

Кинетика и катализ, 2019, T. 60, № 3, стр. 340-345
Влияние диаметра пор цеолитов на величину энергии активации образования 4-алкил-1,3-диоксанов по реакции Принса
И. В. Вакулин 1, *, П. А. Пасько 1, **, Р. Ф. Талипов 1, Г. Р. Талипова 1, О. Ю. Купова 2
1 ФГБОУ ВО Башкирский государственный университет
450076 Уфа, ул. Заки Валиди, д. 32, Россия
2 ФГБОУ ВО Уфимский государственный нефтяной технический университет
450000 Уфа, ул. Первомайская, д. 14, Россия
* E-mail: vakuliniv@mail.ru
** E-mail: pasko.pav62@gmail.com
Поступила в редакцию 21.11.2018
После доработки 04.12.2018
Принята к публикации 24.12.2018
Аннотация
С использованием зависимости энергии активации образования 4-алкил-1,3-диоксанов от диаметра пор цеолитов теоретически обоснована каталитическая активность последних в реакции Принса. Методами молекулярной динамики изучена стабилизация предреакционного π-комплекса в полостях ряда цеолитов (${\text{Na}}_{x}^{ + }$(H2O)y)[AlaSibOc], AlaPbOc, Cax[H2O]yAlaSibOc). Показано, что зависимость энергии стабилизации π-комплекса и переходного состояния от диаметра полости имеет экстремальный характер. С учетом энергий стабилизации π-комплекса и переходного состояния в полости цеолита определено изменение энергии активации реакции образования 4-алкил-1,3-диоксанов в зависимости от размера пор. Проведено сравнение характера изменений энергии стабилизации переходного состояния и энергии активации от диаметра полости. Продемонстрировано, что зависимость энергии активации от диаметра дает более узкий интервал оптимальных размеров пор.
ВВЕДЕНИЕ
Сведения о применении цеолитов в качестве катализаторов реакции Принса достаточно широко представлены в литературе [1–15]. Было показано, что применение цеолитов в этих целях оправдано, а в ряде случаев исследователям удавалось существенно повысить селективность того или иного канала реакции, например образования 1,3-диоксанов [16–19]. При этом выбор цеолита осуществлялся исключительно эмпирически, без какого либо теоретического анализа. Очевидно, что такой подход не гарантирует применение наиболее эффективного цеолита в исследуемом процессе, поэтому использование теоретических методов для обоснования каталитической активности микропористых веществ в реакции Принса представляется важным и актуальным.
В наших предыдущих исследованиях была продемонстрирована возможность использования предложенной ранее в работах [20–22] концепции “transition state shape selectivity” (TSSS) и молекулярной динамики для теоретического обоснования каталитической активности микропористых веществ в реакции Принса [23–25]. Основная идея использованного нами алгоритма – применение “замороженной” структуры переходного состояния (TS) в качестве зонда. Количественной мерой каталитической активности пористого материала выступает расчетное значение энергии адсорбции зонда в полости [23]. Нами было показано, что зависимость этой энергии от диаметра полости имеет экстремальный характер [23–25]. Важно отметить, что значения диаметра полости, при которых наблюдается максимум энергии адсорбции, и размеры пор цеолитов и углеродных нанотрубок, для которых экспериментально определен наибольший каталитический эффект в реакции Принса, оказываются довольно близки [26].
Для пористых веществ различного строения предложенный нами алгоритм неплохо определяет диапазон диаметра пор, в котором должна проявляться каталитическая активность. Одним из допущений подхода TSSS является исключение из рассмотрения предреакционных комплексов (РC) и их энергии стабилизации в полостях. Очевидно, что в строгом виде влияние пористого вещества на энергию активации представляется следующим образом:
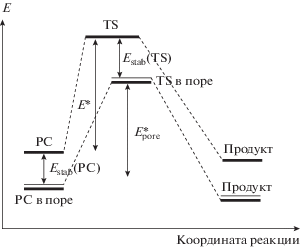
Схема 1 .
