носитель, является одним из путей приготовления эффективных гетерогенных катализаторов [32–35].
Поликаликс[4]резорцинарен, который содержит мономерные звенья в виде чашеобразных полостей, образованных молекулами каликсарена, окруженных гидрофильными группами, принадлежит к новым мезопористым органическим полимерам, поддающимся постсинтетической функционализации для использования их в каталитических целяз [36].
Данная работа является продолжением наших исследований, направленных на разработку безопасных методов органического синтеза [37–42]. Было интересно попытаться ввести группу ‒SO3H в новый мезопористый функционализированный NH2-группами полимер, синтезированный нами на основе каликс[4]резорцинарена. Мы надеялись, что этот катализатор окажется активным в синтезе производных 2,4,6-трехзамещенного пиридина путем однореакторного взаимодействия ацетофенона, арилальдегидов и NH4OAc в мягких условиях и в отсутствие растворителя.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и методы
Исходные материалы, произведенные компаниями “Merck”, “Fluka” и “Aldrich”, использовали без дополнительной очистки. За прохождением реакции наблюдали методом тонкослойной хроматографии (ТСХ). ИК-спектры регистрировали на ИК-Фурье-спектрофотометре BOMEM MB-102 (“Hartmann & Braun”, Канада, KBr). Спектры 1H- и 13C-ЯМР записывали с помощью спектрометра Bruker (“Bruker”, Германия) на частотах 400 и 100 MГц соответственно (растворитель CDCl3, тетраметилсилан как внутренний стандарт). Масс-спектры регистрировали на масс-спектрометре Agilent 5975 (“Agilent,” США). Температуры плавления определяли на приборе Thermo Scientific 9200 (“Thermo Scientific”, США). Значения pH определяли на рН-метре Metrohm 827 pH lab (“Metrohm AG”, Швейцария). Морфологию полимерной матрицы анализировали с помощью атомной силовой микроскопии (АСМ) на микроскопе Wizard II AFM (“JPK instruments”, Германия). Изображения методом полевой эмиссионной сканирующей электронной микроскопии (ПЭ-СЭМ) получали на установке MIRA 3 (“TESCAN”, Чехия). Морфологию и пористость образцов исследовали методом просвечивающей электронной микроскопии (ПЭМ) на приборе Zeiss-EM 10C (Германия) при напряжении 80 кВ. Для оценки термостабильности функционализированного полимера методом термогравиметрического анализа (ТГА) получали ТГ-кривые потери массы, которые регистрировали на дериватографе NETZSCH STA 409 PC/PG (“NETZSCH”, Германия) в атмосфере азота, который подавали со скоростью ~1 л/ч). Элементный анализ выполняли на CHNS-анализаторе Flash EA 1112 (“Thermo Finnigan”, Италия).
Синтез трехмерного полимера 2 на основе каликс[4]резорцинарена
Трехмерный полимер синтезировали путем добавления формальдегида (42 ммоль) к приготовленному каликс[4]резорцинарену 1 (14 ммоль), растворенному в 40 мл 10%-ного водного раствора NaOH при комнатной температуре. Реакционную смесь нагревали до 90°C и выдерживали при этой температуре в течение 20 ч. После этого приготовленный гель отмывали от щелочи холодной водой. Отмытый гель выдерживали при 100°C в течение 1 ч. Затем гель переводили в кислотную форму обработкой 0.1 M раствором HCl. Полученный твердый осадок сушили при 100°C в течение 10 ч [48].
Синтез полимера 3, функционализированного NH2-группами, на основе каликс[4]резорцинарена
Полимер 2, синтезированный по описанному выше методу (1.00 г), и 3-(триэтоксисилил)пропиламин (4.00 г, 18 ммоль) при комнатной температуре помещали в круглодонную колбу объемом 50 мл, содержащую смесь H2O/EtOH (10 мл). Реакционную смесь нагревали до 90°C и перемешивали при этой температуре в течение 20 ч. Целевой полимер 3, функционализированный NH2-группами, собирали на фильтре и промывали несколько раз горячим толуолом, а затем сушили при 100°C [45].
Определение содержания аминогрупп, привитых к полимеру 2
Количество аминогрупп, привитых путем функционализации к полимеру 2, определенное элементным анализом, составило 3.5 ммоль на 1 г сухого полимера [45].
