

Кинетика и катализ, 2019, T. 60, № 3, стр. 315-333
Мультифазное гидродехлорирование 1,3,5-трихлорбензола на палладиевых катализаторах, нанесенных на оксид алюминия: влияние свойств носителя и модификации гетерополикислотой на основе кремния и вольфрама
Е. В. Голубина 1, Е. С. Локтева 1, *, У. Д. Гурбанова 2, А. Н. Харланов 1, Т. Б. Егорова 1, И. А. Липатова 3, М. С. Власкин 3, Е. И. Школьников 3
1 ФГБОУ ВО Московский государственный университет имени М.В. Ломоносова, Химический факультет
119991 Москва, Ленинские горы 1, стр. 3, Россия
2 Филиал Московского государственного университета имени М.В. Ломоносова в г. Баку, Химический факультет
AZ 1144 Баку, Бинагадинский р., пос. Ходжасан, ул. Университетская, 1, Азербайджан
3 ФГБУН Объединенный институт высоких температур РАН
125412 Москва, ул. Ижорская, д. 13, стр. 2, Россия
* E-mail: e.lokteva@rambler.ru
Поступила в редакцию 10.12.2018
После доработки 11.01.2019
Принята к публикации 15.01.2019
Аннотация
Приготовлены каталитические системы 2 мас. % Pd/Al2O3 с использованием в качестве носителей бемита (НП) и двух типов Al2O3, полученного прокаливанием бемита при 600°С (П) и произведенного фирмой “Engelhard” (Е). Для синтеза образцов 2 мас. % Pd/ГПС-Al2O3 эти носители модифицировали пропиткой гетерополисоединением (ГПС) (20 мас. % Н8[Si(W2O7)6] ⋅ 6Н2О). Изучено влияние структуры Al2O3 и его модификации ГПС на физико-химические свойства, активность, селективность и стабильность катализаторов в реакции мультифазного гидродехлорирования (ГДХ) 1,3,5-трихлорбензола (ТХБ). Все катализаторы проявили активность в вышеуказанной реакции с образованием преимущественно бензола, но подвергались дезактивации в реакционной среде. Модификация ГПС приводила к повышению стабильности и была особенно эффективна для катализатора на носителе Al2O3(Е). Методом сканирующей электронной микроскопии (СЭМ) установлены морфологические различия носителей. По данным просвечивающей электронной микроскопии (ПЭМ) все катализаторы содержали Pd в виде частиц размером менее 20 нм, причем размер частиц и ширина распределения по размерам увеличивались в ряду Pd/Al2O3(НП) < Pd/Al2O3(П) < Pd/Al2O3(Е). Модифицирование ГПС способствовало снижению размера частиц Pd. Методом термопрограммированного восстановления водородом (ТПВ-Н2) показано, что в состав всех катализаторов входил гидрид палладия наряду с более прочно связанными с поверхностью формами металла, которые восстанавливаются при повышенных температурах, причем их количество снижается после модификации ГПС и возрастает после каталитических испытаний. По данным инфракрасной спектроскопии диффузного отражения (ИК-ДО) нанесение ГПС приводит к изменению типа льюисовских кислотных центров на поверхности Al2O3 и электронного состояния палладия. Согласно результатам ИК-спектроскопического исследования адсорбированного СО доминирующей формой на поверхности немодифицированных катализаторов является Pd0 в виде относительно крупных частиц. В составе модифицированных ГПС катализаторов присутствуют одиночные катионы Pd+ и Pd2+, а доля Pd0 существенно меньше. Особенности льюисовской кислотности поверхности катализаторов определяют возможность адсорбции и активации ТХБ на носителе и поступления водорода от Pd0 по механизму спилловера. Повышение стабильности катализаторов в результате модификации носителя ГПС можно объяснить появлением новых активных центров при взаимодействии палладия с ГПС или продуктами его термического разложения.
ВВЕДЕНИЕ
Гетерогенно-каталитическое гидродехлорирование (ГДХ) представляет собой эффективный способ переработки хлорированных органических отходов, лишенный опасности выброса диоксиновых соединений. При утилизации значительных количеств отходов ГДХ позволяет выделить и повторно использовать нехлорированную часть молекулы, например, бензол из хлорбензолов [1, 2]. Доказана эффективность ГДХ при детоксификации полихлорированных ароматических соединений [1, 3], включая полихлорированные дибензо-пара-диоксаны/фураны (диоксины) [4, 5], очистке воды от хлорфенолов [6, 7], лекарственных препаратов [8–11], хлорэтанов и хлорэтиленов [12–14]. Наиболее эффективными и стабильными оказались палладийсодержащие катализаторы [1, 15, 16], хотя часто применяют также неблагородные металлы – никель [17], железо [18], кобальт [19] и др., отдельно или в составе биметаллических систем с благородными компонентами [14, 20, 21]. Адсорбция и активация хлорированных органических соединений может происходить как на частицах металла, так и на поверхности носителя, причем второй путь становится основным в том случае, когда поверхность носителя содержит льюисовские кислотные центры [16]. В этом варианте водород, который активируется на частицах активного металла посредством гомо- или гетеролитической диссоциации, поступает к адсорбированной на носителе молекуле по механизму спилловера [16]. Выделяющиеся при ГДХ частицы хлора и хлорид-ионы способны вызывать дезактивацию катализаторов [16], в первую очередь, за счет хлорирования активного металла. Один из подходов, позволяющий ограничить такую дезактивацию, связан с введением модифицирующих добавок, например, второго металла, который снижает степень хлорирования активного компонента [21]. Другой перспективный подход состоит в модификации свойств носителя. Активность, селективность и стабильность каталитических систем можно менять за счет регулирования природы, текстуры, кристаллической структуры носителей [15, 22].
В настоящей работе с целью изменения кислотных свойств поверхности и предотвращения сильного взаимодействия палладия с носителем – оксидом алюминия – проводили его модификацию введением гетерополисоединения (ГПС) кремния и вольфрама. Ранее мы показали [23], что модификация носителя добавлением гетерополисоединений молибдена и вольфрама позволяет улучшить каталитическое действие никелевых и палладий-никелевых катализаторов, нанесенных на оксид алюминия, в реакции гидродехлорирования хлорбензола за счет изменения дисперсности металла и степени его взаимодействия с носителем. Однако причины изменений, происходящих при модифицировании гетерополисоединениями нанесенных металлсодержащих катализаторов, изучены недостаточно.
В представленной работе исследовано влияние структуры оксида алюминия и его модификации гетерополисоединением Н8[Si(W2O7)6] ⋅ Н2О на физико-химические свойства, а также активность, селективность и стабильность катализаторов Pd/Al2O3 в реакции ГДХ 1,3,5-трихлорбензола в мультифазных условиях.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление и модифицирование носителей
В работе использовали три типа носителя на основе оксида алюминия: 1) Al2O3(Е) обозначен γ-Al2O3 производства “Engelhard” (США; удельная поверхность (SБЭТ) – 185 м2/г; 2) Al2O3(НП) – бемит, синтезированный гидролизом изопропилата алюминия под действием изопропилового спирта с последующей сушкой; 3) Al2O3(П) – γ-Al2O3, полученный прокаливанием образца Al2O3(НП) при 600°C.
Модифицирование носителей гетерополикислотой Н8[Si(W2O7)6] ⋅ 6Н2О (ч. д. а., “Диа-М”, Россия) проводили методом пропитки. Навеску носителя сушили (1 ч, 110°С), затем смачивали дистиллированной водой; к суспензии носителя при постоянном перемешивании добавляли по каплям рассчитанное количество раствора гетерополикислоты (с целью нанесения 20 мас. % ГПС на Al2O3), перемешивали еще 10 мин и упаривали на водяной бане. Сушили на воздухе 12 ч, затем 1 ч при 110°С и прокаливали в статической атмосфере на воздухе 1 ч при 200°С и еще 3 ч при 400°С. Таким образом были получены три модифицированных носителя, обозначенные как ГПС-Al2O3(Е), ГПС-Al2O3(НП), ГПС-Al2O3(П).
Приготовление катализаторов
Палладий (2 мас. %) наносили на Al2O3 методом пропитки. Навеску сухого носителя смачивали дистиллированной водой и перемешивали 10 мин на магнитной мешалке. Рассчитанное количество раствора Pd(NO3)2 (содержание палладия – 10 мас. %, азотной кислоты – 10 мас. %, чистота по другим металлам – 99.999% (“Aldrich”, США)) немного разбавляли дистиллированной водой и прикапывали к суспензии носителя при перемешивании в течение 10 мин. Перемешивание полученной суспензии продолжали еще 10 мин, а затем упаривали на водяной бане, сушили на воздухе 12 ч при комнатной температуре и 1 ч при 110°С и прокаливали в статической атмосфере на воздухе (быстрый нагрев до 200°С, выдерживание 1 ч, далее нагрев до 400°С, прокаливание 3 ч). PdO/Al2O3 восстанавливали в кварцевом реакторе в токе водорода (скорость потока Н2 – 10 мл/мин) при 500°С в течение 3 ч. После охлаждения катализатора до 50°С реактор 30 мин продували азотом, содержащим следы кислорода, с целью поверхностного окисления частиц палладия и предотвращения их дальнейшего окисления при хранении на воздухе.
