

Кинетика и катализ, 2019, T. 60, № 4, стр. 532-543
Исследование закономерностей формирования и свойств Ni–Ce–La–O-катализаторов риформинга
Е. В. Матус a, b, *, Д. В. Нефедова b, c, О. Б. Сухова a, И. З. Исмагилов a, В. А. Ушаков a, С. А. Яшник a, А. П. Никитин c, М. А. Керженцев a, З. Р. Исмагилов a, c
a ФГБУН “Федеральный исследовательский центр “Институт катализа им. Г.К. Борескова СО РАН”
630090 Новосибирск, просп. акад. Лаврентьева, 5, Россия
b ФГБОУ ВО Новосибирский государственный технический университет
630073 Новосибирск, просп. К. Маркса, 20, Россия
c Институт углехимии и химического материаловедения ФИЦ УУХ СО РАН
650000 Кемерово, просп. Советский, 18, Россия
* E-mail: matus@catalysis.ru
Поступила в редакцию 20.12.2018
После доработки 13.03.2019
Принята к публикации 18.03.2019
Аннотация
C целью стабилизации в матрице носителя высокодисперсных форм Ni активного компонента, отличающихся термостабильностью и высокой устойчивостью к зауглероживанию в условиях реакций риформинга углеводородного сырья, проведен синтез Ni–Ce–La–O-катализаторов при варьировании состава (La/Ce = 0, 0.25, 1, 4), способа приготовления (метод сложноэфирных полимерных предшественников и пропитка по влагоемкости) и температуры прокаливания (300–900°С) образцов. Изучены закономерности формирования материалов и выполнено сравнительное исследование их физико-химических свойств комплексом методов (низкотемпературная адсорбция азота, рентгенофазовый анализ, спектроскопия комбинационного рассеяния, просвечивающая электронная микроскопия и термопрограммируемое восстановление водородом). Показано, что при La : Ce = 0–4 материалы после прокаливания при 300–500°С представляют собой твердые растворы на основе диоксида церия (Ni–Ce–La–O, Ce–La–O) с нанесенными частицами оксида никеля. Использование метода сложноэфирных полимерных предшественников по сравнению с пропиткой обеспечивает более высокую удельную поверхность и дефектность структуры материала, меньший средний размер кристаллитов твердого раствора и стабилизацию никеля преимущественно в составе твердого раствора на основе диоксида церия. После высокотемпературного прокаливания при 700–900°С различия как в текстурных, так и структурных характеристиках катализаторов, полученных различными способами, становятся менее выраженными, что связано с частичным разрушением твердого раствора Ni–Ce–La–O. Установлено, что увеличение дисперсности и термостабильности Ni-содержащей фазы, а также снижение степени зауглероживания Ni–Ce–La–O-катализаторов в реакции автотермического риформинга этанола достигается при увеличении мольного соотношения La : Ce и использовании для синтеза метода сложноэфирных полимерных предшественников.
ВВЕДЕНИЕ
Никельсодержащие катализаторы являются эффективными каталитическими системами для процессов риформига углеводородного сырья [1–5]. К их ключевым преимуществам относят низкую стоимость и высокую производительность, сравнимую с производительностью катализаторов на основе благородных металлов [5–7]. Однако в условиях каталитического процесса под воздействием реакционной среды наблюдаются спекание, фазовые превращения и зауглероживание активного компонента, что приводит к постепенной дезактивации Ni-катализаторов [8–10]. Преодолеть вышеуказанные недостатки и достигнуть улучшения показателей процесса возможно путем стабилизации высокодисперсных частиц никеля в матрице носителя. Для материалов с наночастицами активного металла по сравнению с грубодисперсными системами отмечается увеличение удельной поверхности активного компонента, формирование развитой межфазной металл-оксидной поверхности, повышение активности, а также снижение скорости образования углеродистых отложений [11–16]. Установлено, что резкое уменьшение степени зауглероживания наблюдается для катализаторов со средним размером Ni металлических частиц менее 3–5 нм [13–15].
Разработаны различные подходы для создания в матрице носителя нанодисперсных форм активного компонента, отличающихся высокой термостабильностью и устойчивостью к зауглероживанию: введение структурного промотора [15, 17]; оптимизация состава предшественника металла [18, 19] или условий активации катализаторов [20, 21]; модифицирование состава носителя [22, 23] и усовершенствование метода приготовления [24, 25] с целью реализации сильного взаимодействия металл–носитель. Показана предпочтительность использования нитрата и ацетата никеля вместо хлорида и формиата никеля [18] и эффективность применения металлсодержащих олигосилсесквиоксанов [19] в качестве соединений-предшественников при синтезе нанесенных катализаторов.
Сложные Ni-содержащие оксиды со структурой перовскита LaNi1 – xMexO3 [26–29], флюорита Ce1–xNixOy [30–32], шпинели (Ni0.75Mg0.25)Al2O4 [33] или слоистые двойные гидроксиды [Mg1 – x – yNixAly(OH)2](CO3)y/2 ⋅ mH2O [34, 35] являются перспективными предшественниками Ni-катализаторов. В этом случае катионы активного компонента встроены в структуру оксида/гидроксида. Активация таких материалов в восстановительной или реакционной среде приводит к разрушению исходной совместной фазы и позволяет получить стабилизированные на оксидной поверхности носителя Ni-содержащие наночастицы и кластеры. Состав оксидной матрицы (La2O3, CeO2, MgO–Al2O3) оказывает влияние как на свойства активного компонента, так и на эффективность участия носителя в каталитическом процессе. Оксиды, отличающиеся высокой кислородной емкостью и подвижностью кислорода кристаллической решетки, имеют преимущество в реакциях риформинга благодаря высокой способности предотвращать образование углеродистых отложений и ускорять их окисление в условиях реакции. В этой связи представляют интерес Се-содержащие системы, характеризующиеся достаточным количеством центров активации H2O/О2 и являющиеся источником активного кислорода, который участвует в окислении углерода на границе раздела металл–носитель [36, 37]. Отмечена положительная роль La в составе носителей, связанная со стабилизацией Ni в высокодисперсном состоянии и улучшением стабильности работы катализатора в реакциях риформинга метана и этанола [3, 27]. Как и носители на основе диоксида церия, La-содержащие материалы способны участвовать в окислении углерода (C) на поверхности активного компонента (Ni) по реакции La2O2CO3 + C–Ni → La2O3 + 2CO + + Ni [30].
