

Кинетика и катализ, 2019, T. 60, № 4, стр. 450-462
Активация и пути превращения кислорода в высокотемпературных реакциях окисления легких алканов: кажущаяся простота кинетического описания
a ФГБУН Институт химической физики им. Н.Н. Семенова РАН
119991 Москва, ул. Косыгина, 4, Россия
* E-mail: sinev@yandex.ru
Поступила в редакцию 11.03.2019
После доработки 15.03.2019
Принята к публикации 18.03.2019
Аннотация
Рассмотрены кинетические закономерности процессов каталитического окисления, протекающих по стадийному окислительно-восстановительному (ОВ) механизму. Анализируются основные допущения модели Марса–ван-Кревелена, широко используемой при описании процессов такого рода, соответствие им реальных каталитических систем – типичных катализаторов процессов парциального окисления легких алканов (окислительной конденсации метана, окислительного дегидрирования C2+- и окислительного крекинга C3+-алканов). Особенности состава и условий функционирования этих катализаторов требуют некоторого пересмотра существующих представлений об активации молекулярного кислорода, включая ключевые факторы, которые определяют его взаимодействие с активными центрами и участие в каталитическом цикле. Особое внимание уделено: термодинамическим и кинетическим условиям протекания каталитических процессов по ОВ-механизму (термохимия оксидных систем, время жизни кислорода в связанном состоянии); реализации механизмов ОВ-типа в присутствии оксидов, не содержащих ионы с переменной степенью окисления; химическим и фазовым превращениям, являющимся частью каталитического ОВ-цикла; интерпретации результатов кинетического эксперимента и влиянию стадий с участием свободных радикалов на величины определяемых эмпирически кинетических параметров при изучении процессов гетерогенно-гомогенного характера.
ВВЕДЕНИЕ
Все классические определения химической кинетики, так или иначе, характеризуют ее как дисциплину, устанавливающую не только закономерности протекания химических процессов во времени, но и связь этих закономерностей с механизмом превращений. В свою очередь, катализ – как по определению, так и по сути – представляет собой совокупность кинетических явлений, поскольку связан с изменением скоростей и направлений химических превращений. При этом в конкретных кинетических исследованиях далеко не всегда ставится задача установить связь между наблюдаемыми скоростями и механизмом процесса. Так, для решения сугубо практических задач бывает достаточно получить набор уравнений, с заданной точностью описывающих связь скорости превращения реагентов и/или образования продуктов с измеряемыми параметрами (температура, давление, концентрации веществ – участников реакции, условия переноса тепла и массы и пр.). Кинетические модели такого типа иногда называют “глобальными”, “феноменологическими” и даже “инженерными”, а по сути их можно считать сугубо математическими, поскольку они в пределе могут не иметь какого-либо химического и физического содержания. Как правило, они пригодны для использования в том диапазоне изменения параметров, в котором проводился базовый эксперимент, положенный в основу построения аппроксимирующих уравнений. Их использование вне этого диапазона невозможно ввиду полной неопределенности результатов.
Противоположным является такой подход к построению кинетических моделей, который основан на представлениях о механизме процесса, реализованных в форме стадийных схем и кинетических уравнений. Как правило, они также содержат параметры, получаемые при обработке экспериментальных данных, но им приписывается вполне определенный физико-химический смысл. Такие модели обладают более существенной предсказательной силой, т.е. могут использоваться для прогноза поведения системы вне диапазона изменения параметров базового эксперимента. Границы применимости таких моделей определяются уже тем, в каких пределах заложенные в описание модельные представления можно считать работоспособными, а определяемые из эксперимента параметры – неизменными.
К сожалению, далеко не всегда указанное ограничение принимается во внимание, т.е. приходится сталкиваться с тем, что некоторые широко известные модели используются для описания систем, не соответствующих им по существу, а получаемым результатам приписывается неадекватный смысл. Примером такого рода является применение модели активированного комплекса к описанию неэлементарных реакций, усугубляющееся тем, что полученные значения “энергии активации” и “предэкспонента” (по сути – эмпирические “подгоночные” параметры) используются для вычисления термодинамических функций “активированного комплекса” с далеко идущими выводами.