Влияние размера пор на изменение энергии активации при переходе от реакции в газовой фазе к реакции в полости должно описываться инкрементом Δstab, который некоторым образом будет связан с диаметром полости:
(1)
${{D}_{{{\text{stab}}}}} = {{E}_{{{\text{stab}}}}}({\text{TS}})--{{E}_{{{\text{stab}}}}}({\text{PC}}).$В зависимости от величин энергий стабилизации (адсорбции) TS и PC итоговая энергия активации ($E_{{{\text{pore}}}}^{*}$) может увеличиваться или уменьшаться, при этом минимумы зависимости $E_{{{\text{pore}}}}^{*}$ от диаметра полости, найденные по инкременту Δstab и при использовании подхода TSSS, могут различаться.
Целью настоящей работы было сравнение двух вариантов анализа результатов теоретического моделирования каталитической активности пористых веществ в реакции Принса (схема 2 ) – непосредственной корреляции диаметра поры с энергией стабилизации переходного состояния в полости и корреляции инкремента Δstab, вычисляемого по уравнению (1), с размером поры.
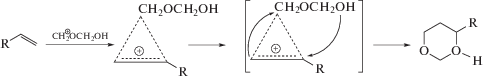
Схема 2 .
МЕТОДЫ РАСЧЕТА
Ранее нами было показано, что образование 1,3-диоксанов по реакции Принса преимущественно протекает с участием олигомеров формальдегида [27]. Первая стадия взаимодействия алкена с протонированным олигомером формальдегида – формирование π-комплекса, который в последующем трансформируется непосредственно в 1,3-диоксановую структуру без образования промежуточного σ-катиона [28, 29]. Таким образом, π-комплекс является предреакционным комплексом (PC) и именно его необходимо рассматривать при расчете инкремента Δstab.
Полуэмперические вычисления проводили с использованием приближения AM1 [30]. Неэмперические расчеты осуществляли с применением валентно-расщепленного базисного набора 6-31G [31] с включением d-поляризационной функции для всех тяжелых атомов. Электронную корреляцию учитывали по теории возмущений Меллера–Плесе второго порядка в варианте “замороженные остовные электроны” [32].
Предварительный поиск переходных состояний осуществлен с помощью процедуры SADDLE в программе MOPAC [33] в приближении AM1 с последующим уточнением геометрии в процедуре TS. Полученную геометрию переходных состояний оптимизировали в программе FireFly v7.1 [34] в приближении MP2(fc)/6-31G(d) [35] с пересчетом матрицы Гесса на каждом шаге.
Установление истинности переходного состояния осуществляли решением колебательной задачи и анализом вычисленных частот в ИК-спектре. Также истинность найденного переходного состояния подтверждалась с помощью процедуры IRC [36], которая проверяет соответствие переходного состояния реагентам и продуктам.
Для оценки энергии взаимодействия π-комплексов с полостями цеолитов были проанализированы следующие структуры π-комплексов, полученные в работе [29]:
| Реагирующий алкен | π-комплекс | R | TS1,3D |
| Этен |  |
R1 = R2 = R3 = H (I) R1 = CH3, R2 = R3 = H (II) R1 = C2H5, R2 = R3 = H (III) R1 = R2 = CH3, R3 = H (IV) R1 = R3 = CH3, R2 = H (V) |
TS1,3D (I) этен |
| Пропен | TS1,3D (II) пропен | ||
| Бутен-1 | TS1,3D (III) бутен-1 | ||
| Изобутен | TS1,3D (IV) изобутен | ||
| транс-2-Бутен | TS1,3D (V) бутен-2 |
Энергии взаимодействия переходных состояний с полостями цеолитов взяты из работы [24]. Были рассмотрены следующие цеолиты с наноразмерными порами:
| Структурный тип | Цеолиты |
| ANA, NAT, TON, MTW, LOS, EUOMEL, MEI, KFI | ${\text{Na}}_{x}^{ + }$ (H2O)y)[AlaSibOc] |
| APC, AFN, AFO, ATO, AEI, AFT, AET | AlaPbOc |
| PAR, GIS, EPI, LAU, LEV, CHA | Cax[H2O]yAlaSibOc |
За энергию стабилизации переходных состояний и предреакционного комплекса в полостях цеолитов принимали энергию адсорбции, которую рассчитывали в программе Accelrys Material Studio 5.5 с применением модуля Adsorption Locator. [37–39]. В качестве адсорбатов использовали “замороженные” структуры переходных состояний и предреакционного комплекса, вычисленные на предыдущем этапе. Программа Material Studio обладает широкими возможностями для моделирования микропористых и мезопористых веществ на основе библиотеки элементарных ячеек. Для получения фрагмента цеолита, рассматриваемого в качестве адсорбента, соответствующую элементарную ячейку с применением встроенных программных средств путем 3-кратной трансляции по осям X, Y и Z превращали в наноразмерный фрагмент, содержащий порядка 9 каналов. При этом структура самого фрагмента в последующем моделировании полагалась неизменной.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Особенности стабилизации π-комплексов в полостях цеолитов
На рис. 1а–1в приведены зависимости энергий стабилизации π-комплексов в полостях рассматриваемых цеолитов.