Исследование катализаторов физико-химическими методами
Изотермы адсорбции–десорбции азота регистрировали при 77 К с использованием автоматизированного сорбтометра Autosorb-1 (“Quantachtome”, США). Удельную площадь поверхности (SБЭТ), объем (Vпор) и средний диаметр пор (dпор) образцов рассчитывали методами БЭТ и BJH по изотермам адсорбции–десорбции азота при 77 К с помощью программного обеспечения прибора.
Электронные микрофотографии поверхности образцов получали на сканирующем электронном микроскопе JEOL JSM-6390 (“JEOL Ltd”, Япония), оснащенном приставкой для локального энергодисперсионного анализа (ЭДА); снимки просвечивающей электронной микроскопии высокого разрешения (ПЭМ ВР) – на приборе JEM 2100F-UHR (“JEOL Ltd”, Япония) с приставкой для локального энергодисперсионного анализа (ЭДА) при ускоряющем напряжении 200 кВ.
Температурно-программированное восстановление водородом (ТПВ-Н2) проводили на анализаторе хемосорбции УСГА-101 (ООО “Унисит”, Россия), пропуская через кварцевый реактор, содержащий 50 мг невосстановленного предшественника катализатора, смесь 5% H2/Ar (30 мл/мин) при нагревании от 30 до 800°С со скоростью 10°С/мин. Перед анализом образец выдерживали при 300°С в течение 30 мин в потоке аргона и охлаждали до 30°С. Изменение состава газовой смеси во время анализа фиксировали с помощью детектора по теплопроводности. С целью количественного анализа данных о поглощении/выделении водорода сигнал детектора предварительно калибровали в ходе ТПВ-Н2 стандартного образца NiO.
Инфракрасные спектры диффузного отражения (ИК ДО) регистрировали на ИК-Фурье-спектрометре EQUINOX 55/S (“Bruker”, Германия). Порошок исследуемого образца помещали в кварцевую ампулу с окошком из CaF2 и прокаливали при температуре 550°С (1 ч на воздухе и 2.5 ч под вакуумом не хуже 5 × 10–5 Торр). Газообразный СО очищали перепусканием через ловушку с жидким азотом и длительным выдерживанием над прокаленным цеолитом. Дифференциальные спектры адсорбированного СО получали вычитанием фонового спектра из экспериментального спектра образца, содержащего адсорбированный СО, с последующей коррекцией базовой линии в программе OPUS 6.0 (“Bruker”).
Рентгенофазовый анализ образцов выполняли на порошковом дифрактометре STOE STADI-P (“STOE & Cie GmbH”, Германия) с использованием CuKα-излучения в диапазоне углов 2θ от 10° до 85°.
Мультифазное гидродехлорирование 1,3,5-трихлорбензола
Мультифазное ГДХ 1,3,5-трихлорбензола (ТХБ) проводили при 50°C и атмосферном давлении в трехгорлой колбе, оснащенной термостатированной “рубашкой”, магнитной мешалкой, обратным холодильником и системой подачи водорода. В реактор, нагретый до температуры опыта (50°С), помещали 100 мг катализатора и восстанавливали оксидную пленку на поверхности частиц палладия в токе водорода (5 мл/мин) в течение 1 ч. Далее загружали 3 мл изооктана, 2 мл 0.13 М раствора Аликват-336 в изооктане, перемешивали 5 мин, приливали 6 мл 20% водного раствора КОН и раствор 63.6 мг (3.5 × 10–4 моль) ТХБ в 5 мл изооктана. Момент загрузки раствора ТХБ в реактор считали временем начала реакции. Реакцию осуществляли при постоянном перемешивании и постоянной подаче водорода со скоростью 5 мл/мин (2.24 × 10–4 моль/мин).
Для анализа реакционной смеси перемешивание останавливали и после расслоения отбирали пробу из органической фазы. Продукты реакции анализировали на газовом хроматографе Agilent 6890N (“Agilent”, США, капиллярная колонка DB-WAX, 30 м, диаметр 0.25 мм, пламенно-ионизационный детектор).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Каталитическая активность и влияние реакционной среды на катализатор
Полученные в работе катализаторы испытывали в гидродехлорировании 1,3,5-трихлорбензола (ТХБ) в мультифазной системе [24] в реакторе периодического действия при температуре 50°С. Принято считать, что реакции ГДХ полихлорированных органических соединений и, в частности, ТХБ, протекают последовательно в соответствии со схемой:
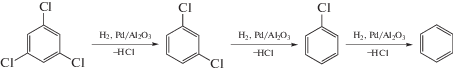
На рис. 1а и 1б приведены кинетические кривые, описывающие зависимости концентраций исходного ТХБ и продуктов реакции ГДХ от времени ее проведения в присутствии немодифицированных (рис. 1а) и модифицированных (рис. 1б) катализаторов на носителях Al2O3(НП), Al2O3(П) и Al2O3(Е). Видно, что во всех случаях основным продуктом является бензол. Он наряду с 1,3-дихлорбензолом (ДХБ) обнаруживается уже в первых пробах, взятых через 5 мин после начала реакции. При использовании немодифицированных катализаторов хлорбензол (ХБ) найден в незначительных количествах, а на катализаторах, модифицированных ГПС, его было несколько больше (~1% на Pd/ГПС-Al2O3(П), ~2% на Pd/ГПС-Al2O3(Е)). Концентрации обоих интермедиатов не проходят через максимумы, а остаются приблизительно постоянными на протяжении всего времени проведения реакции. Содержание ДХБ в реакционной смеси на образце Pd/Al2O3(П) составляет примерно 12%, на катализаторе Pd/Al2O3(Е) – чуть менее 10%. Оно немного выше в начальный период реакции в присутствии модифицированных систем (~15%), а далее заметно снижается – до 10% на образце Pd/ГПС-Al2O3(П) и до 5% на Pd/ГПС-Al2O3(Е)
Рис. 1.
Кинетические кривые реакции гидродехлорирования 1,3,5-ТХБ в присутствии немодифицированных (а) и модифицированных ГПС (б) образцов катализаторов и зависимость конверсии 1,3,5-трихлорбензола от времени реакции (в).
Среди трех исследованных немодифицированных катализаторов выделяется Pd/Al2O3(НП): в случае его применения содержание ДХБ и ХБ примерно такое же, как для Pd/Al2O3(П) и Pd/Al2O3(Е), а содержание бензола более низкое, что коррелирует с невысокой конверсией ХБ. Модифицированный Pd/ГПС-Al2O3(НП), хотя и увеличивает конверсию по сравнению с Pd/Al2O3(НП), все равно менее активен по сравнению с немодифицированными Pd/Al2O3(П) и Pd/Al2O3(Е).
Наблюдаемый вид кинетических кривых может быть обусловлен двумя основными причинами: 1) реализацией в системе принципа квазистационарности, когда промежуточные продукты быстро подвергаются дальнейшей трансформации и, следовательно, не накапливаются в реакционной смеси; 2) сильной адсорбцией промежуточных соединений, когда они практически не десорбируются с поверхности катализатора до тех пор, пока не произойдет образование бензола. Учитывая, что Al2O3 является хорошим адсорбентом, более вероятно, что реализуется второй вариант. Таким образом, можно полагать, что происходит сильная адсорбция ТХБ на поверхности обоих типов катализаторов, как немодифицированных, так и включающих ГПС; превращение ТХБ до бензола протекает, в основном, без десорбции интермедиатов. В пользу этого свидетельствует тот факт, что содержание промежуточных соединений в реакционной смеси практически одинаково для катализаторов на различных видах Al2O3.
На рис. 1в приведены кривые зависимости конверсии ТХБ в реакционной смеси от времени для всех образцов. Каждая кинетическая кривая имеет два характерных участка. В начальный период (первые 10 мин опыта) скорость реакции на всех катализаторах высокая, далее под действием реакционной среды она заметно снижается, по-видимому, в результате дезактивации части активных центров катализатора. Длительность первого периода примерно одинакова для всех немодифицированных катализаторов (рис. 1в), что косвенно может указывать на сходную природу их дезактивации. Экспериментальные точки второго участка кинетической кривой хорошо описываются линейной зависимостью (R2 > 0.9, где R – коэффициент корреляции), т.е. порядок реакции на этом отрезке близок к нулевому, что весьма вероятно в случае, когда все активные центры поверхности заполнены реагентом. Вследствие полного насыщения поверхности скорость реакции не зависит от концентрации реагента. Тогда вполне логичной представляется наблюдаемая постоянная концентрация интермедиатов в реакционной смеси: активные центры практически не освобождаются вплоть до образования конечного продукта – бензола.
Чтобы сравнить активность на начальном и втором участке на кинетических кривых, мы рассчитали каталитическую активность (TOF) (turnover frequency) на обоих участках по формуле:
где ΔСТХБ – изменение концентрации ТХБ в реакционной смеси за время Δt, n – содержание Pd в навеске катализатора в реакторе (2.8 × 10–5 моль). Полученные результаты приведены в табл. 1.Таблица 1.