Приготовление полимера 4, синтезированного на основе каликс[4]резорцинарена и модифицированного N-пропилсульфаминовой кислотой
К смеси, состоящей из полимера, функционализированного NH2-группами и описанного в предыдущем разделе (5 г), и CHCl3 (20 мл), при перемешивании на магнитной мешалке при 0°С по каплям в течение 2 ч добавляли хлорсульфоновую кислоту (5 мл, 75.2 ммоль). Затем смесь перемешивали еще в течение 3 ч до полного удаления HCl из реакционной смеси, фильтровали, промывали этанолом (30 мл) и сушили при комнатной температуре.
Определение содержания функциональных групп сульфаминовой кислоты, привитых на катализаторе 4
Содержание функциональных групп сульфаминовой кислоты, привитых на полимерный носитель катализатора, определяли элементным анализом. Было установлено, что 1 г сухого катализатора 4 на полимерном носителе содержит 1.55 ммоль H+-ионов.
Дополнительно кислотную емкость катализатора определяли методом pH-метрии следующим образом. К 1 М водному раствору NaCl (25 мл) с pH, равным 6.01, добавляли навеску катализатора 4 (50 мг) и полученную смесь перемешивали в течение 2 ч. После этого pH раствора снизился до 2.49. Такое снижение означает, что 1 г сухого полимера содержит 1.62 ммоль SO3H [46].
Общая методика синтеза производных 2,4,6-трехзамещенного пиридина (8a–g)
Смесь ароматического альдегида (1 ммоль), ацетофенона (2 ммоль), NH4OAc (1.3 ммоль, 0.103 г) и катализатора (0.05 ммоль, 0.025 г) выдерживали при перемешивании в запаянной трубке при 60°C. После завершения реакции (контроль с помощью ТСХ) в реакционную смесь добавили горячий этанол и нерастворимый катализатора отфильтровали. Чистые продукты получали перекристаллизацией из горячего этанола.
2,4,6-Трифенилпиридин (8a): белое кристаллическое вещество, выход 90%, т. пл. 134–136°C (из EtOH). m/z: 307 [M+]. 1H-ЯМР (400 МГц, CDCl3), δ, м. д.: 7.47–7.77 (m, 11H, Ar–H), 8.22–8.24 (m, 4H, Ar–H), 7.92 (s, 2H, H1). 13C-ЯМР (100МГц, CDCl3), δ, м. д.: 117.2 (C3, C5), 127.2, 128.7, 129.0, 129.1 (ArC), 139.1, 139.5 (ArCq), 150.1 (C4), 157.4 (C2, C6).
Повторное использование катализатора 4
По окончании реакции катализатор отфильтровывали, осадок промывали последовательно CH2Cl2 (5 мл), затем Et2O (5 мл), EtOH (5 мл) и н-гексаном (5 мл). После высушивания катализатора при 60°C в течение 4 ч его можно было использовать вновь без дальнейшей очистки.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Прививка фрагмента, содержащего кислотную группу –SO3H, к высокоэффективным мезопористым материалам создает систему, которая совмещает свойства гомогенных и гетерогенных кислотных катализаторов. В данной работе основное внимание направлено на каталитическую активность нового пористого органического полимера на основе каликс[4]резорцинарена, функционализированного N-пропилсульфаминовой кислотой. Модельной реакцией служило получение производных 2,4,6-трехзамещенного пиридина. Многоступенчатый процесс функционализации в приготовлении катализатора показан на схеме 1 .
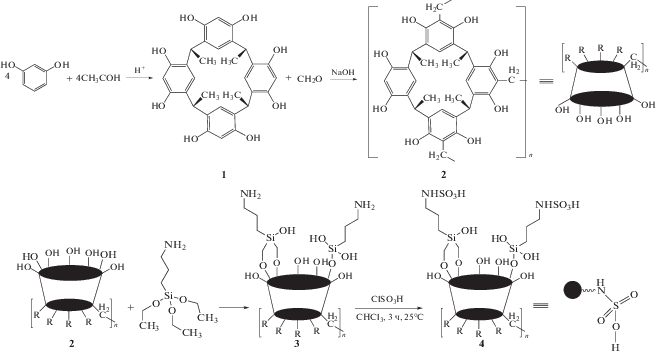
Схема 1 . Синтез каликс [4]резорцинарена 1, трехмерного полимера 2, полимера 3, функционализированного аминогруппами, и полимерного катализатора 4, модифицированного N-пропилсульфаминовой кислотой.