Рис. 1.
Зависимость энергии стабилизации π-комплексов от диаметра полости цеолитов групп ${\text{Na}}_{x}^{ + }$(H2O)y)[AlaSibOc] (а), AlaPbOc (б) и Cax[H2O]yAlaSibOc (в).
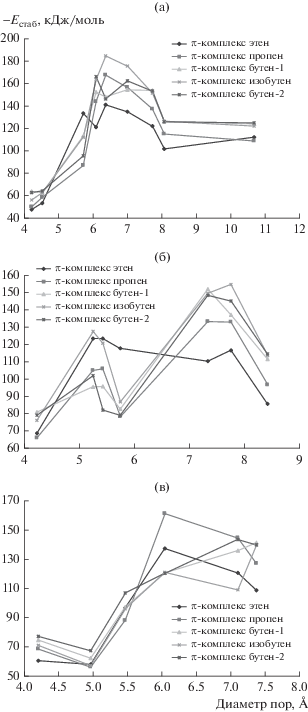
Все представленные зависимости имеют экстремальный характер. В целом, интервал диаметров полостей, при котором в координатах “–Естаб/диаметр пор” наблюдаются наибольшие значения энергии стабилизации, составляет 5.2–7.8 Å (рис. 1а–1в). Однако для каждой группы цеолитов точные значения указанного диапазона и максимальные значение энергии стабилизации несколько отличаются.
Так, для цеолитов группы ${\text{Na}}_{x}^{ + }$(H2O)y)[AlaSibOc] этот интервал составляет 6.08–7.00 Å, для цеолитов группы AlaPbOc – 5.25–7.75 Å, а для цеолитов группы Cax[H2O]yAlaSibOc – 6.04–7.37 Å. Очевидно, это связано с различием элементного состава и формы каналов цеолитов рассмотренных групп. Важно, что представленные зависимости также отражают влияние размера участвующего в реакции алкена на Estab. Так, в случае π-комплексов, образованных из этилена и пропилена, максимум энергии стабилизации наблюдается при меньших значения диаметров полостей из оптимального диапазона.
Такой характер зависимости энергии стабилизации π-комплексов от диаметра полости цеолитов совпадает с полученными ранее аналогичными зависимостями для переходных состояний [24]. С учетом экзотермического характера превращения π-комплекса в 1,3-диоксановую структуру отмеченное совпадение хорошо согласуется с постулатом Крама–Хэммонда.
Оценка влияния диаметра полости цеолитов на энергию активации по величине вклада Δstab
Вклады Δstab в энергию активации реакции образования 1,3-диоксанов в полостях цеолитов изученных групп представлены на рис. 2а–2в.
Рис. 2.
Влияние диаметра полости цеолитов групп ${\text{Na}}_{x}^{ + }$(H2O)y)[AlaSibOc] (а), AlaPbOc (б) и Cax[H2O]yAlaSibOc (в) на Δstab и на энергию стабилизации переходного состояния.