Для приготовления сложных Ni-содержащих оксидов применяются различные способы синтеза: термическое разложение, соосаждение, цитратный золь–гель-метод, метод сложноэфирных полимерных предшественников (метод Пекини), сжигание нитратов металлов в присутствии восстановителей или метод обратной микроэмульсии [26–35, 38–40]. Высокая гомогенность распределения металлов в оксидной матрице достигается при использовании золь–гель-методов, когда смешение катионов металлов как активного компонента, так и носителя происходит уже на молекулярном уровне – в исходном растворе, который затем превращается в гель и далее осуществляется фиксация всех катионов в полимерной матрице.
Таким образом, Се–La-оксидная система является уникальной матрицей для Ni-наночастиц, сочетающей в себе как возможность стабилизации высокодисперсных структур активного компонента, так и способность к генерации и транспорту активных форм кислорода для окисления С-содержащих промежуточных соединений, образующихся на Ni-центрах. В настоящей работе с целью разработки устойчивого к дезактивации Ni-катализатора риформинга проведено сравнительное исследование закономерностей формирования Ni–Ce–La–О-катализаторов в зависимости от способа их синтеза (метод сложноэфирных полимерных предшественников, пропитка по влагоемкости). Исследовано влияние мольного соотношения La : Ce и температуры прокаливания на физико-химические свойства и функциональные характеристики материалов в реакции автотермического риформинга этанола (АТР С2H5OH).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методика синтеза катализаторов
Ni–Ce–La–O-катализаторы синтезировали методами сложноэфирных полимерных предшественников (P-серия) и пропитки по влагоемкости (I-серия). Содержание никеля было фиксировано – 10 мас. %, мольное соотношение La : Ce варьировали от 0 до 4, при этом массовая доля лантана в составе материала составляла 0, 15, 35, 60 и 75%.
Для получения образцов методом сложноэфирных полимерных предшественников лимонную кислоту (ЛК) растворяли в этиленгликоле (ЭГ) при температуре 70°С. К приготовленному раствору последовательно добавляли водный раствор с необходимой концентрацией нитрата церия, нитрата лантана и нитрата никеля (Сe + La + + Ni), этилендиамин (ЭД). При этом концентрация нитрата никеля в водном растворе была постоянной и составляла 33 мгNi/мл, а нитратов церия и лантана зависела от состава материала и варьировалась в диапазоне от 0 до 238 мгCe/мл и от 0 до 248 мгLa/мл соответственно. Мольное соотношение (Сe + La + Ni) : ЛK : ЭГ : ЭД было фиксировано и составляло 1 : 4 : 12 : 4. Образовавшийся гель перемешивали и затем сушили под ИК-лампой в течение 72 ч для удаления избытка растворителя. Полученное смолообразное вещество прокаливали в муфельной печи в режиме ступенчатого подъема температуры со скоростью 2 град/мин от 50 до 250°C с выдерживанием в течение 30 мин при фиксированной температуре каждые 50°С. Далее, если не оговорено особо, термообработку образовавшегося пеплообразного вещества осуществляли при 500°С в течение 4 ч. Для обозначения образцов P-серии использовали формулу ((Ce1 – xLax)0.8Ni0.2Oy, x = 0, 0.2, 0.5, 0.8, 1), где коэффициенты обозначают мольную долю (округленную до десятых единиц) данного элемента в составе материала.
Для приготовления образцов катализаторов методом пропитки по влагоемкости Се1 – xLaxОy-носители (x = 0, 0.2, 0.5, 0.8, 1) пропитывали водным раствором нитрата никеля с заданной концентрацией (57 мгNi/мл), образцы сушили под ИК-лампой и прокаливали в муфельной печи, если не оговорено особо, при температуре 500°С в течение 4 ч. Отметим, что синтез Се1 – xLaxОy-носителей осуществляли методом сложноэфирных полимерных предшественников по методике, аналогичной описанной выше для получения образцов (Ce1 – xLax)0.8Ni0.2Oy. Основные характеристики Се1 – xLaxОy-носителей представлены в [41]. Для обозначения образцов I-серии использовали формулу Ni/Ce1 – xLaxOy, где коэффициенты показывают мольную долю соответствующего элемента в составе носителя.
В отдельных, особо оговоренных случаях термообработку катализаторов проводили при 300, 700 или 900°С в течение 4 ч. Перед началом реакции катализаторы активировали в потоке 30 об. % H2/Не при 600°С в течение часа.
Физико-химические методы исследования катализаторов
Содержание металлов в исследуемых катализаторах определяли рентгеноспектральным флуоресцентным методом на анализаторе ARL ADVANT’X с Rh-анодом рентгеновской трубки (“ThermoTechno Scientific”, Швейцария). Химический анализ образцов выполняли после их прокаливания при температуре 900°С в течение 4 ч.
Текстурные характеристики катализаторов (удельная поверхность (SБЭТ), объем пор (Vпор) и средний диаметр пор (Dпор)) исследовали на автоматизированной волюмометрической установке ASAP 2400 (“Micromeritics”, США) путем измерения и обработки изотерм низкотемпературной адсорбции азота при 77 K.
Рентгенофазовый анализ (РФА) образцов проводили на дифрактометре HZG-4С (“Freiberger Prazisionmechanik”, Германия) в монохроматизированном CоKα-излучении (λ = 1.79021 Å). Фазовый состав определяли по дифракционным картинам, полученным путем сканирования области углов 2θ = 10°–80° с шагом 0.1 град и временем накопления 6–15 с. Эксперименты по исследованию фазового состава катализаторов после активации осуществляли ex situ. Размеры области когерентного рассеяния (ОКР) твердого раствора на основе диоксида церия рассчитывали из уширения дифракционного пика 1.1.1. фиксируемых фаз, имеющих кубическую структуру типа флюорита; OKР NiO – пика 2.0.0. фазы NiO; ОКР Ni – пика 2.0.0. фазы Ni0. Термостабильность Ni-содержащей фазы определяли по характеру влияния условий термообработки на размер ОКР.
Регистрацию спектров комбинационного рассеяния (CКР) проводили в спектральном диапазоне частотного смещения 120–900 см–1 на КР-спектрометре Renishaw Invia (“Renishaw plc.”, Великобритания) с использованием в качестве источника возбуждения аргонового лазера с длиной волны 514.5 нм, дифракционной решетки 1800 ш/мм и объектива L50x. Мощность лазерного излучения, попадающая на образец, не превышала 2 мВт, а время накопления полезного сигнала составляло 120 с.
Снимки просвечивающей электронной микроскопии высокого разрешения (ПЭМВР) получали на электронном микроскопе JEM–2010 (“JEOL”, Япония) с разрешением по решетке 0.14 нм при ускоряющем напряжении 200 кВ. Локальный элементный анализ образцов проводили методом спектрометрии характеристического рентгеновского излучения с дисперсией по энергиям (EDX) на спектрометре EDAX (“EDAX Inc.”, США), оснащенном Si(Li)-детектором с энергетическим разрешением не менее 130 эВ. Образцы для ПЭМВР наносили на перфорированные углеродные подложки, закрепленные на медных сетках.