Классическим примером кинетической модели второго типа, подтвердившей свою обоснованность, работоспособность и даже универсальность (в смысле возможности применения для различных катализаторов), может служить модель реакции синтеза аммиака, на которой основано классическое уравнение Темкина–Пыжева [1]. Она основана на нескольких основных положениях: об обратимости процесса, его лимитировании диссоциативной адсорбцией азота на поверхности металлического катализатора и линейном росте энергии активации хемосорбции азота при уменьшении теплоты его адсорбции.
Еще один пример кинетической модели второго типа – широко используемая в исследованиях по каталитическому окислению модель Марса–ван-Кревелена, впервые предложенная для описания кинетики окисления ароматических соединений на оксидном ванадиевом катализаторе [2]. В основе этой модели лежит представление о том, что в катализаторе присутствует активная форма кислорода, которая непосредственно участвует во взаимодействии с окисляемым субстратом, удаляется из катализатора в стадии восстановления:
где [O] и [ ] – активный центр катализатора в окисленном и восстановленном состоянии, и восполняется (регенерируется) при реакции с окислителем, например, с кислородом (стадия реокисления):В простейшем случае скорости реакций (I) и (II) имеют первый порядок по реагенту A (окисляемому субстрату) и активным центрам, а при стационарном протекании процесса эти скорости равны. При этих условиях скорость образования продукта AO описывается уравнением:
(1)
${{W}_{{{\text{AO}}}}} = {{k}_{{{\text{в о с }}}}}{{k}_{{{\text{о к }}}}}{{P}_{{\text{A}}}}{{P_{{{{{\text{O}}}_{2}}}}^{n}} \mathord{\left/ {\vphantom {{P_{{{{{\text{O}}}_{2}}}}^{n}} {\left( {{{k}_{{{\text{в о с }}}}}{{P}_{{\text{A}}}} + {{k}_{{{\text{о к }}}}}P_{{{{{\text{O}}}_{2}}}}^{n}} \right)}}} \right. \kern-0em} {\left( {{{k}_{{{\text{в о с }}}}}{{P}_{{\text{A}}}} + {{k}_{{{\text{о к }}}}}P_{{{{{\text{O}}}_{2}}}}^{n}} \right)}},$Приведенное уравнение в литературе называют по именам авторов работы [2] уравнением Марса–ван-Кревелена. Показана его применимость для описания кинетики большого числа реакций каталитического окисления – в основном, селективного парциального – различных органических соединений (олефинов, спиртов, альдегидов), а также – для высокотемпературного окисления CO на оксидах переходных металлов и SO2 до SO3.
Формально схема (I)–(II) является частным случаем так называемой стадийной схемы, а уравнение (1) можно считать предельным случаем уравнения двухстадийной обратимой реакции с одним промежуточным веществом (в терминах М.И. Темкина [3]):
(2)
$r = \frac{{{{k}_{1}}{{P}_{{{{{\text{A}}}_{1}}}}}{{k}_{2}}{{P}_{{{{{\text{A}}}_{2}}}}} - {{k}_{{ - 1}}}{{P}_{{{{{\text{B}}}_{1}}}}}{{k}_{{ - 2}}}{{P}_{{{{{\text{B}}}_{2}}}}}}}{{{{k}_{1}}{{P}_{{{{{\text{A}}}_{1}}}}} + {{k}_{2}}{{P}_{{{{{\text{A}}}_{2}}}}} + {{k}_{{ - 1}}}{{P}_{{{{{\text{B}}}_{1}}}}} + {{k}_{{ - 2}}}{{P}_{{{{{\text{B}}}_{2}}}}}}},$Легко заметить, что главное отличие между двумя уравнениями – (1) и (2) – определяется тем, что в модели Марса–ван-Кревелена обе стадии рассматриваются как практически необратимые. Иными словами, в отличие от модели М.И. Темкина [3], в модели Марса–ван-Кревелена не учитывается в явном виде принцип микроскопической обратимости, т.е. она не является термодинамически согласованной.