Полимер 2 с трехмерным каркасом был получен взаимодействием резорцина с ацетальдегидом с последующей поликонденсацией каликс- [4]резорцинарена 1 с формальдегидом (схема 1 ). Структура соединения 1 была однозначно установлена спектроскопическими методами (ИК, 1H ЯМР, 13C ЯМР) (см. Electronic Supporting materials). Образование полимера 2 было также подтверждено с применением атомной силовой микроскопии (АСМ), рентгеновской дифрактометрии (РД) и сканирующей электронной микроскопии (СЭМ (см. Electronic Supporting materials)). Далее, в соответствии со схемой 1 , отображающей первый этап постсинтетической функционализации, приготовленный пористый полимер 2 был функционализован по реакции с 3-(триэтоксисилил)пропиламином в качестве органического спейсера. Таким образом можно подготовить благоприятные позиции для закрепления кислотных групп на поверхности полимера. Структура синтезированного полимера 3, функционализированного NH2-группами, охарактеризована с применением ряда аналитических методов, таких как ИК-спектроскопия, атомная силовая микроскопия (АСМ), полевая эмиссионная сканирующая электронная микроскопия (ПЭ-СЭМ), термический гравиметрический анализ (ТГА) и энергодисперсионная рентгеновская спектроскопия (ЭДС) (см. Electronic Supporting materials). На следующем этапе SO3H-группы были закреплены на полимерной матрице 3 путем введения хлорсульфоновой кислоты в реакционную смесь.
Новый гетерогенный катализатор 4, функционализированный сульфаминовой кислотой, был приготовлен реакцией между предварительно синтезированным функционализированным NH2-группами полимером 3 и хлорсульфоновой кислотой в хлороформе. Можно считать, что сильная основность функциональных аминогрупп по отношению к кислотным реагентам, таким как хлорсульфоновая кислота, является движущей силой кислотно-основного взаимодействие, играющего решающую роль в приготовлении катализатора 4.
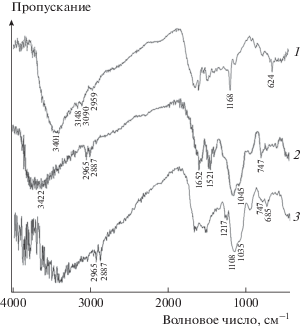
ИК-спектр кислотного катализатора 4 содержит полосу валентных колебаний связей N–S при 685 см–1. Симметричные и антисимметричные валентные колебания группировок Si–O–Si наблюдаются при 1035 и 747 см–1. Полосы при 1217 и 1124 см–1 могут отвечать присутствию сульфонильных фрагментов в катализаторе 4. Присутствие в спектре полос деформационных колебаний функциональных аминогрупп указывает на наличие аминогрупп, не участвовавших в превращении в сульфаминовые кислоты (рис. 1).
Рис. 1.
ИК-Фурье-спектры полимерного каркаса 2 (1), функционализированного аминогруппами полимера 3 (2) и катализатора 4 (3).
По данным элементного анализа на присутствие серы, содержание протонов в полимерной матрице составляет 1.55 ммоль H+/г (табл. 1).
Таблица 1.
Результаты элементного анализа полимера 2, функционализированного NH2-группами полимера 3 и функционализированного сульфаминовой кислотой катализатора 4
| Образец | C, мас. % |
H, мас. % |
N, мас. % |
S, мас. % |
|---|---|---|---|---|
| Полимер 2 | 45.720 | 4.960 | 0.030 | – |
| NH2-функционализированный полимер 3 | 45.500 | 5.020 | 4.950 | – |
| Функционализированный сульфаминовой кислотой катализатор 4 | 44.429 | 6.071 | 5.001 | 4.782 |
Чтобы определить, какая часть аминогрупп превратилась в сульфокислотные группы, с помощью рН-метра была определена кислотность катализатора при 25°C. Оказалось, что 1 г твердого кислотного катализатора содержит 1.62 ммоль SO3H. Это значение представляет концентрацию протонов в катализаторе и находится в хорошем согласии с результатом, полученным элементным анализом.