Термопрограммируемое восстановление водородом (ТПВ-H2) осуществляли в проточном реакторе (внутренний диаметр 5 мм) по методике, описанной в [42].
Термический анализ (ТА) (дифференциальный термический анализ (ДТА), термогравиметрический анализ (ТГА) и дифференциальный термогравиметрический анализ (ДТГ)) образцов выполняли на термоанализаторе STA 449 C (”NETZSCH-Geratebau GmbH”, Германия) в интервале температур 25–900°С со скоростью подъема температуры 10°С/мин в атмосфере воздуха.
Степень зауглероживания катализаторов определяли по количеству углеродистых отложений, образующихся на поверхности катализатора в ходе реакции АТР С2Н5OH. Для этого образцы катализаторов после реакции исследовали методом термического анализа. Потеря веса образца в области температур 400–900°С, обусловленная окислением углеродсодержащих компонентов образца, соответствовала количеству углеродистых отложений.
Методика исследования активности катализаторов
Исследование активности катализаторов в реакции АТР С2Н5OH проводили в проточном кварцевом реакторе (внутренний диаметр 11 мм) по методике, описанной в [43], при атмосферном давлении, температуре 200–600°C, скорости газового потока 320 млN/мин и мольном соотношении реагентов C2H5OH : H2O : O2 : He = 1 : 3 : 0.4 : 0.7. Активность катализаторов в реакции АТР С2Н5OH характеризовали общей конверсией этанола, выходом водорода (${{Y}_{{{{{\mathbf{H}}}_{{\mathbf{2}}}}}}}$), селективностью образования C-продуктов реакции (S). Выход водорода рассчитывали по формуле:
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Физико-химические свойства
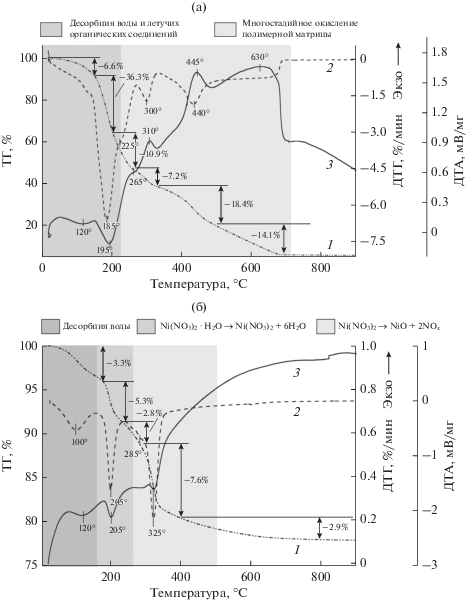
Ni–Ce–La–O-катализаторы одинакового химического состава (10 мас. % Ni; La : Ce = 0, 0.25, 1, 4) синтезировали двумя способами, обеспечивающими различную степень взаимодействия металл–носитель: методом сложноэфирных полимерных предшественников (P-серия, (Ce1 – xLax)0.8Ni0.2Oy) и методом пропитки по влагоемкости (I-серия, Ni/Ce1 – xLaxOy). Термический анализ исходного геля образцов P-cерии показал, что процесс деградации органической матрицы является многостадийным. Как видно из рис. 1a, потеря веса образца за счет окисления цитратных комплексов металлов и фрагментов полимерного каркаса сопровождается экзотермическими эффектами с максимумами при T = = 210, 445 и 690°С. Следует отметить, что параметры термического генезиса (т.е. положение пиков на ДТГ- и ДТА-кривых) слабо зависят от мольного соотношения катионов La : Ce в образце, что, вероятно, обусловлено существенным избытком органической составляющей геля. Отметим, что исследование прокаленных при 500°С образцов P-серии методом ТА показало отсутствие остатков органических веществ. Для высушенных катализаторов I-серии разложение нанесенного гексагидрата нитрата никеля происходит в две стадии и сопровождается эндоэффектами при T = 205 и 325°С (x = 0.5) (рис. 1б). В этом случае наблюдается зависимость параметров термического генезиса от состава носителя: с увеличением мольной доли La в составе материала (0 → 0.5 → 1) температура разложения соли с образованием оксида никеля увеличивается (305→ 325 →365°С).
Рис. 1.
Термический анализ предшественников катализаторов P-серии (а) и I-серии (б), (La : Ce = 1): 1 – ТГ, 2 – ДТГ, 3 – ДТА.
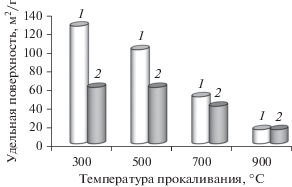
По данным рентгеноспектрального флуоресцентного анализа химический состав катализаторов соответствует расчетному. Текстурные и структурные свойства полученных образцов представлены в табл. 1. Из результатов низкотемпературной адсорбции азота следует, что синтезированные образцы являются мезопористыми материалами. Наблюдается IV тип изотермы адсорбции с петлей гистерезиса типа H3. Гистерезис при парциальном давлении Р/Р0 = 0.7–0.9 указывает на присутствие преимущественно текстурной мезопористости [44]. Видно (табл. 1, рис. 2), что независимо от состава катализаторов удельная поверхность выше для образцов, полученных методом сложноэфирных полимерных предшественников. Этот эффект более выражен для образцов 1) с низким содержанием лантана (до 35 мас. %) и 2) прокаленных при температуре 300–500°С. В частности, в случае пары (Ce0.8La0.2)0.8Ni0.2O1.7 и Ni/Ce0.8La0.2O1.9, прокаленных при 300 и 700°С, удельная поверхность составляет 125 vs. 60 м2/г и 50 vs. 40 м2/г, а для пары (Ce0.5La0.5)0.8Ni0.2O1.6 и Ni/Ce0.5La0.5O1.75 – 60 vs. 55 м2/г и 40 vs. 30 м2/г соответственно. Для обеих серий катализаторов SБЭТ снижается с увеличением содержания лантана (табл. 1) и ростом температуры прокаливания (рис. 2).
Таблица 1.