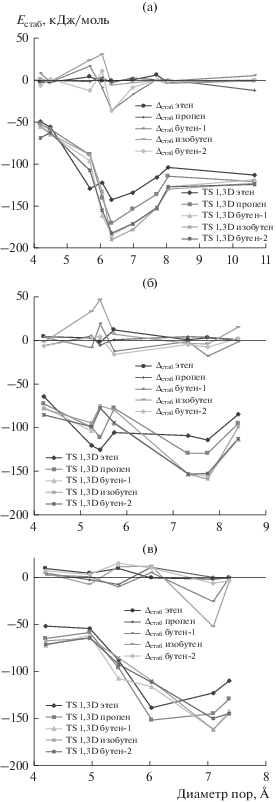
Во всех рассмотренных случаях инкремент Δstab не имеет ярко выраженной зависимости от диаметра пор. Диаметры пор, при которых инкремент обладает положительным значением, соответствуют ингибирующему влиянию цеолита, т.к. цеолиты с таким размером полостей способны стабилизировать π-комплексы лучше, чем переходное состояние. С другой стороны, отрицательные значения Δstab обуславливают уменьшение энергии активации. При этом по абсолютной величине инкремент Δstab ожидаемо оказывается меньше, чем энергия стабилизации TS в полости цеолитов.
Сравнение зависимостей энергии стабилизации TS и Δstab от диаметра пор (рис. 2а–2в) показывает, что интервалы оптимальных значений диаметров (при которых должно наблюдаться наибольше снижение энергии активации образования алкил-1,3-диоксанов по реакции Принса) в существенной степени перекрываются. В зависимости от элементного состава цеолита оценка эффективного диаметра с использованием инкремента Δstab предсказывает тот же оптимальный диапазон или интервал в области меньших значений диаметра по сравнению с оценкой на основе энергии стабилизации TS. При этом в отличие от подхода TSSS, инкремент Δstab оказывается более чувствителен к строению алкена, участвующего в реакции.
ЗАКЛЮЧЕНИЕ
Таким образом, обоснование каталитической активности цеолитов в реакции Принса и определение оптимальных диаметров пор, при которых должно наблюдаться наибольшее снижение энергии активации образования 4-алкил-1,3-диоксанов, может быть осуществлено с использованием молекулярной динамики.
Показано, что при определении диапазона оптимальных диаметров пор цеолитов использование подхода “transition state shape selectivity”, оперирующего только энергией стабилизации переходного состояния в полости без учета энергии стабилизации предреакционного комплекса, дает зависимость энергии стабилизации от диаметра пор цеолитов с явно выраженным экстремальным характером.
При этом область значений диаметров, близких к оптимальному, достаточно велика и составляет 1–2 Å в зависимости от структурного типа цеолитов.
Применение инкремента Δstab, который учитывает как энергию стабилизации переходного состояния в полости, так и стабилизацию предреакционного комплекса (уравнение (1)), позволяет точнее определить область оптимальных диаметров по сравнению с подходом “transition state shape selectivity”. В этом случае оценка оптимальных диаметров полостей цеолитов дает интервал 0.4 Å.
Список литературы
Venuto P.B. // Micropor. Mater. 1994. V. 2. № 5. P. 297.
Okachi T., Onaka M. // J. Americ. Chem. Soc. 2004. V. 126. P. 2306.
Camblor M., Corma A., Iborra S., Miquel S., Primo J., Valencia S. // J. Catal. 1997. V. 172. P. 76.
Aramendía M.A., Borau V., Jiménez C., Marinas J.M., Romero F.J., Urbano F.J. // Catal. Lett. 2001. V. 73. P.203.
Ratnasamy P., Kumar R. // Catal. Today 1991. V. 9. P. 329.
Venuto P.V., Landis P.S. // Adv. Catal. 1968. V. 18. P. 259.
Fu H., Xie S., Fu A., Ye T. // Computat. Theoret. Chem. 2012. V. 982. P. 51.
Sangthong W., Probst M., Limtrakul J. // J. Mol. Struct. 2005. V. 748. P. 119.
Kim H.J., Seo G., Kim J.-N., Choi K.H. // Bull. Korean Chem. Soc. 2004. V. 25. P. 1726.