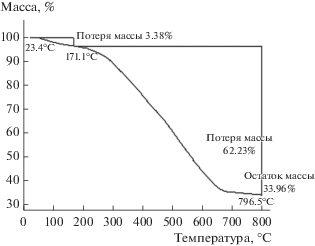
Для оценки термостабильности полученного катализатора был проведен термогравиметрический анализ в атмосфере азота. При нагревании образца катализатора до 172°C потеря массы составляет 3% за счет удаления воды и следов растворителей, которые остались в порах в процессе синтеза. Нагревание от 172 до 796°С сопровождается потерей ~62% массы, относящейся, главным образом, к отщеплению привитых линкеров и других органических фрагментов. Остальная масса образца (34%), которая относится к сохранившемуся полимерному каркасу, остается стабильной вплоть до 800°C. Таким образом, термогравиметрические данные свидетельствуют о высокой стабильности синтезированной структуры (рис. 2).
Методы ПЭ-СЭМ и ПЭМ были использованы для изучения морфологии новой полученной полимерной системы. Как видно на рис. 3, изучаемая структура состоит из мезопор и сферических частиц, которые сохраняют морфологию исходного полимерного каркаса (рис. 3a, 3б).

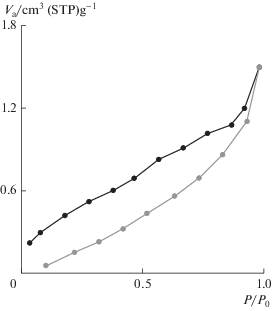
Как показано на рис. 4, изотерма адсорбции–десорбции азота на новом катализаторе, функционализированном сульфаминовой кислотой, близка к изотерме типа IV по величинам площади поверхности, объема пор и среднего диаметра пор, равным 199 м2/г, 0.023 см3/г и 4.67 нм соответственно.
Рис. 4.
Изотерма адсорбции–десорбции азота на полимерном катализаторе 4, функционализированном N-пропилсульфаминовой кислотой.
Для исследования эффективности каталитического действия нового разработанного нами катализатора в качестве модельной реакции было выбрано превращение трехкомпонентной реакционной смеси, состоящей из бензальдегидов, ацетофенона и NH4OAc (схема 2 ). Чтобы убедиться в необходимости использования для этой реакции катализатора, вначале была проведена реакция в отсутствие катализатора. Как и ожидалось, реакция не протекала должным образом (табл. 2, опыт 6). Дальнейшие наши усилия были направлены на оптимизацию условий проведения реакции в присутствии нового катализатора. Основные результаты оптимизации параметров приведены в табл. 2.

Схема 2 . Синтез 2,4,6-трехзамещенных пиридинов.
Далее различные параметры реакции, такие как масса навески катализатора, время и температура реакции, были оптимизированы. Мы обнаружили, что увеличение массы навески катализатора выше 0.025 г практически не влияет на выход продуктов. Поэтому навеска 0.025 г была принята в качестве оптимальной (табл. 2, опыт 2). Далее было показано, что оптимальные температуры реакции находятся в интервале 60–70°C. Наилучший выход продуктов был получен в следующих условиях: 5 мол. % катализатора 4, время реакции 10 мин, температура 60–70°C, в отсутствие растворителя (табл. 2, опыт 5).
Неожиданно, что реакция завершается за короткий период времени и приводит к высоким выходам продуктов. Этот результат можно отнести за счет пористой структуры и большой площади поверхности катализатора, что облегчает перемещение реактантов к доступным активным центрам, способствуя повышению выхода продуктов. Кроме того, гидрофобные ароматические полости поликаликсареновых мономерных блоков образуют “карманы”, в которых накапливаются реактанты, что промотирует эффективность реакции.