Физико-химические свойства образцов1
| Образец | Текстурные характеристики | Данные РФА | Данные CКР |
Результаты ТПВ-H2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| после прокаливания | после активации2 | |||||||||||
| SБЭТ, м2/г |
Vпор, см3/г |
Dпор, нм |
фазовый состав |
размер ОКР (параметр ячейки) CeO2, нм |
размер ОКР NiO, нм |
фазовый состав |
размер ОКР (параметр ячейки) CeO2, нм |
размер ОКР Ni, нм |
I570/I465 | ν(H2)3, ммольH2/гкат | H2/Ni4 | |
| Катализаторы, полученные методом сложноэфирных полимерных предшественников | ||||||||||||
| Ce0.8Ni0.2O1.8 | 105 | 0.23 | 8.8 | CeO2* | 5.5 (0.541) | – | CeO2*, Ni | 18.0 (0.541) | 7.8 | 1.0 | 2.4 | 1.4 |
| (Ce0.8La0.2)0.8Ni0.2O1.7 | 100 | 0.31 | 12.3 | CeO2* | 6.0 (0.547) | – | CeO2* | 14.0 (0.549) | – | 1.4 | 2.3 | 1.4 |
| (Ce0.5La0.5)0.8Ni0.2O1.6 | 50 | 0.18 | 15.6 | CeO2* | 4.0 (0.555) | – | CeO2* | 11.0 (0.559) | – | – | 4.7 | 2.8 |
| (Ce0.2La0.8)0.8Ni0.2O1.4 | 35 | 0.15 | 18.1 | CeO2* | 4.0 (0.566) | – | CeO2* La(OH)3 |
(0.586)** | – | – | 3.5 | 2.1 |
| La0.8Ni0.2O1.4 | 25 | 0.10 | 17.1 | La2O2CO3 | – | – | La2O3, La(OH)3 |
– | – | – | 5.6 | 3.3 |
| Катализаторы, полученные методом пропитки по влагоемкости | ||||||||||||
| Ni/CeO2 | 75 | 0.19 | 10.1 | CeO2, NiO | 11.0 (0.541) | 25.0 | CeO2, Ni | 25.0 (0.541) | 25.0 | 0.1 | 2.9 | 1.7 |
| Ni/Ce0.8La0.2O1.9 | 60 | 0.18 | 11.4 | CeO2*, NiO | 7.7 (0.547) | 11.0 | CeO2*, Ni | 15.0 (0.549) | 30.0 | 0.1 | 2.3 | 1.4 |
| Ni/Ce0.5La0.5O1.75 | 40 | 0.11 | 11.7 | CeO2* | 5.0 (0.556) | – | CeO2* | 11.0 (0.558) | – | 1.2 | 2.9 | 1.7 |
| Ni/Ce0.2La0.8O1.6 | 15 | 0.06 | 17.9 | CeO2* | 3.5 (0.562) | – | CeO2* La(OH)3 |
(0.583)** | – | – | 3.2 | 1.9 |
| Ni/La2O3 | 10 | 0.05 | 19.5 | La2O2CO3 | – | – | La2O3, La(OH)3 |
– | – | – | 4.1 | 2.4 |
1 Если не оговорено особо, образцы после прокаливания при 500°С на воздухе. 2Образцы после восстановления при 600°С в 30% H2/Не. 3Общее количество водорода, поглощенного за эксперимент ТПВ-Н2. 4Мольное соотношение общего количества поглощенного водорода к количеству никеля. * Твердый раствор на базе структуры диоксида церия. ** Из-за наложения линий второй фазы определить размер ОКР не представляется возможным. Примечание: прочерки в таблице означают отсутствие соответствующего параметра для образца.
Рис. 2.
Влияние температуры прокаливания на удельную поверхность катализаторов P-серии (1) и I-серии (2). (La : Ce = 0.25).
На основании данных рентгенофазового анализа катализаторов установлено, что после прокаливания при 300–500°С для P-серии образцов (Ce1 – xLax)0.8Ni0.2Oy (x ≤ 0.8) дифракционная картина и значения параметра решетки (табл. 1) соответствуют флюоритоподобному твердому раствору кубического типа на основе диоксида церия. Отсутствие на рентгенограммах дифракционных максимумов, относящихся к Ni-содержащей фазе, может свидетельствовать о ее высокодисперсном состоянии или встраивании катионов никеля в структуру твердого раствора [30, 45]. Напротив, для I-серии катализаторов Ni/Ce1 – xLaxOy (x ≤ 0.8) на фоне дифракционной картины носителя, представляющего собой твердый раствор Сe–La–O [46], зафиксированы пики, обусловленные наличием оксида никеля в составе образца. Как было показано ранее [46], c увеличением мольной доли La в составе Ce1 – xLaxOy-носителя повышается степень взаимодействия между нанесенным Ni и компонентами носителя, что проявляется в возрастании дисперсности Ni активного компонента: при x = 0–0.2 фаза NiO имеет ОКР 11–25 нм, а при x = 0.5–0.8 наблюдается высокодисперсная фаза NiO (ОКР менее 5 нм). Следует отметить, что средний размер кристаллитов твердого раствора на основе диоксида церия также уменьшается с увеличением содержания La в составе материала, что характерно для формирования твердых растворов Се–M–O (M = Ni, La, Mg, Gd) [3, 31]. ОКР СеО2 в немодифицированном и модифицированном La образцах ниже для образцов P-серии, чем для I-серии (11 → 3.5 нм vs. 5.5 → 4 нм) (табл. 1).
С повышением температуры прокаливания обнаружены изменения структурных параметров образцов, характер которых контролируется методом приготовления и составом материала (табл. 1, 2). Для образцов P-серии происходит ухудшение дисперсности фаз, а также изменение фазового состава: при высоких температурах прокаливания (700–900°С) в дополнении к фазе твердого раствора на основе диоксида церия наблюдается Ni-содержащая фаза, представляющая собой либо фазу оксида никеля NiO (x = 0–0.5), либо фазу перовскита La2NiO4 (x = 0.8). Для катализаторов I-серии с x = 0–0.5 фазовый состав сохраняется, но изменяется дисперсность фаз. Происходит увеличение размера кристаллитов, как носителя, так и активного компонента. А для образцов с x = 0.8 помимо фазы твердого раствора на основе диоксида церия появляется фаза La–Ni–O-перовскита. Фазовый состав Ni–La–O-катализаторов (образцы без Сe) не зависит от метода приготовления и включает только фазу оксикарбоната лантана (прокаливание при 300–500°С, табл. 1) или смесь фаз La2O3 + La2NiO4 (прокаливание при 700–900°С).
Таблица 2.