Обозначения и свойства катализаторов. Величины каталитической активности приведены для 1-го (начального) и 2-го участков кинетической кривой
| Образец | Тип Al2O3 | Содержание ГПС, мас. % | Sуд, м2/г | dпор, нм | t50а, мин | t30б, мин | TOF, ч–1 | |
|---|---|---|---|---|---|---|---|---|
| 1-й участок | 2-й участок | |||||||
| Pd/Al2O3(Е) | Engelhard | –* | 173 ± 17 | 4.1 | 180 | 0 | 11в | 0.6 |
| Pd/Al2O3(П) | Прокален при 600°С | –* | 263 ± 26 | 5.5 | 250 | 4 | 30 | 0.7 |
| Pd/Al2O3(НП) | Не прокален | –* | 220 ± 22 | –** | –*** | 65 | 25.6 | 0.3 |
| Pd/ГПС-Al2O3(Е) | Engelhard | 20 | 177 ± 18 | 4.1 | 62 | 5 | 18.2 | 0.9 |
| Pd/ГПС-Al2O3(П) | Прокален при 600°С | 20 | 241 ± 24 | 6.7 | 105 | 8 | 34.6 | 0.7 |
| Pd/ГПС-Al2O3(НП) | Не прокален | 20 | 215 ± 21 | –** | 176 | 1 | 31 | 0.6 |
Видно, что начальная активность Pd/Al2O3(E) несколько меньше по сравнению с Pd/Al2O3(П) и Pd/Al2O3(НП), что связано с задержкой при взятии первой пробы для анализа. На втором участке кинетических кривых активность катализаторов резко падает и становится сравнимой для всех трех систем, хотя дезактивирующее действие реакционной среды наиболее сильно сказывается на катализаторе Pd/Al2O3(НП) – его активность уменьшается с 25.6 до 0.3 ч–1. Последнее значение примерно в два раза ниже, чем для двух других немодифицированных образцов на соответствующих участках кинетических кривых.
Модификация ГПС вызывает три эффекта (рис. 1в): возрастание начальной активности катализаторов на всех носителях, существенное увеличение (до 100 мин) продолжительности начального периода и незначительное повышение активности на втором участке кинетических кривых, особенно заметное для Pd/ГПС-Al2O3(НП).
Pd/ГПС-Al2O3(E) устойчивее в реакционной среде по сравнению с Pd/Al2O3(E), что видно по несколько более высокой активности на втором участке кинетических кривых. Активность модифицированного и немодифицированного катализаторов на носителе Al2O3(П) на втором участке кинетических кривых одинаковая, а различие в конверсии за сравнимые промежутки времени реакции обусловлено высокой активностью Pd/ГПС-Al2O3(П) в начальный период реакции.
Таким образом, по эффективности в ГДХ 1,3,5-трихлорбензола катализаторы можно расположить в следующий ряд: Pd/ГПС-Al2O3(Е) > > Pd/ГПС-Al2O3(П) > Pd/ГПС-Al2O3(НП) ≈ ≈ Pd/Al2O3(Е) ≈ Pd/Al2O3(П) > Pd/Al2O3(НП). Все изученные катализаторы теряют активность под действием реакционной среды, хотя для модифицированных образцов дезактивация происходит заметно медленнее, а наиболее устойчив к ней катализатор на носителе ГПС-Al2O3(Е).
В связи с относительно низкой эффективностью образца Pd/Al2O3(НП) и учитывая, что при высокотемпературных обработках в ходе нанесения ГПС бемит должен превращаться в оксид алюминия, что делает такой носитель аналогом Al2O3(П), физико-химические свойства катализаторов, приготовленных с использованием Al2O3(НП), изучали менее подробно по сравнению с другими образцами.
Исследование текстуры и морфологии поверхности образцов
Текстуру и морфологию поверхности образцов исследовали методами низкотемпературной адсорбции–десорбции азота и СЭМ-ЭДА. Значения площади удельной поверхности представлены в табл. 1. Видно, что SБЭТ и dпор катализатора Pd/Al2O3(П) заметно выше, чем в случае Pd/Al2O3(Е), а величины для Pd/Al2O3(НП) имеют промежуточное значение. Нанесение ГПС не приводит к существенному изменению SБЭТ. Анализ кривых распределения пор по размерам показал, что введение ГПС снижает количество малых пор: так, доля пор размером менее 4 нм заметно падает в катализаторе Pd/ГПС-Al2O3(E), а в образце Pd/ГПС-Al2O3(П) они исчезают. Средний размер пор после модификации не изменяется в случае Al2O3(E), а для Al2O3(П) немного повышается от 5.5 до 6.7 нм. Таким образом, при модифицировании обоих типов оксида алюминия (Е и П) устья малых пор закрываются частицами модификатора.
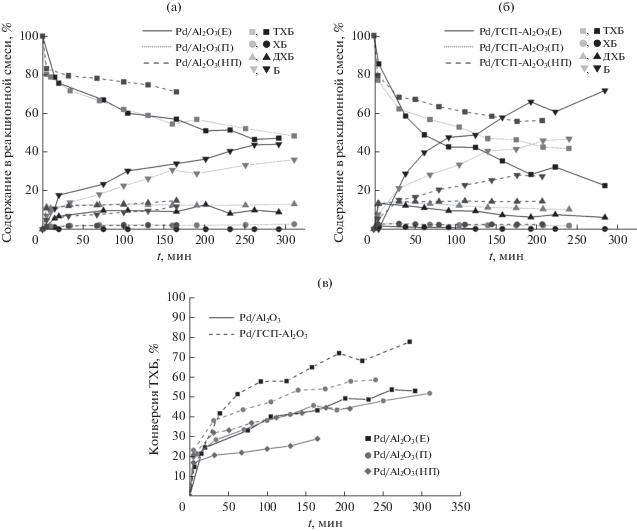
Сравнение микрофотографий, полученных методом сканирующей электронной микроскопии (СЭМ) (рис. 2), показывает, что все немодифицированные образцы включают окристаллизованные частицы неправильной формы со сглаженными ребрами и с широким распределением по размерам. Средний размер частиц Pd/Al2O3(E) составляет 25 мкм. Для Pd/Al2O3(E) и Pd/Al2O3(П) характерно присутствие очень малых частиц на поверхности более крупных, причем такой мелкой фазы больше в Pd/Al2O3(Е), а в образце Pd/Al2O3(НП) она практически отсутствует. Этот катализатор, в отличие от двух других, образован более крупными (в среднем 80 мкм) и лучше окристаллизованными частицами, а распределение Pd по поверхности, по данным ЭДА, в нем менее однородное, чем в других образцах.

Модифицирование ГПС приводит к значительным изменениям морфологии частиц: они приобретают более округлую форму и рыхлую поверхность. По-видимому, это объясняется присутствием на поверхности Al2O3 модифицирующего ГПС или продуктов его разложения в ходе термической обработки.
Анализ карт распределения элементов, полученных методом СЭМ-ЭДА (рис. 2), свидетельствует, что и палладий, и ГПС равномерно распределяются по поверхности Al2O3: на картах отсутствуют места концентрации W или Si, или области, не содержащие этих элементов. Поскольку в ходе приготовления модифицированных образцов сначала наносили ГПС, а затем соль палладия, разумно полагать, что частицы палладия находятся преимущественно на поверхности ГПС. Следовательно, в модифицированных ГПС образцах взаимодействие палладия с Al2O3 должно быть существенно слабее, чем в немодифицированных аналогах.
Исследование методами рентгенофазового анализа и просвечивающей электронной микроскопии
На дифрактограммах всех образцов имеются сильно уширенные пики, что говорит о низком размере ОКР. По данным РФА в состав Pd/Al2O3(НП) и Pd/ГПС-Al2O3(НП) входит кристаллический AlO(OH) (карта PDF № 21-1307), его содержание в Pd/Al2O3(НП) составляет 36 об. %. В образцах, полученных на основе Al2O3(П) и Al2O3(Е), присутствует кристаллический γ-Al2O3 (карта PDF № 10-425). Его доля в составе Pd/Al2O3(П) равна 67 об. %. Остальная часть материала находится в рентгеноаморфном состоянии. Кристаллическая структура Al2O3 типов (П) и (Е) существенно не различается, примерно совпадает и размер ОКР, рассчитанный по формуле Шеррера–Селякова из данных РФА (около 5 нм). Модификация ГПС приводит к снижению кристалличности и размера ОКР (4.3–4.5 нм для Pd/ГПС-Al2O3(П) и Pd/ГПС-Al2O3(Е), 3 нм для Pd/ГПС-Al2O3(НП)). Ни в одном из образцов не найдено признаков присутствия кристаллических фаз, включающих палладий (Pd, PdO, PdHx), по-видимому, вследствие малого размера таких частиц. По той же причине в катализаторах, содержащих ГПС, не обнаружены кристаллические фазы, в состав которых входит W или Si. В специально приготовленном образце ГПС, прокаленном при 450°С, найдены кристаллические фазы оксидов кремния, что свидетельствует о по меньшей мере частичном разложении ГПС, но кристаллические фазы на основе вольфрама (исходная гетерополикислота, WO3, HWO4) не идентифицированы, вероятно, по причине их рентгеноаморфности.
На рис. 3 представлены данные ПЭМ для Pd/Al2O3(П), Pd/Al2O3(НП) и Pd/Al2O3(Е). Видно, что морфология частиц носителей П, НП и Е существенно различается. В образце Pd/Al2O3(П) (рис. 3д) содержатся хорошо окристаллизованные образования сферической и столбчатой формы, столбики имеют длину до 50 нм и толщину до 5 нм, что характерно для γ-Al2O3, полученного прокаливанием при 500–700°С бемита после осаждения из изопропоксида алюминия [25]. На снимке Pd/Al2O3(НП) (рис. 3и) также заметны столбчатые структуры, хотя их меньше и они в среднем короче. В образце Pd/Al2O3(Е) (рис. 3а) форма частиц оксида алюминия округлая или близка к таковой. Более высокая кристалличность Al2O3(П) по сравнению с Al2O3(НП) (67 и 36% соответственно) подтверждена методом РФА.