На следующем этапе была предпринята попытка оценить возможности и ограничения предложенной методики (табл. 3). Реакцию проводили в оптимизированных условиях для различных замещенных ароматических альдегидов, и в каждом случае были получены соответствующие 2,4,6-трехзамещенные пиридины с хорошим или превосходным выходом. Интересно отметить, что ароматические альдегиды, несущие электроноакцепторные группы (такие как NO2 и галоиды) (табл. 3, опыты 5, 6 и 7) в пара-положениях ароматического кольца, обеспечивают более высокие выходы, чем альдегиды с электронодонорными группами (такие как CH3, OCH3 и N(CH3)2) (табл. 3, опыты 2, 3 и 4 соответственно) в этом же положении. В то же время природа заместителя, будь то электроноакцепторная или электронодонорная группа, не оказывает заметного влияния на выход продуктов (табл. 3). Все продукты хорошо известны и были охарактеризованы нами сравнением температур плавления и спектральных данных, полученных методами ИК- и 1H-ЯМР, с литературными данными [2, 47].
Таблица 3.
Однореакторный синтез 2,4,6-трехамещенных пиридинов*
| № опыта |
R1 | R2 | Продукт | Выход, %** |
Время, мин |
Тпл, °C (Тпл, °C [лит. ссылка]) |
|---|---|---|---|---|---|---|
| 1 | H | H | 8a | 90 | 10 | 134–136 (134–135 [47]) |
| 2 | 4-Метил | H | 8b | 85 | 15 | 122–124 (123–124 [47]) |
| 3 | 4-Метокси | H | 8c | 82 | 20 | 99–101 (100–103 [47]) |
| 4 | 4-(N,N-диметиламин) | H | 8d | 88 | 5 | 141–143 (138–140 [47]) |
| 5 | 4-Хлор | H | 8e | 98 | 5 | 126–128 (125–127 [47]) |
| 6 | 4-Бром | H | 8f | 96 | 10 | 166–168 (164–166 [2]) |
| 7 | 4-Нитро | H | 8g | 95 | 5 | 198–200 (195–197 [47]) |
Следует также отметить, что конечные продукты реакции легко очищаются перекристаллизацией без какой-либо необходимости использовать дорогостоящие и небезопасные хроматографические методы.
Возможный механизм образования производных 2,4,6-триарилпиридина приведен на схеме 3 . Образование целевого продукта 8 включает ряд процессов, таких как альдольная конденсация, присоединение по Михаэлю, циклизация и окисление. Вначале активированный арилальдегид 5 в присутствии катализатора 4 подвергается нуклеофильному присоединению енольной формы ацетофенона 6 с образованием продукта альдольной конденсации A. Затем вторая молекула ацетофенона в присутствии ацетата аммония превращается в молекулу енамина B, который после этого атакует A и путем присоединения по Михаэлю дает интермедиат C. На следующем этапе в процессе циклизации образуется дигидропиридин D, и, наконец, окисление D воздухом приводит к образованию конечного продукта 8 (схема 3 ).
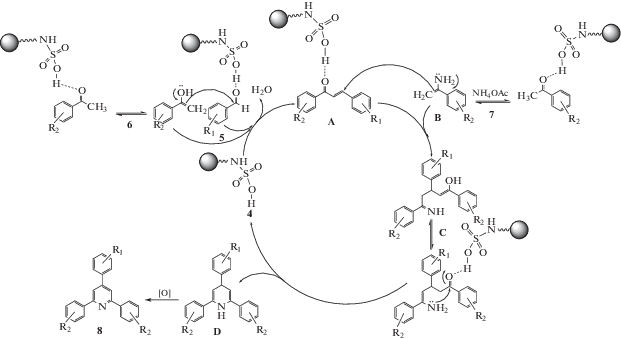
Схема 3 . Возможный механизм синтеза 2,4,6-трехзамещенных пиридинов в присутствии катализатора 4.
Как видно в табл. 4, катализатор 4 выступает в качестве более эффективного катализатора по сравнению с ранее известными каталитическими системами в категориях выхода целевых продуктов, времени и условий реакции.
Таблица 4.
Сравнение катализатора данной работы с другими катализаторами в синтезе триарилпиридинов
В заключительной части работы была изучена возможность многоразового использования катализатора. Для этого по завершении реакции катализатор был отделен обычной фильтрацией через пористый стеклянный фильтр, промыт дихлорметаном, диэтиловым эфиром, этанолом и н-гексаном с последующей сушкой и вновь использован в 5 последовательных циклах реакции без значительной потери каталитической активности (рис. 5).