Влияние температуры прокаливания на фазовый состав катализаторов*
| Образец | Фазовый состав образцов, прокаленных при температуре | ||
|---|---|---|---|
| 300°С | 700°С | 900°С | |
| Катализаторы, полученные методом сложноэфирных полимерных предшественников | |||
| Ce0.8Ni0.2O1.8 | CeO2** (5.5; 0.541) | CeO2**(13.0; 0.541) | CeO2** (50.0; 0.541) NiO (50.0) |
| (Ce0.8La0.2)0.8Ni0.2O1.7 | CeO2** (5.0; 0.547) | CeO2** (9.5; 0.547) NiO (8.0) |
CeO2** (50.0; 0.548) NiO (50.0) |
| (Ce0.5La0.5)0.8Ni0.2O1.6 | CeO2** (4.0; 0.555) | CeO2** (9.0; 0.558) NiO (6.5) |
CeO2** (50.0; 0.556) NiO (50.0) |
| (Ce0.2La0.8)0.8Ni0.2O1.4 | CeO2** (4.0; 0.567) | CeO2** (7.0; 0.566) La2NiO4 |
CeO2** (50.0; 0.560) La2NiO4 |
| La0.8Ni0.2O1.4 | La2O2CO3 | La2O3 La2NiO4 |
La2O3 La2NiO4 |
| Катализаторы, полученные методом пропитки по влагоемкости | |||
| Ni/CeO2 | CeO2 (11.0; 0.541) NiO (15.0) |
CeO2 (14.0; 0.541) NiO (25.0) |
CeO2 (50.0; 0.541) NiO (50.0) |
| Ni/Ce0.8La0.2O1.9 | CeO2** (8.0; 0.547) NiO (11.0) |
CeO2** (11.0; 0.547) NiO (18) |
CeO2** (25.0; 0.547) NiO (50.0) |
| Ni/Ce0.5La0.5O1.75 | CeO2** (4.5; 0.556) | CeO2** (7.5; 0.556) NiO (9.0) |
CeO2** (22.0; 0.556) NiO (50.0) |
| Ni/Ce0.2La0.8O1.6 | CeO2** (3.0; 0.562) | CeO2** (6.0; 0.567) La2NiO4 |
CeO2** (12.0; 0.564) La2NiO4 |
| Ni/La2O3 | La2O2CO3 | La2O3 La2NiO4 |
La2O3 La2NiO4 |
Согласно результатам исследования катализаторов методом ПЭМВР частицы (Ce1 – xLax)0.8Ni0.2Oy представлены кристаллитами с высокой дефектностью структуры и размером не более 5 нм (рис. 3). Полученные значения межплоскостных расстояний соответствуют значениям, характерным для твердых растворов на основе диоксида церия [47]. Локальный элементный анализ различных участков образцов показывает равномерность распределения элементов Ni, Сe и La в частицах материала. Высокодисперсные кристаллиты образуют поликристаллические агрегаты, на поверхности которых наблюдаются единичные частицы оксида никеля размером не более 3 нм (рис. 3). В отличие от катализаторов P-серии, для катализаторов I-серии, изучение которых методом электронной микроскопии было проведено нами ранее [46, 48], на поверхности поликристаллического носителя Се1 – хLaxOy (x = = 0–0.8) присутствуют частицы и кластеры оксида никеля. Размер NiO-частиц зависит от состава носителя и изменяется в пределах от 2 до 50 нм [46, 48].
Рис. 3.
Микроснимки ПЭМВР катализаторов Ce0.8Ni0.2O1.8 (а), (Ce0.8La0.2)0.8Ni0.2O1.7 (б) и (Ce0.2La0.8)0.8Ni0.2O1.4 (в) и результаты локального элементного анализа (ат. %) различных участков образцов.

Сравнительный анализ данных КР-спектроскопии для образцов P- и I-серий свидетельствует о более высокой дефектности структуры материалов, полученных методом сложноэфирных полимерных предшественников. При одинаковом составе катализаторов значение соотношения интенсивности полос при 570 и 465 см–1 (I570/I465), используемое для оценки разупорядоченности кислородной подрешетки (концентрации кислородных вакансий) твердого раствора типа флюорита [49], гораздо выше для образцов P-серии (табл. 1), что может служить дополнительным подтверждением вхождения катионов никеля в структуру твердого раствора. В отличие от Ce1 – xNixOy [31] для Ni–Ce–La–О-системы (при x = 0.5–0.8 для P-серии и x = 0.8 для I-серии) наблюдается более существенная перестройка структуры материала и уменьшение размеров кристаллитов, что приводит к уширению и заметному сдвигу полос в СКР и, соответственно, затрудняет корректную оценку дефектности образцов при высоком содержании La.
Фазовый состав катализаторов после активации в атмосфере 30 об. % H2/Не зависит от метода приготовления и состава образцов (табл. 1). Так, для P-серии катализаторов Ni-содержащая фаза по-прежнему отсутствует в составе материалов, исключение составляет только образец, не содержащий La (La : Ce = 0). Активация катализаторов I-серии (La : Ce = 0; 0.25) приводит к восстановлению фазы NiO и образованию фазы Ni0 (табл. 1). Из сравнения данных для образцов Ce0.8Ni0.2O1.8 и Ni/CeO2 видно, что средний размер частиц Ni0 существенно ниже для образца P-серии. По данным метода ПЭМ на поверхности Ni0-частиц обнаружена оксидная пленка, т.е. при контакте образца с воздухом происходит его частичное реокисление. В остальных активированных образцах методом ex situ РФА формирования Ni0 не обнаружено, что, по-видимому, связано с реокислением высокодисперсных металлических никелевых частиц при контакте с воздухом. По сравнению с остальными катализаторами образцы с x = 0–0.2 (Ce0.8Ni0.2O1.8, Ni/CeO2 и Ni/Ce0.8La0.2O1.9) характеризуются меньшей дисперсностью оксида никеля и более низкой температурой его восстановления, а, следовательно, взаимодействие металл–носитель в них слабее, что обеспечивает восстановление NiO до металлического никеля в процессе их активации. Следует отметить, что интенсивность реокисления возрастает с увеличением дисперсности и степени взаимодействия металл–носитель [50, 51]. Для обеих серий катализаторов при La/Ce ≤ 1 происходит повышение среднего размера кристаллитов твердого раствора на основе диоксида церия, а при большем значении La : Ce = 4 – формирование La-содержащей фазы (табл. 1).