Selvaraj M., Kawi S. // J. Mol. Catal. A: Chem. 2006. V. 246. P. 218.
Wang J., Jaenicke S., Chuah G.K., Hua W., Yue Y., Gao Z. // Catal. Commun. 2011. V. 12. P. 1131.
Telalović S., Ng J.F., Maheswari R., Ramanathan A., Chuah G. K., Hanefeld U. // Chem. Commun. 2008. V. 7345. P. 4631.
Tateiwa J., Hashimoto K., Yamauchi T., Uemura S. // Bull. Chem. Soc. Jpn. 1996. V. 69. P. 2361.
Reddy S.S., Raju D.B., Kumar S.V., Padmasri A.H., Narayanan S., Rao R.K.S. // Catal. Commun. 2007. V. 8. P. 261.
Yadav J. S., Reddy B. V. S., Kumar G.M. // Tetrahedr. Lett. 2001. V. 42. P. 89.
Dimitriu E., Gongescu D., Hulea V. // Stud. Surf. Sci. Catal. 1993. V. 78. P. 669.
Dimitriu E., Trong O.D., Kaliaguine S. // J. Catal. 1997. V. 170. P. 150.
Dimitriu E., Hulea V., Chelaru C., Hulea T., Kaliaguine S. // Zeolites and Related Microporous Materials: State of the Art. 1994. V. 84. P. 1997.
Dimitriu E., Hulea V., Fechete I., Catrinescu C., Auroux A., Lacaze J. F., Guimon C.// Appl. Catal. A: General. 1999. V. 181. P. 15.
Smit D., Maesen T.L.M. // NATURE. 2008. V. 451 P. 671
Henriques C. Catalysis by zeolites. EUCHEME, 2012.
Csicsery S.M. // Zeolites. 1984. V. 4. P. 202
Vakulin I.V., Talipov R.F., Pasko P.A., Talipova G.R., Kupova O.Yu // Micropor. Mesopor. Mater. 2018. V. 270. P. 30.
Пасько П.А., Вакулин И.В., Талипов Р.Ф. // Вестник Башкирского университета. 2017. Т. 22. № 4. С. 966.
Пасько П.А., Вакулин И.В., Талипов Р.Ф. // Бутлеровские сообщения. 2017. Т. 52. №. 10. С. 45.
Vakulin I.V., Talipov R.F., Tukhvatshin V.S., Talipova G.R., Pasko P.A. // 5th international School-Conference on catalysis for young scientists. Catalytic design. 2018. P. 259.
Kupova O.Y., Vakulin I.V., Talipov R.F., Talipova G.R., Morozkin N.D. // Reaction kinetics, mechanisms and catalysis. 2013. V. 110. № 1. P. 41.
Вакулин И.В., Купова О.Ю., Талипов Р.Ф. // Вестник Башкирского университета. 2011. Т. 16. № 3. С. 694.
Kupova O.Y., Vakulin I.V., Talipov R.F. // Computational and Theoretical Chemistry. 2013. V. 1013. P. 57.
Dewar M., Thiel W. // J. Amer. Chem. Soc. 1977. V. 99. P. 4499.
Ditchfield R., Hehre W.J., Pople J.A. // J. Chem. Phys. 1971. V. 54. P. 724.
Kestutis A., Mikkelsen K.V., Stephan P.A. // J. Chem. Theory Comput. 2008. V. 4. P. 267.
Stewart J.J.P. // Quant. Chem. Prog. Exch. 1985. V. 5. P. 62.
Granovsky A.A. // http://classic. chem. msu. su/gran/gamess/index. html
Su K.H., Wang Y.B., Wen Z.Y. // Acta Phys. Chim. Sin. 1998. V. 14. P. 856.
Gonzales C., Schlegel H.B. // J. Chem. Phys. 1989. V. 90. P. 2154.
Delley B. // J. Chem. Phys. 1990. V. 92. P. 508.
Delley B. // J. Chem. Phys. 2000. V. 113. P. 7756.
Materials Studio Version 6.0., 2011. Accelrys Inc., San Diego.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