Рис. 3.
Микрофотографии ПЭМ образцов в режиме светлого (а, д, и) и темного (б, е, к) поля, распределение частиц по размерам (а, д, и), карты распределения Pd (в, ж, л) и микрофотографии высокого разрешения (г, з, м): Pd/Al2O3(Е) (а–г); Pd/Al2O3(П) (д–з); Pd/Al2O3(НП) (и–м).

На рис. 3 приведены также микрофотографии ПЭМ высокого разрешения, снимки, полученные в режиме темного поля, карты распределения палладия в образце и диаграммы распределения его частиц по размерам.
В режиме темного поля хорошо заметны светлые частицы палладия. Его присутствие также подтверждено методом локального ЭДА в ходе исследования ПЭМ. Во всех образцах частицы Pd имеют нанометровые размеры, причем ни в одном из них не найдено частиц размером >20 нм. Минимальный размер и самое узкое распределение частиц Pd по размеру характерны для Pd/Al2O3(НП) (рис. 3м): >50% частиц имеет диаметр 4 нм. Образец Al2O3(П) содержит более крупные частицы, преимущественно в интервале от 4 до 8 нм. В катализаторе Pd/Al2O3(Е) распределение частиц Pd по размерам еще шире; основная их доля имеет диаметр от 4 до 11 нм, но встречаются и крупнее. По данным ПЭМ ВР все образцы содержат кристаллические частицы Pd округлой формы с межплоскостными расстояниями 0.226 нм, характерными для Pd (111).
Модификация ГПС приводит к существенному изменению структуры, наблюдаемой методом ПЭМ (рис. 4 и рис. 5): на микроснимках образцов Pd/ГПС-Al2O3(Е) и Pd/ГПС-Al2O3(П) имеются однородные по размеру (примерно 0.5 нм) небольшие темные области, равномерно расположенные на поверхности Al2O3. Учитывая их обилие, можно предположить, что они относятся к вольфрамсодержащим частицам, поскольку содержание ГПС (20%) значительно выше, чем палладия (2%). Сравнение микрофотографий, полученных в режиме темного поля, и результатов локального элементного анализа в различных областях (см. рис. 4 и рис. 5) показывает, что светлые точки соответствуют частицам палладия, тогда как вольфрам распределен по поверхности Al2O3 равномерно, его соединения не образуют крупных частиц. Из рис. 5 хорошо видно, что в области расположения наиболее светлых точек присутствуют одновременно Pd и W, тогда как в менее светлой области наблюдается только вольфрам. Вероятно, палладий при нанесении на модифицированный Al2O3 локализуется на участках, покрытых ГПС. Судя по нескольким темнопольным изображениям, размеры частиц палладия составляют от 2 до 8 нм.
Рис. 4.
Микрофотографии ПЭМ ВР образца Pd/ГПС-Al2O3(Е) в режиме светлого поля (а), в режиме темного поля (б, г), результаты ЭДА-анализа (в) и карты распределения Pd и W (д, е).
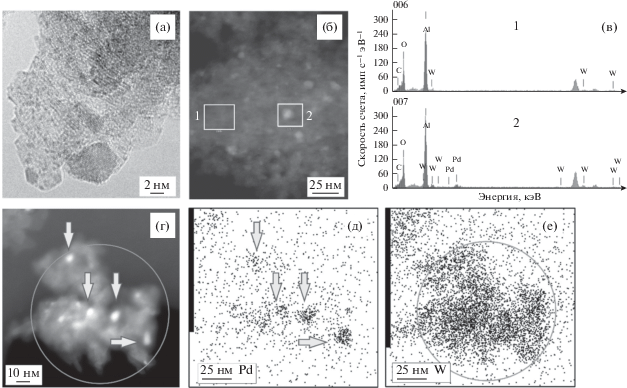
Рис. 5.
Микрофотографии ПЭМ ВР образца Pd/ГПС-Al2O3(П) в режиме светлого поля (а), в режиме темного поля (б, г), результаты ЭДА-анализа (в) и карты распределения Pd и W (д, е).
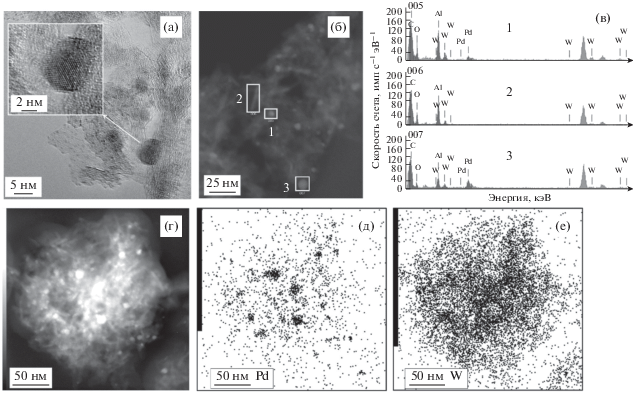
Отметим, что малый диаметр частиц ГПС или продуктов его разложения, определенный методом ПЭМ, не позволил провести анализ дисперсности методом РФА.
Исследование катализаторов методом ТПВ-Н2
На рис. 6 приведены профили ТПВ-Н2 прекурсоров приготовленных в работе катализаторов (до восстановления). Для всех образцов наблюдается отрицательный пик с максимумом примерно при 76°С, соответствующий выделению водорода при разложении гидрида β-PdHх. Следовательно, уже в ходе предварительной продувки водородом коммуникаций установки ТПВ-Н2 при 30°С происходит восстановление некоторой части палладия из оксида и насыщение частиц Pd0 водородом с получением β-PdHх, который разлагается с выделением водорода в ходе нагревания после запуска анализа. Атомарно-диспергированный палладий [26] и его мелкие частицы не способны образовывать гидрид: согласно Будару [27], растворимость водорода в палладии стремится к нулю при приближении дисперсности частиц к 100%. Появление β-PdHх при низкотемпературной обработке предшественников катализаторов водородом свидетельствует не только о способности по меньшей мере части палладия в прекурсоре катализатора легко восстанавливаться из оксида уже при температуре 30°С, но и фомировании в ходе восстановления достаточно крупных (в нанометровом диапазоне) частиц металла.
Рис. 6.
Профили ТПВ-Н2 невосстановленных предшественников катализаторов и катализаторов после испытаний в реакции ГДХ (обозначены _гдх).
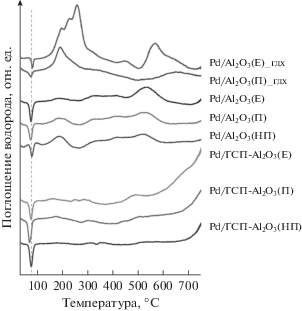
Положение максимума пика выделения водорода совпадает для всех образцов, кроме Pd/Al2O3(НП), для которого он немного сдвинут в низкотемпературную область, и Pd/ГПС-Al2O3(П), в профиле ТПВ-Н2 которого максимум, наоборот, смещен к повышенным температурам. Дрейф в сторону высоких температур может объясняться увеличенным размером частиц PdO [28]. Наблюдаемое различие соответствует данным ПЭМ, свидетельствующим о меньшем размере частиц Pd в Pd/Al2O3(НП) по сравнению с Pd/Al2O3(П).
Профили ТПВ-Н2 всех немодифицированных образцов содержат также широкие пики поглощения водорода с размытыми максимумами при 180, 310, 410 и 530°С, что говорит о присутствии частиц PdO разного размера [28, 29], сильно и слабо связанных с поверхностью [28]. Так, в [29] пики поглощения водорода при Т = 363 и 355°C относят к восстановлению частиц PdO относительно небольших размеров и гидроксильных групп на поверхности оксида алюминия. Хотя Al2O3, не содержащий палладия, способен восстанавливаться только при Т > 700°С, в присутствии каталитически активного металла гидроксильные группы на его поверхности восстанавливаются уже при температурах ниже 400°С [30].
Крупные кристаллиты PdO первыми восстанавливаются до металла, который способен обеспечивать адсорбцию и диссоциацию молекул Н2. Образовавшиеся атомы или ионы по механизму спилловера перетекают на поверхность Al2O3 и более мелких частиц палладия и способствуют их восстановлению при повышенных температурах. Сверхстехиометрическая адсорбция водорода за счет спилловера на носитель также может вызывать поглощение водорода при Т > 300°C [28]. Кроме того, возможно, что присутствие высокотемпературных пиков обусловлено восстановлением атомарно диспергированного палладия.
Пик при 180°С, по-видимому, связан с парциальным восстановлением PdO до Pd2O [20]:
Интенсивность пика при 180°С максимальна для образца Pd/Al2O3(НП), ниже в профиле Pd/Al2O3 (П) и минимальна для Pd/Al2O3(E). Для последнего образца характерна наибольшая площадь пика при 530°С (данные о поглощении водорода приведены в табл. 2). Можно полагать, что в составе прекурсора Pd/Al2O3(E) основная часть PdO сильно связана с носителем.
Таблица 2.