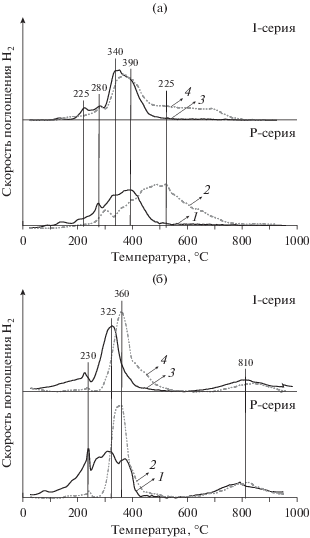
Наряду с изменением текстурных и структурных характеристик, варьирование метода синтеза приводит к разной восстанавливаемости материала (табл. 1, рис. 4). Для катализатора Ce0.8Ni0.2O1.8, прокаленного при 300–500°С, на кривой ТПВ-H2 наблюдается пик с максимумом при T = 230°C, широкий пик в области температур 250–450°С и пик при T = 810°C. Считается [52–54], что для Ni–Ce–O-катализаторов 1) в низкотемпературной области (<250°С) происходит восстановление структур [Ni–O–Ce], локализованных на поверхности твердого раствора или в местах контакта высокодисперсных частиц оксида никеля с кристаллитами диоксида церия; 2) в интервале температур 250–550°С – восстановление катионов никеля как в составе частиц и кластеров оксида никеля, так и в составе твердого раствора; 3) в высокотемпературной (>550°С) – восстановление катионов Ce4+, локализованных в объеме частиц. Варьирование метода введение никеля в матрицу носителя, при прочих равных условиях, приводит к существенному изменению профиля кривой ТПВ-H2 (рис. 4): для образцов I-серии по сравнению с катализаторами P-серии: снижается интенсивность поглощения водорода в низкотемпературной области, а пик в средней области сужается и имеет выраженный максимум. Это свидетельствует об уменьшении доли структур [Ni–O–Ce] и росте вклада частиц NiO в формы стабилизации никеля в составе катализаторов, что коррелирует с данными структурных методов. При повышении температуры прокаливания до 900оС различия в профилях ТПВ-H2 образцов, полученных различными методами, становятся менее заметными. Для катализаторов обеих серий снижается интенсивность поглощения в низкотемпературной области, а максимумы пиков в области 250–550°С сдвигаются в высокотемпературную область, что может быть обусловлено ростом размера частиц оксида никеля и хорошо согласуется с результатами фазового анализа образцов (табл. 1, 2). Увеличение мольного соотношения La : Ce, как и повышение температуры прокаливания, приводит к смещению пиков восстановления катионов никеля в высокотемпературную область (рис. 4). Значение мольного соотношения H2 : Ni > 1 указывает на вклад носителя в поглощение водорода в ходе ТПВ-H2 [55, 56].
Рис. 4.
ТПВ водородом Ni–Се–La–О-катализаторов: влияние мольного соотношения La : Ce (а) и температуры прокаливания (б). Условия эксперимента: а – La : Ce = 0.25 (1, 3) и 4 (2, 4), температура прокаливания 500°С; б – температура прокаливания 300 (1, 3) и 900°С (2, 4), La : Ce = 0.
На основании результатов физико-химических методов исследования выбраны 1) минимальная температура (Тmin) прокаливания катализаторов (500°С), обеспечивающая высокую дисперсность Ni-содержащей фазы и удаление органической матрицы и 2) Тmin активации катализаторов (600°С), определяемая максимальной температурой проведения каталитической реакции и результатами ТПВ-H2, согласно которым бóльшая часть активного компонента после восстановления при 600°С находится в восстановленном состоянии.
Активность катализаторов в реакции АТР С2H5OH
Для выявления влияния физико-химических характеристик Ni–Ce–La–O-катализаторов на их функциональные свойства была исследована активность образцов в реакции автотермического риформинга этанола. В основе метода автотермического риформинга лежит сочетание экзотермической реакции парциального окисления с эндотермическими реакциями парового риформинга и конверсии водяного газа, что обеспечивает снижение энергопотребления и более гибкое регулирование состава синтез-газа. Исследования показали, что основными продуктами автотермического риформинга этанола являются водород, оксиды углерода и метан. Конверсия и распределение продуктов в АТР этанола в присутствии Ni–Ce–La–O-катализаторов зависят от температуры реакции: при ее повышении от 200 до 600°С конверсия этанола возрастает, выход монооксида углерода и водорода увеличиваются, выход СО2 выходит на плато, а выход метана уменьшается, что хорошо согласуется с литературными данными [57 ] . Из анализа температурных кривых конверсии этанола следует, что для обеих серий образцов температура достижения полной конверсии этанола снижается с сокращением содержания La в составе катализатора и, например, для Ce0.8Ni0.2O1.8 и (Ce0.5La0.5)0.8Ni0.2O1.6 составляет 300 и 600°С соответственно. Аналогично полученным нами ранее результатам для Ni/CeO2 [31], максимальный выход водорода в присутствии Ni–Ce–La–O-образцов наблюдается при температуре реакции 600°С и слабо зависит от состава оксидной матрицы и метода приготовления (табл. 3). С ростом температуры прокаливания материалов от 500 до 900°С выход водорода несколько падает. В меньшей степени этот эффект проявляется для образцов с мольным соотношением La/Ce > 0, что, по-видимому, связано с большей термостабильностью La-содержащих систем.
Таблица 3.
Активность* катализаторов в реакции автотермического риформинга этанола
| Образец | Tемпература прокаливания катализатора, °C |
Продолжи-тельность реакции, ч | ${{Y}_{{{{{\text{H}}}_{{\text{2}}}}}}},$ % | S, % | ||
|---|---|---|---|---|---|---|
| CO | CO2 | CH4 | ||||
| Ce0.8Ni0.2O1.8 | 500 | 6 | 57 | 27 | 59 | 14 |
| 900 | 6 | 43 | 20 | 69 | 11 | |
| (Ce0.8La0.2)0.8Ni0.2O1.7 | 500 | 6 | 48 | 24 | 64 | 12 |
| 500 | 24 | 49 | 25 | 60 | 15 | |
| 900 | 6 | 44 | 28 | 59 | 13 | |
| (Ce0.5La0.5)0.8Ni0.2O1.6 | 500 | 6 | 43 | 27 | 60 | 13 |
| (Ce0.2La0.8)0.8Ni0.2O1.4 | 500 | 6 | 49 | 29 | 56 | 15 |
| La0.8Ni0.2O1.4 | 500 | 6 | 44 | 28 | 62 | 9 |
| Ni/CeO2 | 500 | 6 | 50 | 23 | 65 | 12 |
| 500 | 24 | 33 | 28 | 59 | 13 | |
| Ni/Ce0.8La0.2O1.9 | 500 | 6 | 47 | 28 | 58 | 14 |
| 500 | 24 | 48 | 28 | 59 | 13 | |
| Ni/Ce0.5La0.5O1.75 | 500 | 6 | 45 | 25 | 61 | 14 |
| Ni/Ce0.2La0.8O1.6 | 500 | 6 | 45 | 28 | 58 | 14 |
| Ni/La2O3 | 500 | 6 | 44 | 27 | 58 | 15 |
Исследование стабильности работы катализаторов показало, что выход водорода в присутствии Ni/CeO2 снижается с течением времени. Напротив, образцы (Ce0.8La0.2)0.8Ni0.2O1.7 и Ni/Ce0.8La0.2O1.9 одинакового состава, но полученные разными методами, обеспечивают высокий (~50%) и стабильный выход водорода. Это указывает, что введение Lа в состав образцов улучшает стабильность их работы. Другой важной характеристикой катализатора является его устойчивость к образованию углеродистых отложений в ходе каталитического процесса. После длительных испытаний количество углеродистых отложений, определенное методом термического анализа, составило 31.3, 29.1 и 4.3 мас. % для Ni/CeO2, Ni/Ce0.8La0.2O1.9 и (Ce0.8La0.2)0.8Ni0.2O1.7 соответственно, что свидетельствует о более высокой устойчивости к зауглероживанию образцов, полученных методом сложноэфирных полимерных предшественников.