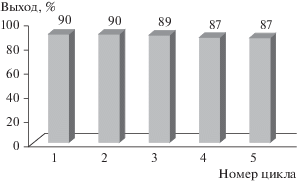
Заметное синтетическое преимущество этого мало затратного однореакторного метода проведения каталитической реакции в отсутствие растворителя состоит в том, что его можно осуществлять с доступными исходными материалами, он прост в исполнении и очистке продуктов. Синтез приводит к целевым продуктам с очень высоким выходом и не нуждается в каком-либо разделении, а также привлечении дорогостоящих материалов и колоночной хроматографии. Это главные преимущества предложенной экологически безопасной многокомпонентной методики синтеза производных триарилпиридина. Еще одним достоинством методики является возможность повторного использования катализатора, по меньшей мере, в пяти последовательных циклах без заметных потерь в выходах продуктов.
ЗАКЛЮЧЕНИЕ
Таким образом, использование нового пористого органического полимера в качестве гетерогенной каталитической системы в трехкомпонентном синтезе мультизамещенных пиридинов позволило провести эту реакцию за короткий промежуток времени в отсутствие растворителей и получить продукты с очень высоким выходом. Кроме того, простота выделения продуктов, мягкие условия реакции, соблюдение экологических требований, а также возможность регенерации катализатора являются наиболее примечательными особенностями этой новой методики.
Список литературы
Safari J., Zarnegar Z., Borjian Borujeni M. // Chem. Pap. 2013. V. 67. P. 688.
Mouradzadegun A., Kiasat A.R., Asareh H. // Rev. Roum. Chim. 2015. V. 60. P. 943.
Kannan V., Sreekumar K. // Modern Res. Catal. 2013. V. 2. P. 42.
Lesanko A.U., Hall D.G. // Curr. Opin. Chem. Biol. 2005. V. 9. P. 266.
Penta S., Vedula R.R. // J. Heterocycl. Chem. 2013. V. 50. P. 859.
Barluenga J., Jiménez-Aquino A., Fernández M.A., Aznar A., Valdés C. // Tetrahedron. 2008. V. 64. P. 778.
Banerjee S., Sereda G. // Tetrahedron Lett. 2009. V. 50. P. 6959.
Maleki B., Azarifar D., Veisi H., Hojati S.F., Salehabadi H., Nejat Yami R. // Chinese Chem. Lett. 2010. V. 21. P. 1346.
Thapa P., Karki R., Yun M., Kadayat T.M., Lee E., Kwon H.B., Na Y., Cho W.J., Kim N.D., Jeong B.S., Kwon Y., Lee E.S. // Eur. J. Med. Chem. 2012. V. 52. P. 123.
Bringmann G., Reichert Y., Kane V.V. // Tetrahedron. 2004. V. 60. P. 3539.
Pillai A.D., Rathod P.D., Franklin P.X., Patel M., Nivsarkar M., Vasu K.K., Padh H., Sudarsanam V. // Biochem. Biophys. Res. Commun. 2003. V. 301. P. 183.
Liu G., Wang Y., Shen C., Jua Z., Yuan D. // J. Mater. Chem. A. 2015. V. 3. P. 3051.
Dawson R., Cooper A.I., Adams D.J. // Prog. Polym. Sci. 2012. V. 37. P. 530.
Wu D., Xu F., Sun B., Fu R., He H., Matyjaszewski K. // Chem. Rev. 2012. V. 112. P. 3959.
Chen L., Honsho Y., Seki S., Jiang D.L. // J. Am. Chem. Soc. 2010. V. 132. P. 6742.
Liu X.M., Xu Y.H., Jiang D.L. // J. Am. Chem. Soc. 2012. V. 134. P. 8738.
Feng X., Ding X., Jiang D. // Chem. Soc. Rev. 2012. V. 41. P. 6010.
Luo Y., Li B., Wang W., Wu K., Tan B. // Adv. Mater. 2012. V. 24. P. 5703.
Ghanem B.S., Msayib K.J., McKeown N.B., Harris K.D., Pan Z., Budd P.M., Butler A., Selbie J., Book D., Walton A. // Chem. Commun. 2007. V. 1. P. 67.
Weber J., Thomas A. // J. Am. Chem. Soc. 2008. V. 130. P. 6334.