Поглощение водорода на разных участках профилей ТПВ-Н2
| Образец | Поглощение водорода в области температур, мкмоль/г | |||
|---|---|---|---|---|
| 76°Са | 190°С | 290–450°С | 530°С | |
| Pd/Al2O3(E) | –18 | 6 | 27 | 63 |
| Pd/Al2O3(П) | –16 | 26 | 152 | – |
| Pd/Al2O3(НП) | –10 | 34 | 134 | – |
| Pd/ГПС-Al2O3(E) | –17 | 5 | 11 | 15 |
| Pd/ГПС-Al2O3(П) | –23 | 6 | 7 | 8 |
| Pd/ГПС-Al2O3(НП) | –21 | 0 | 3 | 4 |
| Pd/Al2O3(E) после ГДХ | –5 | 264б | – | 128 |
| Pd/Al2O3(П) после ГДХ | –7 | 106 | – | – |
На основании результатов ТПВ-Н2 для восстановления палладия выбрали температуру 500°C, так как при ней в изотермическом режиме возможно восстановление палладия практически из всех форм, присутствующих в прекурсорах.
После модификации ГПС катализаторов на носителях Al2O3(П) и Al2O3(НП) интенсивность пиков поглощения водорода в температурном интервале 180–530°С заметно снижается (табл. 2), а в профиле катализатора Pd/ГПС-Al2O3(E) – практически не меняется, но все они сдвигаются в сторону меньших температур. Наблюдаемые изменения можно связать с ослаблением взаимодействия PdO с поверхностью Al2O3. В результате большая часть PdO восстанавливается при относительно малых температурах. Действительно, площадь низкотемпературного пика выделения водорода, соответствующего разложению гидрида палладия, в профилях образцов Pd/ГПС-Al2O3(НП) и Pd/ГПС-Al2O3(П) увеличивается (табл. 2), но для Pd/ГПС-Al2O3(Е) такого роста не наблюдается. Полученные данные дают основание считать, что пик при Т = 500–530°С характеризует восстановление PdO в виде хорошо структурированных относительно крупных частиц и/или частиц, сильно связанных с поверхностью Al2O3. Это предположение находит подтверждение в результатах ПЭМ, согласно которым распределение частиц палладия по размерам наиболее широкое для Pd/Al2O3(Е), а доля крупных частиц в нем максимальная по сравнению с образцами Pd/Al2O3(П) и Pd/Al2O3(НП).
Очень сильное падение интенсивности широких пиков в температурном интервале 300–450°С в результате модификации ГПС может объясняться не только снижением степени взаимодействия палладия с поверхностью Al2O3, но и уменьшением доли доступных для восстановления гидроксильных групп на поверхности Al2O3 в результате равномерного (по данным СЭМ и ПЭМ) распределения модификатора на поверхности.
Рост поглощения водорода при температурах выше 650°С вызван началом восстановления ГПС [31]. В случае Pd/ГПС-Al2O3(Е) этот процесс начинается существенно раньше, при 550°С, по-видимому, из-за менее тесного контакта ГПС с носителем, что связано с особенностями морфологии частиц Al2O3(Е).
Полученные методом ТПВ-Н2 данные подтверждают снижение степени взаимодействия палладия с носителем при его модификации ГПС, а также изменение состава гидроксильного покрова на поверхности Al2O3, и, соответственно, кислотных свойств поверхности.
Исследование катализаторов методом ИК-спектроскопии диффузного отражения с Фурье-преобразованием адсорбированного СО
На рис. 7 представлены спектры ИК ДО адсорбированного СО для немодифицированных и модифицированных образцов Al2O3(Е) и Al2O3(П). Поглощение в области 2250–2150 см–1 обусловлено комплексами СО с катионами алюминия на поверхности носителя.
Рис. 7.
Спектры ИК ДО CO, адсорбированного на оксидах алюминия Al2O3(П) (а) и Al2O3(Е) (б) и на модифицированных ГПС оксидах алюминия ГПС-Al2O3(П) (в) и ГПС- Al2O3(Е) (г). Давление СО: 5 (кривые 1), 20 (кривые 2) и 50 Торр (кривые 3).
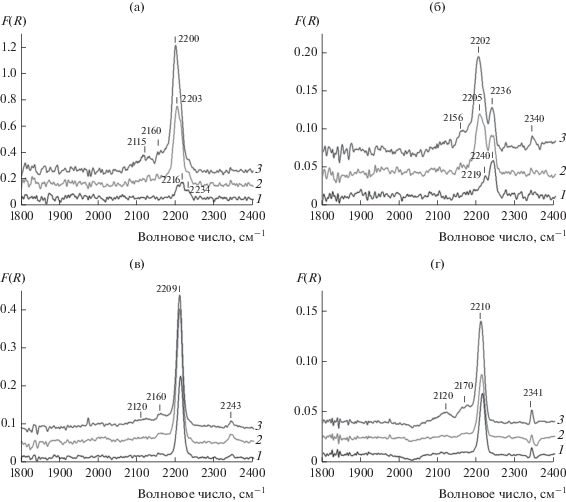
В спектре исходных образцов Al2O3 присутствует полоса поглощения (п. п.) с максимумом 2200 см–1, которая относится к комплексам СО с координационно-ненасыщенными катионами Al3+ в тетраэдрической координации на участках поверхности с регулярной структурой – льюисовскими кислотным центрам (ЛКЦ) типа L3 по классификации Мортерры [32–34]. Интенсивность этой п. п. возрастает с повышением давления СО от 5 до 55 Торр, что связано с заполнением свободных ЛКЦ-L3. Также для обоих типов Al2O3 в спектре имеется плечо в области 2216–2219 см–1, которое может соответствовать комплексам с ЛКЦ типа L2, в которых катионы алюминия находятся вблизи дефектов поверхности [32, 33].
В спектре Al2O3(E) также есть п. п. около 2240 см–1, характерная для карбонильных комплексов с ЛКЦ типа L1 [32, 33]. При низком давлении СО образуются комплексы преимущественно с таким типом ЛКЦ. Указанные центры заполняются уже при небольшом давлении СО, и последующее его повышение до 55 Торр не сказывается на интенсивности этой полосы. При увеличении давления СО постепенно заполняются ЛКЦ-L3, что хорошо видно по возрастанию интенсивности п. п. в области 2190–2220 см–1.
Вид ИК-спектра в диапазоне 2250–2150 см–1 для модифицированных образцов меняется. После нанесения ГПС п. п. 2235–2240 и 2002–2205 см–1 исчезают, а интенсивность п. п. 2209–2215 см–1 растет. Вероятно, ГПС блокирует большую часть ЛКЦ регулярной поверхности, заполняя их координационную сферу, что приводит к ослаблению или полному исчезновению ЛКЦ типа L1 и L3. Однако количество ЛКЦ-L2, наоборот, повышается. Возможно, модификация ГПС способствует переходу части катионов алюминия в новое координационное состояние, совпадающее по координационному числу с центрами типа L2. Под влиянием новой координационной сферы электроноакцепторная способность центров увеличивается. Альтернативное объяснение состоит в формировании ЛКЦ новой природы при нанесении ГПС. Об образовании дополнительных ЛКЦ в результате адсорбции оксидов вольфрама на поверхности Al2O3 сообщалось в [35–37].
Электронное состояние частиц палладия на поверхности катализаторов исследовали этим же методом по изменению полос поглощения адсорбированного СО (рис. 8). В спектре Pd/Al2O3(Е) (рис. 8а) наблюдаются п. п., характерные для адсорбции СО на Pd0. Полоса поглощения при 2091–2095 см–1 соответствует линейным комплексам Pd0–CO, п. п. при 1990 см–1 с плечом при 1950 см–1 может быть отнесена к мостиковым комплексам СО с двумя расположенными рядом атомами палладия (Pd0)2–CO; поглощение в области 1800–1900 см–1 указывает на вклад мостиковых комплексов СО, координированного тремя атомами палладия (Pd0)3–CO [38–41].
Рис. 8.
Спектры ИК ДО CO, адсорбированного на поверхности Pd/Al2O3(Е) (а), Pd/Al2O3(П) (б), Pd/ГПС-Al2O3(Е) (в) и Pd/ГПС-Al2O3(П) (г). Давление СО: 5 (кривые 1), 20 (кривые 2) и 50 Торр (кривые 3).
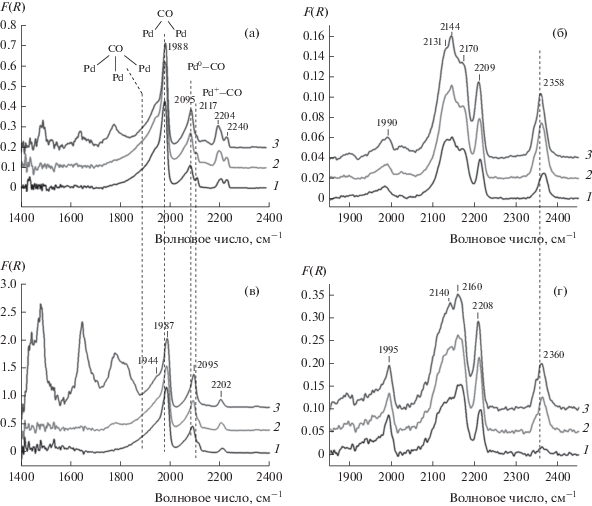
Интенсивность п. п., отвечающей линейному комплексу, примерно в три раза меньше, чем для мостиковых комплексов. Согласно литературным данным, такое соотношение интенсивностей характерно для молекул, занимающих два участка в элементарной ячейке [39], что согласуется с обнаружением окристаллизованных частиц палладия методом ПЭМ. В случае атомарно диспергированного на поверхности палладия (размер частиц менее 2 нм) в спектре имеется интенсивная полоса в интервале 2070–2110 см−1, отвечающая линейным комплексам [42, 43]. Широкий пик п. п. мостиковых комплексов с размытым плечом в области меньших волновых чисел указывает на адсорбцию молекул СО на различных гранях частиц палладия.