ЗАКЛЮЧЕНИЕ
Методами сложноэфирных полимерных предшественников и пропитки по влагоемкости синтезированы две серии Ni–Cе–La–O-катализаторов при варьировании мольного соотношения La : Ce в широком диапазоне (0, 0.25, 1, 4). Исследованы закономерности формирования материалов, их физико-химические свойства и активность в реакции автотермического риформинга этанола. Обобщая полученные данные, можно заключить, что генезис, физико-химические и функциональные свойства Ni–Ce–La–O-катализаторов определяются их составом и методом приготовления. Материалы с La : Ce = 0–4 после прокаливания при 300–500°С представляют собой твердый раствор на основе диоксида церия (Ni–Ce–La–O, Ce–La–O) с нанесенными частицами оксида никеля. При этом метод сложноэфирных полимерных предшественников обеспечивает стабилизацию никеля преимущественно в составе твердого раствора на основе диоксида церия, а метод пропитки – в виде частиц NiO. В целом, образцы P-серии по сравнению с образцами I-серии отличаются большей удельной поверхностью (SВЭТ = 100 vs. 60 м2/г при La/Ce = 0.25), дефектностью структуры (I570/I465 = 1.4 vs. 0.1) и меньшим средним размером кристаллитов твердого раствора (OKР составляет 6 vs. 8 нм). C повышением температуры прокаливания до 700–900°С различия как в текстурных, так и структурных характеристиках катализаторов P- и I-серий становятся менее выраженными, что связано с частичным разрушением твердого раствора Ni–Ce–La–O и формированием Ni-содержащей фазы в образцах P-серии. Независимо от метода приготовления при мольном соотношении La : Ce = 4 после высокотемпературного прокаливания формы стабилизации никеля определяются присутствием в системе лантана – наблюдается формирование фазы перовскита La2NiO4. Для обеих серий катализаторов с мольным соотношением La : Ce = 0–1 термостабильность материалов возрастает с увеличением содержания лантана в их составе. Важно отметить, что после активации катализаторов в восстановительной среде дисперсность Ni-фазы существенно выше для образцов P-серии. В автотермическом риформинге этанола для обеих серий образцов наблюдались сравнимые значения активности, но различная стабильность работы. Степень зауглероживания снижалась в ряду Ni/CeO2 > > Ni/Ce0.8La0.2O1.9$ \gg $ (Ce0.8La0.2)0.8Ni0.2O1.7. Полученный ряд устойчивости к зауглероживанию коррелирует с повышением дисперсности компонентов катализатора (уменьшением среднего размера кристаллитов твердого раствора на основе CeO2 и Ni активного компонента) и увеличением концентрации кислородных вакансий, что хорошо согласуется с литературными данными [11–16, 36, 37].
Список литературы
Horn R., Schlögl R. // Catal. Lett. 2015. V. 45. P. 23.
Enger B.C., Lødeng R., Holmen A. // Appl. Catal. A: Gen. V. 346. P. 1.
Ismagilov Z.R., Matus E.V., Ismagilov I.Z., Sukhova O.B., Yashnik S.A., Ushakov V.A., Kerzhentsev M.A. // Catal. Today. 2019. V. 323. P. 166.
Усачев Н.Я., Харламов В.В., Беланова Е.П., Старостина Т.С., Круковский И.М. // Рос. Хим. Журн. 2008. Т. LII. № 4. С. 22.
De S., Zhang J., Luque R., Yan N. // Energy Environ. Sci. 2016. V. 9. P. 3314.
Ananikov V.P. // ACS Catal. 2015. V. 5. P. 1964.
Jones G., Jakobsen J.G., Shim S.S., Kleis J., Andersson M.P., Rossmeisl J., Abild-Pedersen F., Bligaard T., Helveg S., Hinnemann B., Rostrup-Nielsen J.R., Chorkendorff I., Sehested J., Nørskov J.R. // J. Catal. 2008. V. 259. P. 147.
Moulijn J.A., van Diepen A.E., Kapteijn F. // Appl. Catal. A: Gen. 2001. V. 212. P. 3.
Bartholomew C.H. // Appl. Catal. A: Gen. 2001. V. 212. P. 17.
Arora S., Prasad R. // RSC Adv. 2016. V. 6. P. 108668.
Han J.W., Park J.S., Choi M.S., Lee H.H. //Appl. Catal. B: Environ. 2017. V. 203. P. 625.
Sharma Y.C., Kumar A., Prasad R., Upadhyay S.N. // Renew. Sustain. Energy Rev. 2017. V. 74. P. 89.
Aramouni N.A.K., Zeaiter J., Kwapinski W., Ahmad M.N. // Energy Convers. Manage. 2017. V. 150. P. 614.
Bengaard H.S., Nørskov J.K., Sehested J., Clausen B.S., Nielsen L.P., Molenbroek A.M., Rostrup-Nielsen J.R. // J. Catal. 2002. V. 209. P. 365.
Ligthart D.A.J.M., Pieterse J.A.Z., Hensen E.J.M. // Appl. Catal. A: Gen. 2011. V. 405. P. 108.
Li X., Li D., Tian H., Zeng L., Zhao Z., Gong J. // Appl. Catal. B: Environ. 2017. V. 202. P. 683.
Hou Z., Yokota O., Tanaka T., Yashima T. // Appl. Surf. Sci. 2004. V. 233. P. 58.
Urasaki K., Tanpo Y., Nagashima Y., Kikuchi R., Satokawa S. // Appl. Catal. A: Gen. 2013. V. 452. P. 174.
Ismagilov I.Z., Matus E.V., Kuznetsov V.V., Yashnik S.A., Kerzhentsev M.A., Gerritsen G., Abbenhuis H.C.L., Ismagilov Z.R. // Eurasian Chem.-Technol. J. 2017. V. 19. P. 3.
Somacescu S., Florea M., Osiceanu P., Calderon-Moreno J.M., Ghica C., Serra J.M. // J. Nanopart. Res. 2015. V. 17. P. 426.
Juan-Juan J., Roman-Martınez M.C., Illan-Gomez M.J. // Appl. Catal. A: Gen. 2009. V. 355. P. 27.