Kuhn P., Antonietti M., Thomas A. // Angew. Chem. Int. Ed. 2008. V. 47. P. 3450.
Uribe-Romo F.J., Doonan C.J., Furukawa H., Oisaki K., Yaghi O.M. // J. Am. Chem. Soc. 2011. V. 133. P. 11478.
Luo Y.L., Zhang S.C., Ma Y.X., Wang W., Tan B. // Polym. Chem. 2013. V. 4. P. 1126.
Li B., Guan Z., Yang X., Wang W.D., Wang W., Hussain I., Song K., Tan B., Li T. // J. Mater. Chem. A. 2014. V. 2. P. 11930.
Jiang J.X., Wang C., Laybourn A., Hasell T., Clowes R., Khimyak Y.Z., Xiao J., Higgins S.J., Adams D.J., Cooper A.I. // Angew. Chem. Int. Ed. 2011. V. 50. P. 1072.
Du X., Sun Y., Tan B., Teng Q., Yao X., Su C., Wang W. // Chem. Commun. 2010. V. 46. P. 970.
Gharib A., Noroozi Pesyan N., Vojdani Fard L., Roshani M. // Science PG. 2015. V. 1. P. 6.
Melero J.A., Grieken R.V., Morales G. // Chem. Rev. 2006. V. 106. P. 3790.
Niknam K., Abolpour P. // J. Chem. Sci. 2015. V. 127. P. 1315.
Jetti S.R., Bhatewara A., Kadre T., Jain S. // Chinese Chem. Lett. 2014. V. 25. P. 469.
Rahi T., Baghernejad M., Niknam K. // Chin. J. Catal. 2012. V. 33. P. 1095.
Mohammadi Ziarani G., Lashgari N., Badiei A. // J. Mol. Catal. A. Chem. 2015. V. 397. P. 166.
Niknam K., Jamali A., Tajaddod M., Deris A.S. // Chin. J. Catal. 2012. V. 33. P. 1312.
Sheykhan M., Ma’mani L., Ebrahimi A., Heydari A. // J. Mol. Catal. A. Chem. 2011. V. 335. P. 253.
Kassaee M.Z., Masrouri H., Movahedi F. // Appl. Catal. A. Gen. 2011. V. 395. P. 28.
Бучаченко А.Л. // Успехи химии. 1999. Т. 68. № 2. С. 99.
Mouradzadegun A., Kiasat A.R., Kazemian Fard P. // Catal. Commun. 2012. V. 29. P. 1.
Mouradzadegun A., Abadast F., Elahi S. // Monatsh. Chem. 2014. V. 145. P. 1663.
Mouradzadegun A., Elahi S., Abadast F. // RSC. Adv. 2014. V. 4. P. 31239.
Mouradzadegun A., Elahi S., Abadast F. // Catal. Lett. 2014. V. 144. P. 1636.
Mouradzadegun A., Elahi S., Abadast F. // Synthesis. 2015. V. 47. P. 630.
Mouradzadegun A., Abadast F. // Monatsh Chem. 2013. V. 144. P. 375.
Tunstad L.M., Tucker J.A., Dalcanale E., Weiser J., Bryant J.A., Sherman J.C., Helgeson R.C., Knobler C.B., Cram D.J. // J. Org. Chem. 1989. V. 54. P. 1305.
Altshuler H., Ostapova E., Fedyaeva O., Sapozhnikova L., Altshuler O. // Macromol. Symp. 2002. V. 181. P. 1.
Mouradzadegun A., Mostafavi M.A. // RSC. Adv. 2016. V. 6. P. 42522.
Karimi B., Zareyee D. // Org. Lett. 2008. V. 10. P. 3989.
Adib M., Tahermansouri H., Aali Koloogani S., Mohammadia B., Bijanzadeh H.R. // Tetrahydron Lett. 2006. V. 47. P. 5957.
Kannan V., Sreekumar K. // Sci. Res. 2013. V. 2. P. 42.
Montazeri N., Ayoubi S.F., Pourshamsian K., Bashtini F. // Orient. J. Chem. 2012. V. 28. P. 303.
Rajput P., Subhashini N.J.P., Raj S. // J. Sci. Res. 2010. V. 2. P. 337.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