Полоса поглощения при 2120–2117 см–1 может быть отнесена к линейным комплексам СО с окисленным палладием Pd+–CO [44]. Авторы [38] отмечают, что поглощение в области 2100–2150 см–1 характерно для комплексов СО с частично окисленным палладием, а п. п., отвечающие комплексам (Pd2+)–CO, лежат в области более высоких значений волновых чисел (2215–2145 cм–1). Особенно четко п. п. Pd+–CO выражена в спектре, полученном при низком давлении СО. По-видимому, эти центры одними из первых заполняются адсорбированными молекулами СО.
При повышении давления СО с 5 до 55 Торр п. п., соответствующая линейному комплексу Pd0–CO, сдвигается от 2070 к 2095 см–1. Авторы [40] связывают п. п. при 2070 см–1 с линейными комплексами СО с атомами палладия, локализованными на ребрах, ступеньках и террасах граней (100) и (111). Таким образом, при низких давлениях СО происходит заполнение наиболее дефектных участков поверхности палладия. Дальнейшая адсорбция СО проявляется в ИК-спектре в незначительном сдвиге соответствующей п. п. Большая полуширина п. п. в области 2070–2095 см–1 [40] и небольшое плечо в области 2050 см–1 [41], свидетельствующее о присутствии центров как минимум двух типов, указывают на неоднородность поверхности палладия.
При давлении СО 55 Торр в области 1700–1200 см−1 ИК-спектра Pd/Al2O3(E) имеются п. п., относящиеся к карбонатным структурам на поверхности Al2O3. Образование карбонатов происходит при взаимодействии молекул СО с ОН-группами на поверхности Al2O3 [45, 46].
В отличие от Pd/Al2O3(Е) в спектре ИК ДО для СО, адсорбированного на Pd/Al2O3(П) (рис. 8), практически отсутствует п. п., соответствующая комплексу (Pd+)–CO, т.е. почти весь палладий находится в металлическом состоянии. Кроме того, не так ярко выражена п. п., характерная для комплексов (Pd0)3–CO. Это может быть обусловлено меньшим размером частиц в указанном образце, что подтверждается методом ПЭМ. При давлении 55 Торр также отмечается существенно больший вклад карбонатных структур по сравнению со спектром Pd/Al2O3(Е).
Таким образом, на поверхности немодифицированных катализаторов после термовакуумной обработки присутствует Pd0, для образца Pd/Al2O3(E) характерен также заметный вклад частично окисленного палладия. Полученные данные ИК-спектроскопии согласуются с результатами ПЭМ относительно распределения частиц Pd по размерам.
Модификация носителей ГПС приводит к существенному изменению вида ИК-спектров адсорбированного СО палладиевых катализаторов (рис. 8в и 8г). Так, в спектрах обеих систем Pd/ГПС/Al2O3(П) и Pd/ГПС/Al2O3(Е) имеется п. п. при 2360 см–1, которая возникает в присутствии молекул СО2, координированных на поверхности Al2O3 без образования карбоната. В литературе нет однозначного ответа на вопрос, на каких центрах происходит такая адсорбция, однако можно предположить, что они включают катионы палладия. Наблюдаемые п. п. при 2340 см–1 и 2365–2358 см–1 мы относим к комплексам СО2 с металлическим Pd (Pd0…CO2) [47].
Суперпозиция полос в диапазоне 2180–2050 см–1 может быть обусловлена присутствием п. п. 2170–2172 см–1, характерной для карбонильных комплексов Pd2+–CO, и п. п. при 2144 и 2134–2131 см–1, которые соответствуют карбонильным комплексам Pd+–CO. По-видимому, п. п. 2157 см–1 относится к комплексам Pd2+–CO. Присутствует также малоинтенсивная п. п. при 1984–1990 см–1, характерная для комплексов с двумя атомами (Pd0)2–CO. Таким образом, в отличие от образцов, не содержащих ГПС, в модифицированных образцах основной формой палладия на поверхности являются катионы Pd2+ (меньше) и Pd+ (больше). Металлического палладия немного, при этом имеется как металлическая фаза (основная форма в образцах без ГПС), так и отдельные атомы.
Полоса поглощения при 1900 см–1 соответствует мостиковой форме адсорбции СО, но не на металлическом палладии. Возможно, СО координируется на соседних катионах палладия и алюминия.
Отличия спектра Pd/ГПС/Al2O3(П) от Pd/ГПС/Al2O3(Е) следующие. В диапазоне 2180–2050 см–1 в спектре Pd/ГПС/Al2O3(П) доминирует п. п. 2164–2159 см–1, т.е. комплексов Pd2+–CO больше, чем комплексов Pd+–CO. Заметен вклад еще одной п. п. при 2100–2111 см–1 (проявляется в виде перегиба, но в спектре образца Pd/ГПС/Al2O3(Е) нет даже перегиба), которую можно отнести к комплексам с одиночными атомами – Pd0–CO. Также присутствуют п. п. 1995 см–1 карбонильных комплексов (Pd0)2–CO и п. п. мостиковой формы адсорбции при 1890 см–1.
Таким образом, результаты ИК-исследования демонстрируют существенные отличия в составе ЛКЦ на поверхности носителей Al2O3(Е) и Al2O3(П), что приводит к различным типам координации палладия в ходе нанесения и разному электронному состоянию в готовых катализаторах. Все это должно влиять на каталитические свойства полученных систем.
Наблюдаются также существенные изменения в организации поверхности в результате модификации ГПС. После термовакуумной обработки на поверхности катализаторов, полученных без ГПС, основной формой является Pd0, преимущественно в виде парных центров адсорбции, т.е. относительно крупных частиц. Напротив, на поверхности модифицированных ГПС катализаторов присутствуют в основном одиночные (для СО) катионы Pd+ и Pd2+. Также свой вклад вносит Pd0, но он не является доминирующим.
Для образца Pd/ГПС/Al2O3(Е) отмечено подавление наиболее сильных ЛКЦ поверхности носителя, которые в значительном количестве имелись в Al2O3(Е) и отсутствовали в Al2O3(П).
На поверхности модифицированных катализаторов отмечается появление СО2, координированного поверхностью и образовавшегося в результате окисления СО, но поверхностных карбонатов в достаточных для уверенной регистрации количествах не обнаружено. Напротив, на поверхности немодифицированных катализаторов наблюдается образование большого количества монодентатных, бидентатных, а также “органических” карбонатов, возникающих в результате окисления СО поверхностью. Это указывает на полимодальность распределения основных свойств анионов поверхности (кислорода), а также на присутствие на поверхности центров, активных в реакции окисления СО.
Обсуждение результатов
В литературе отмечается [48], что структурная чувствительность реакции ГДХ может быть обусловлена повышенной устойчивостью более крупных (в нанометровом диапазоне) частиц палладия к воздействию НСl, в то время как мелкие частицы легко хлорируются в реакционной среде. В случае благородных металлов, нанесенных на Al2O3, в результате взаимодействия с ЛКЦ на поверхности носителя малые частицы металла становятся электронодефицитными и сильнее подвергаются хлорированию адсорбированным хлором (или HCl). Однако кинетические кривые превращения ТХБ на немодифицированных ГПС катализаторах Pd/Al2O3(Е) и Pd/Al2O3(П) практически не различаются, несмотря на существенные различия в значениях дисперсности активного компонента и удельной поверхности, составе ЛКЦ и имеющихся на поверхности формах палладия. По-видимому, это объясняется присутствием в обоих образцах нанометровых частиц палладия, которые подвержены частичному окислению под действием компонентов реакционной среды.
Причиной дезактивации катализаторов в реакционной среде может быть окисление Pd при взаимодействии с выделяющимися частицами хлора и хлорид-ионов, а также при взаимодействии с носителем. Такое окисление должно приводить к изменению профилей ТПВ-Н2 дезактивированных образцов после катализа. Профили ТПВ-Н2 образцов Pd/Al2O3(E) и Pd/Al2O3(П) после каталитического испытания приведены на рис. 6, а количества поглощенного/выделившегося водорода – в табл. 2.
Видно, что после каталитических испытаний интенсивность отрицательного пика в профиле ТПВ-Н2 обоих образцов уменьшается приблизительно в три раза. Существенно возрастает интенсивность группы пиков в интервале температур от 135 до 335°С, которые характеризуют различные формы палладия, предположительно хлорид и оксид, слабо и сильно связанные с поверхностью носителя. Форма и ширина вышеуказанной группы пиков различны для этих двух образцов: в профиле Pd/Al2O3(E) после ГДХ пик восстановления расположен в интервале от 135 до 450°С и имеет три плохо разделенных максимума; в профиле Pd/Al2O3(П) эта группа выглядит как один широкий пик с максимумом при 185°С.
Хлорид палладия может образовываться при взаимодействии частиц активного компонента (Pd0) с частицами хлора на поверхности, появляющимися в результате диссоциативной адсорбции ТХБ, а его оксид – в результате контакта палладия с носителем Al2O3.