Ismagilov I.Z., Matus E.V., Kuznetsov V.V., Kerzhentsev M.A., Yashnik S.A., Prosvirin I.P., Mota N., Navarro R.M., Fierro J.L.G., Ismagilov Z.R. // Int. J. Hydrogen Energy. 2014. V. 39. P. 20992.
Abreu A.J., Lucrédio A.F., Assaf E.M. // Fuel. Process. Technol. 2012. V. 102. P. 140.
Zhang L., Wang X., Tan B., Ozkan U.S. // J. Mol. Catal. A. 2009. V. 297. P. 26.
Li H., Xu H., Wang J. // J. Natural. Gas Chem. 2011. V. 20. P. 1.
Ismagilov I.Z., Matus E.V., Kuznetsov V.V., Yashnik S.A., Prosvirin I.P., Kerzhentsev M.A., Ismagilov Z.R., Mota N., Navarro R.M., Fierro J.L.G. // Appl. Catal. A. 2014. V. 481. P. 104.
Mota N., Ismagilov I.Z., Matus E.V., Kuznetsov V.V., Kerzhentsev M.A., Ismagilov Z.R., Navarro R.M., Fierro J.L.G. // Int. J. Hydrogen Energy. 2016. V. 41. P. 19373.
Pavlova S., Kapokova L., Bunina R., Alikina G., Sazonova N., Krieger T., Ishchenko A., Rogov V., Gulyaev R., Sadykov V., Mirodatos C. // Catal. Sci. Technol. 2012. V. 2. P. 2099.
Deng J., Cai M., Sun W., Liao X., Chu W., Zhao X.S. // ChemSusChem. 2013. V. 6. P. 2061.
Liu F., Zhao L., Wang H., Bai X., Liu Y. // Int. J. Hydrogen Energy. 2014. V. 39. P. 10454.
Матус Е.В., Шляхтина A.C., Сухова О.Б., Исмагилов И.З., Ушаков В.А., Яшник С.А., Никитин А.П., Bharali P., Керженцев М.А., Исмагилов З.Р. // Кинетика и катализ. 2019. Т. 60. № 2. С. 245.
Tan J., Lee D., Ahn J., Kim B., Kim J., Moon J. // J. Mater. Chem. A. 2018. V. 6. P. 18133.
Misture S.T., McDevitt K.M., Glass K.C., Edwards D.D., Howe J.Y., Rector K.D., Hec H., Vogel S.C. // Catal. Sci. Technol. 2015. V. 5. P. 4565.
Li D., Koike M., Chen J., Nakagawa Y., Tomishige K. // Int. J. Hydrogen Energy. 2014. V. 39. P. 10959.
Hernández W.Y., Lauwaert J., Van Der Voort P., Verberckmoes A. // GreenChem. 2017. V. 19. P. 5269.
Zhu T., Flytzani-Stephanopoulos M. // Appl. Catal. A: Gen. 2001. V. 208. P. 403.
Nahar G., Dupont V. // Renew. Sust. Energ. Rev. 2014. V. 32. P. 777.
Bukhtiyarova M.V. // J. Solid State Chem. 2019. V. 269. P. 494.
Sadykov V., Pavlova S., Smal E., Arapova M., Simonov M., Mezentseva N., Rogov V., Glazneva T., Lukashevich A., Roger A.-C., Parkhomenko K., van Veen A., Smorygo O. // Catal. Today. 2017. V. 293–294. P. 176.
Pintona N., Vidal M.V., Signoretto M., Martínez-Arias A., Corberán V.C. // Catal. Today. 2017. V. 296. P. 135.
Керженцев М.А., Матус Е.В., Исмагилов И.З., Ушаков В.А., Стонкус О.А., Ларина Т.В., Козлова Г.С., Bharali P., Исмагилов З.Р.
Исмагилов И.З., Матус Е.В., Нефедова Д.В., Кузнецов В.В., Яшник С.А., Керженцев М.А., Исмагилов З.Р. // Кинетика и катализ. 2015. Т. 56. № 3. С. 397.
Matus E.V., Okhlopkova L.B., Sukhova O.B., Ismagilov I.Z., Kerzhentsev M.A., Ismagilov Z.R. // J. Nanoparticle Res. 2019. V. 21. № 1. P. 11.
Nguyen-Phan T.-D., Song M.B., Kim E.J., Shin E.W. // Micropor. Mesopor. Mater. 2009. V. 119. P. 290.
Ponchel A., D’Huysser A., Lamonier C., Jalowiecki-Duhamel L. // Phys. Chem. Chem. Phys. 2000. V. 2. P. 303.
Матус Е.В., Нефедова Д.В., Кузнецов В.В., Ушаков В.А., Стонкус О.А., Исмагилов И.З., Керженцев М.А., Исмагилов З.Р. // Кинетика и катализ. 2017. Т. 58. № 5. С. 623.
Wang F., Xu L., Yang J., Zhang J., Zhang L., Li H., Zhao Y., Li H.X., Wu K., Xu G.Q., Chen W. // Catal. Today. 2017. V. 281. P. 295.
Kerzhentsev M.A., Matus E.V., Ismagilov I.Z., Sukhova O.B., Bharali P., Ismagilov Z.R. // Eurasian Chem.-Technol. J. 2018. V. 20. № 4. P. 283.
Liyanage A.D., Perera S.D., Tan K., Chabal Y., Balkus K.J. // ACS Catal. 2014. V. 4. P. 577.
Yinghui Z., Linze D., Yunkai Z., Jing Z. // Surf. Sci. 2019. V. 681 P. 47.
Löfberg A., Guerrero-Caballero J., Kane T., Rubbens A., Jalowiecki-Duhamel L. // Appl. Catal. B: Env. 2017. V. 212. P. 159.
Lamonier C., Ponchel A., D’Huysser A., Jalowiecki-Duhamel L. // Catal. Today. 1999. V. 50. P. 247.
Vita A., Italiano C., Fabiano C., Lagana M., Pino L. // Mater. Chem. Phys. 2015. V. 163. P. 337.
Shan W., Luo M., Ying P., Shen W., Li C. // Appl. Catal. A: Gen. 2003. V. 246. P. 1.
Singh K., Kumara R., Chowdhury A. // Mater. Today: Proceed. 2018. V. 5. P. 22993.
Li X., Zhao Z.J., Zeng L., Zhao J., Tian H., Chen S., Li K., Sang S., Gong J. // Chem. Sci. 2018. V. 9. P. 3426.
Zanchet B., Santos J.B.O., Damyanova S., Gallo J.M.R., Bueno J.M.C. //ACS Catal. 2015. V. 5. P. 3841.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