Пик восстановления с максимумом при 530°С, который может соответствовать прочно связанным с носителем формам палладия, после каталитических испытаний сдвигается в область более высоких температур. Это говорит об усилении связи палладия с носителем, например, в результате возникновения на поверхности частиц палладия слоя, включающего оксид и хлорид палладия. Пик восстановления при 530°С в профиле ТПВ Pd/Al2O3(П) после ГДХ сдвинут в область повышенных температур гораздо сильнее, чем в профиле Pd/Al2O3(Е) после ГДХ, однако имеет меньшую интенсивность. Как уже было показано при обсуждении результатов ПЭМ, в образце Pd/Al2O3(E) распределение частиц по размеру значительно шире, чем в Pd/Al2O3(П). Хлорирование и окисление частиц палладия разного размера может приводить к появлению форм, восстанавливающихся в более широком интервале температур.
Отметим, что сильнее всего дезактивации подвергается в реакционной среде катализатор, в котором носителем служит бемит (Pd/Al2O3(НП)). Очевидной причиной может служить очень малый по сравнению с другими катализаторами размер частиц палладия и узкое распределение по размерам. С другой стороны, известно, что бемит при температурах 30°С и выше способен взаимодействовать с основанием, присутствующим в реакционной среде, причем при рН более 7 будет образовываться преимущественно тетрагидроксоалюминат-ион [49]:
В результате такого взаимодействия возможно разрушение поверхности катализатора, приводящее к дополнительному снижению активности.
Добавление основания в реакционную среду давно используют с целью увеличения эффективности катализаторов ГДХ, включая нанесенные на Al2O3; эффективность повышается в результате связывания выделяющегося хлороводорода с образованием соли [11, 50]. Однако в водных средах при повышенных рН дезактивация каталитических систем усиливается: например, при ГДХ 4-хлорфенола константа скорости дезактивации Pd/Al2O3 оказалась в два раза больше, чем при нейтральном рН [51].
Хлорбензолы могут адсорбироваться и активироваться как на металлах, так и на носителях. Вероятность последнего варианта возрастает при использовании носителей, обладающих льюисовской кислотностью. В этом случае водород для восстановления адсорбированного на ЛКЦ субстрата поступает по механизму спилловера с металлических центров диссоциативной адсорбции [16]. Активация хлорбензолов может также происходить на поверхности частично окисленного палладия. Известно, что при жидкофазном ГДХ преимуществами обладают катализаторы, в которых соотношение Pd0/Pdδ+ близко к 1, что создает хорошие условия для активации как хлорбензолов, так и водорода [52]. Изменение этого соотношения в результате нанесения ГПС на поверхность носителя, о чем свидетельствуют полученные нами результаты ИК ДО, обеспечивает наблюдаемое повышение TOF на начальных участках кинетических кривых (табл. 1), но не предотвращает дезактивацию под действием реакционной среды.
Приведенные рассуждения позволяют объяснить образование малых количеств интермедиатов (дихлор- и монохлорбензолов) в реакционной смеси на протяжении всего каталитического эксперимента. Можно предположить, что катализатор содержит достаточно сильные адсорбционные центры, например, Pdδ+ и ЛКЦ на поверхности носителя. На них происходит превращение ТХБ в конечный продукт реакции ГДХ, бензол, без десорбции интермедиатов. Присутствие ДХБ и ХБ в небольших количествах в реакционной среде связано с тем, что адсорбционное равновесие сильно сдвинуто в сторону адсорбированной формы.
Другим возможным объяснением вида кинетических кривых является протекание реакции по последовательной схеме в условиях квазистационарного приближения, когда промежуточные соединения не накапливаются, а сразу реагируют с водородом. В этом случае невысокая концентрация интермедиатов на протяжении всего реакционного цикла вызвана их частичной десорбцией с поверхности катализатора в объем реакционной смеси.
В ходе термической обработки может происходить разложение модификатора Н8[Si(W2O7)6] с образованием оксидов вольфрама и кремния. В работе [53] было показано, что при нанесении на Al2O3, полученный золь–гель-методом или коммерческий, термостабильность ГПС H6P2W18O62 структуры Доусона существенно возрастает: до нанесения разложение ГПС происходило уже при 250°С, а после нанесения структура была устойчива при нагревании до 620°С, а выше этой температуры начинала перестраиваться в структуру Кеггина. В работе [54], напротив, наблюдалась деградация аниона [PW12O40]3– с образованием вольфрамдифосфата уже при сушке нанесенной на Al2O3 системы при 70°С. Однако в [55] после прокаливания при 350°С системы H4SiW12O40Н2О/Al2O3 (это же соединение использовано в качестве модификатора в настоящей работе) на поверхности были обнаружены анионы ГПС H4SiW12O40 типа Кеггина, после прокаливания при 450°С – W3O13, а при 550°С и выше – фаза WO3 и отдельные анионы WO4.
В представленной работе прокаливание Al2O3 после добавления ГПС проводили при 400°С, что гарантирует сохранение структуры Кеггина в подложке, на которую наносили палладий. Однако все катализаторы восстанавливали при 500°С, поэтому нельзя исключить присутствия в модифицированных катализаторах оксидов вольфрама и кремния. Действительно, анализ методом РФА гетерополисоединения Н8[Si(W2O7)6] ⋅ Н2О, прокаленного на воздухе при 450°С, показал наличие кристаллических оксидов кремния различной природы, однако оксиды вольфрама этим методом обнаружены не были, вероятно, в связи с аморфным состоянием и малым размером частиц. Оксид кремния на поверхности может частично блокировать палладиевые центры, а оксиды вольфрама при взаимодействии с водородом в ходе восстановления способны образовывать вольфрамовые бронзы, которые известны своей активностью в гидрировании [56].
Методом ИК-спектроскопии установлено, что модификация поверхности Al2O3 ГПС приводит к ослаблению или полному исчезновению ЛКЦ типа L1 и L3, с одновременным увеличением количества ЛКЦ типа L2, что может быть связано с модификацией ГПС или продуктами его разложения. Изменение кислотности поверхности Al2O3 в присутствии оксидов вольфрама описано в литературе [35–37]. Так, в [36] было показано, что изолированные тетраэдрические частицы WO4 обеспечивают появление дополнительных ЛКЦ на поверхности модифицированных катализаторов. Наличие на поверхности катализатора Pd–WOx(5%)/Al2O3 частиц WO4 оказывает превосходный промотирующий эффект в реакции гидрогенолиза глюкозы. Этот катализатор обладал хорошей стабильностью (продолжительность работы более 200 ч).
В работе [37] было обнаружено, что повышение количества ЛКЦ связано с присутствием координационно ненасыщенных катионов Wn+, а кислотные центры Бренстеда возникают из частично гидратированных вольфрамсодержащих частиц, включая ОН-группы, координированные на участках с W=O–, W–O–W- и W–O–Al-связями в кластерах WOx. Более того, было установлено, что электронное взаимодействие между кластерами Pt и WOx приводит к образованию частиц Ptδ+, которые являются активными центрами для окисления пропана. Активация С–Н-связи происходит на границе контакта платины и оксида вольфрама.
Возможно, похожее промотирующее действие наблюдается при модифицировании оксида алюминия ГПС в исследованных в настоящей работе катализаторах. В пользу этого предположения свидетельствует явное изменение соотношения разных типов ЛКЦ после добавления ГПС, обнаруженное методом ИК-спектроскопии. Тесный контакт палладия и нанесенного ГПС способствует образованию новых активных центров на линии контакта или даже биметаллических центров, которые, как было описано в [57], проявляют повышенную эффективность в ГДХ. Отметим, что смещение пиков поглощения в профиле ТПВ-Н2 катализатора Pd/ГПС-Al2O3(E) в результате модификации может объясняться взаимодействием Pd с вольфрамом в составе ГПС или продуктов его разложения. Именно для этого катализатора характерна бóльшая стабильность работы по сравнению с катализаторами на других носителях, т.е. новые типы активных центров несколько устойчивее к воздействию реакционной среды при ГДХ.
Как хорошо видно из данных ПЭМ и ТПВ, равномерное распределение ГПС и/или продуктов его разложения по поверхности носителя (бемита или Al2O3) снижает степень взаимодействия палладия с носителем, облегчает восстановление частиц палладия при температуре реакционной среды, что способствует увеличению стабильности катализаторов.
ЗАКЛЮЧЕНИЕ
В настоящей работе показано, что структура носителя влияет на каталитические свойства 2%Pd/Al2O3-катализаторов в мультифазном гидродехлорировании 1,3,5-трихлорбензола. В ряду Pd/Al2O3(НП) < Pd/Al2O3(П) < Pd/Al2O3(Е) снижается дисперсность частиц палладия и увеличивается ширина их распределения по размерам в диапазоне до 20 нм. Согласно результатам ИК-спектроскопии диффузного отражения адсорбированного СО (ИК-ДО) на поверхности всех этих образцов преобладает Pd0 в виде относительно крупных частиц.
Модифицирование ГПС (20 мас. % Н8[Si(W2O7)6] ⋅ 6Н2О) способствует уменьшению размера частиц Pd. Методом термопрограммированного восстановления водородом (ТПВ-Н2) показано, что в состав всех катализаторов входит гидрид палладия наряду с более прочно связанными с поверхностью окисленными формами металла, которые восстанавливаются при повышенных температурах, причем их количество снижается после модификации ГПС и возрастает после каталитических испытаний.
По данным ИК-ДО адсорбированного СО нанесение ГПС приводит к изменению типа льюисовских кислотных центров на поверхности Al2O3 и электронного состояния палладия. На поверхности модифицированных ГПС практически полностью исчезают ЛКЦ типа L1 и L3 с одновременным увеличением количества центров L2. В составе модифицированных ГПС катализаторов присутствуют одиночные катионы Pd+ и Pd2+, а доля Pd0 существенно меньше. Особенности льюисовской кислотности поверхности катализаторов определяют возможность адсорбции и активации ТХБ на носителе и поступления водорода от Pd0 по механизму спилловера. Проведенное в работе комплексное исследование свидетельствует о возможности образования новых активных центров при взаимодействии палладия с ГПС или продуктами его термического разложения (оксиды вольфрама и катионы WO4 на поверхности). Перечисленные структурные особенности способствуют повышению стабильности каталитических систем, модифицированных ГПС, в мультифазном гидродехлорировании 1,3,5-трихлорбензола с образованием, преимущественно, бензола.
Список литературы
Perosa A., Selva M., Maschmeyer T. // Chemosphere. 2017. V. 173. P. 535.
Dai C., Zhou Y., Peng H., Huang S., Qin P., Zhang J., Yang Y., Luo L., Zhang X. // J. Ind. Eng. Chem. 2018. V. 62. P. 106.
Zinovyev S.S., Shinkova N.A., Perosa A., Tundo P. // Appl. Catal. B: Env. 2005. V. 55. P. 39.
Tundo P., Perosa A., Selva M., Zinovyev S.S. // Appl. Catal. B: Env. 2001. V. 32. P. L1.
Cobo M., Quintero A., Correa C.M.D. // Catal. Today. 2008. V. 133–135. P. 509.
Wu Y., Gan L., Zhang S., Song H., Lu C., Li W., Wang Z., Jiang B., Li A. // J. Hazard. Mater. 2018. V. 356. P. 17.
Cheng L., Jin Z., Wang X. // Catal. Commun. 2013. V. 41. P. 60.
Han B., Liu W., Li J., Wang J., Zhao D., Xu R., Lin Z. // Water Research. 2017. V. 120. P. 199.
Nieto-Sandoval J., Munoz M., de Pedro Z.M., Casas J.A. // Chemosphere. 2018. V. 213. P. 141.
De Corte S., Sabbe T., Hennebel T., Vanhaecke L., De Gusseme B., Verstraete W., Boon N. // Water Research. 2012. V. 46. P. 2718.
Wu K., Qian X., Chen L., Xu Z., Zheng S., Zhu D. // RSC Adv. 2015. V. 5. P. 18702.
El-Sharnouby O., Boparai H. K., Herrera J., O’Carroll D.M. // Chem. Eng. J. 2018. V. 342. P. 281.
Mori T., Kubo J., Morikawa Y. // Appl. Catal. A: General. 2004. V. 271. P. 69.
Śrębowata A., Juszczyk W., Kaszkur Z., Karpiński Z. // Catal. Today. 2007. V. 124. P. 28.
Diaz E., Mohedano A.F., Casas J.A., Shalaby C., Eser S., Rodriguez J.J. // Appl. Catal. B: Env. 2016. V. 186. P. 151.
Hashimoto Y., Uemichi Y., Ayame A. // Appl. Catal. A: General. 2005. V. 287. P. 89.
Ordóñez S., Sastre H., Díez F.V. // React. Kinet. Catal. Lett. 2000. V. 70. P. 61.
Lokteva E.S., Erokhin A.V., Kachevsky S.A., Yermakov A.Y., Uimin M.A., Mysik A.A., Golubina E.V., Zanaveskin K.L., Turakulova A.O., Lunin V.V. / Metal-carbon nanocomposite systems as stable and active catalysts for chlorobenzene transformations. In: M. D. S. H. P. A. J. J. A. M. Gaigneaux E.M. and Ruiz P. (Eds.) Studies in Surface Science and Catalysis, Elsevier, 2010. P. 289.
Klokov S.V., Lokteva E.S., Golubina E.V., Chernavskii P.A., Maslakov K.I., Egorova T.B., Chernyak S.A., Minin A.S., Konev A.S. // Appl. Surf. Sci. 2019. V. 463. P. 395.
Babu N.S., Lingaiah N., Kumar J.V., Prasad P.S.S. // Appl. Catal. A: General. 2009. V. 367. P. 70.
Golubina E.V., Lokteva E.S., Lunin V.V., Telegina N.S., Stakheev A.Y., Tundo P. // Appl. Catal. A: General. 2006. V. 302. P. 32.
Srikanth C.S., Kumar V.P., Viswanadham B., Chary K.V.R. // Catal. Commun. 2011. V. 13. P. 69.
Навалихина М.Д., Кавалерская Н.Е., Локтева Е.С., Перистый А.А., Голубина Е.В., Лунин В.В. // Журн. физ. химии. 2012. V. 86. P. 1792.
Tundo P., Perosa A., Zinovyev S. // J. Mol. Catal. A: Chem. 2003. V. 204–205. P. 747.
Mrabet D., Vu M.-H., Kaliaguine S., Do T.-O. // J. Coll. Interface Sci. 2017. V. 485. P. 144.
Sepúlveda J.H., Fígoli N.S. // Appl. Surf.ace Sci. 1993. V. 68. P. 257.
Boudart M., Hwang H.S. // J. Catal. 1975. V. 39. P. 44.
Zheng Q., Farrauto R., Deeba M. // Catalysts. 2015. V. 5. P. 1797.
Bhogeswararao S., Srinivas D. // J. Catal. 2015. V. 327. P. 65.
Hurst N.W., Gentry S.J., Jones A., McNicol B.D. // Catal. Rev. 1982. V. 24. P. 233.
Jin H., Yi X., Sun X., Qiu B., Fang W., Weng W., Wan H. // Fuel. 2010. V. 89. P. 1953.
Morterra C., Magnacca G. // Catal. Today. 1996. V. 27. P. 497.
Morterra C., Bolis V., Magnacca G. // Langmuir. 1994. V. 10. P. 1812.
Busca G., Finocchio E., Escribano V.S. // Appl. Catal. B: Env. 2012. V. 113–114. P. 172.
Barrera A., Montoya J.A., del Angel P., Navarrete J., Cano M.E., Tzompantzi F., López-Gaona A. // J. Phys. Chem. Solids. 2012. V. 73. P. 1017.
Liu C., Zhang C., Sun S., Liu K., Hao S., Xu J., Zhu Y., Li Y. // ACS Catal. 2015. V. 5. P. 4612.
Wu X., Zhang L., Weng D., Liu S., Si Z., Fan J. // J. Hazard. Maters. 2012. V. 225–226. P. 146.
Binet C., Jadi A., Lavalley J.-C. // J. Chim. Phys. 1989. V. 86. P. 451.
Bourguignon B., Carrez S., Dragnea B., Dubost H. // Surface Sci. 1998. V. 418. P. 171.
Bertarione S., Scarano D., Zecchina A., Johánek V., Hoffmann J., Schauermann S., Frank M.M., Libuda J., Rupprechter G., Freund H.-J. // J. Phys. Chem. B. 2004. V. 108. P. 3603.
Lear T., Marshall R., Antonio Lopez-Sanchez J., Jackson S.D., Klapötke T.M., Bäumer M., Rupprechter G., Freund H.-J., Lennon D. // J. Chem. Phys. 2005. V. 123. P. 174706.
Clarke J.K.A., Farren G.M., Rubalcava H.E. // J. Phys. Chem. 1967. V. 71. P. 2376.
van Hardeveld R., Hartog F. / Influence of Metal Particle Size in Nickel-on-Aerosil Catalysts on Surface Site Distribution, Catalytic Activity, and Selectivity. In: Eley D.D., Pines H. and Weisz P.B. (Eds.) Advances in Catalysis, Academic Press, 1972. P. 75.
Hadjiivanov K.I., Vayssilov G.N. / Characterization of oxide surfaces and zeolites by carbon monoxide as an IR probe molecule. In: Advances in Catalysis, Academic Press, 2002. P. 307.
Föttinger K., Emhofer W., Lennon D., Rupprechter G. // Topics in Catal. 2017. V. 60. P. 1722.
Föttinger K., Schlögl R., Rupprechter G. // Chem. Commun. 2008. P. 32.
Давыдов А.А. ИК-спектроскопия в химии поверхности окислов. Новосибирск: Наука, 1984. 245 с.
Coq B., Ferrat G., Figueras F. // J. Catal. 1986. V. 101. P. 434.
Panias D., Asimidis P., Paspaliaris I. // Hydrometallurgy. 2001. V. 59. P. 15.
Yuan G., Keane M.A. // J. Catal. 2004. V. 225. P. 510.
de Pedro Z.M., Diaz E., Mohedano A.F., Casas J.A., Rodriguez J.J. // Appl. Catal. B: Env. 2011. V. 103. P. 128.
Gómez-Sainero L.M., Seoane X.L., Fierro J.L.G., Arcoya A. // J. Catal. 2002. V. 209. P. 279.
Tarlani A., Abedini M., Khabaz M., Amini M.M. // J. Coll. Interface Sci. 2005. V. 292. P. 486
Pizzio L.R., Cáceres C.V., Blanco M.N. // Appl. Catal. A: General. 1998. V. 167. P. 283.
Liu L., Wang B., Du Y., Zhong Z., Borgna A. // Appl. Catal. B: Env. 2015. V. 174–175. P. 1
Hoang-Van C., Zegaoui O. // Appl. Catal. A: General. 1997. V. 164. P. 91.
Srinivas S.T., Jhansi Lakshmi L., Lingaiah N., Sai Prasad P.S., Kanta Rao P. // Appl. Catal. A: General. 1996. V. 135. P. 201.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