На этот ее недостаток указывалось и ранее, как и на не вполне очевидный смысл величины порядка n по кислороду и на то, что в этой модели игнорируется образование каких-либо интермедиатов помимо “активного кислорода” [O], включая адсорбированные формы окисляемых реагентов, промежуточных и конечных продуктов (см., например, [4]).
Действительно, цепочка превращений окисляемого субстрата A полностью выпадает из рассмотрения в модели Марса–ван-Кревелена. С формально-кинетической точки зрения, ее применимость ограничивается процессами, в которых в последовательности, приводящей к образованию окисленного продукта AO, лимитирующей является первая стадия – активация реагента A с участием активного кислорода катализатора [O]. По-видимому, круг такого рода процессов достаточно широк. И в него входят, в том числе, высокотемпературные реакции окисления метана и его ближайших гомологов – легких алканов. В них, как правило, наиболее энергозатратной является стадия активации углеводорода с отрывом первого атома водорода от прочной C–H-связи. Кроме того, при характерных температурах этих реакций (>600°С) метан и его ближайшие гомологи молекулярно не адсорбируются на оксидах – типичных катализаторах рассматриваемых здесь процессов.
Наконец, можно утверждать, что в модели Марса–ван-Кревелена никак не отражена концепция идеальности/неидеалиности поверхности катализатора. Действительно, классический ее вариант по сути оперирует исключительно реакционной способностью активных центров в окисленном и восстановленном состоянии. Она характеризуется двумя константами скорости (kвос и kок), которые по умолчанию считаются постоянными для всех центров и не зависят от глубины восстановления катализатора.
Признавая все эти ограничения, следует подчеркнуть, что ценность подхода Марса и ван-Кревелена состоит в том, что предложенная ими модель позволяет выделить из сложного каталитического процесса свойства активной формы кислорода [O] – его реакционную способность по отношению к молекуле реагента и кинетику регенерации.
В настоящей статье внимание будет сосредоточено на том, в какой степени соответствие наблюдаемых скоростей соотношениям типа уравнения Марса–ван-Кревелена может служить указанием на то, что в системе реализуются представления, заложенные в модель. Иными словами, на примере реакций окисления метана и его гомологов в присутствии оксидных катализаторов мы постараемся проследить соотношение между реальными путями превращения молекулярного кислорода при взаимодействии с катализатором и зависимостями скорости каталитической реакции от экспериментальных параметров. В какой-то степени это может послужить шагом к решению обратной кинетической задачи (поиска перехода от наблюдаемой кинетики к представлениям о механизме процесса и свойствах каталитической системы), которая, как известно, может не иметь однозначного решения при использовании формальных математических процедур [5].
Одним из принципиально важных является вопрос об обратимости стадии хемосорбции кислорода. С несколько иных, чем в [4], позиций он подробно рассмотрен ниже.
ТЕРМОХИМИЯ ОКСИДНЫХ СИСТЕМ
Как правило, применимость модели и уравнения Марса–ван-Кревелена связывают с присутствием в оксидной системе так называемого активного решеточного кислорода, под которым понимается кислород решетки оксида, имеющий повышенную реакционную способность, например, за счет нахождения в положении с пониженной координацией и наличии в ближайших катионных позициях ионов переходных металлов в высших степенях окисления. В этом случае реакция типа (I) связана с возникновением кислородной вакансии (идентична в данном случае состоянию [ ]) и cнижением степени окисления ионов (одного или двух) переходного металла, например:
(III)
$\begin{gathered} {{{\text{M}}}^{{(n}}}{{^{{\text{ }}}}^{{ + {\text{ 1}}) + }}}{\kern 1pt} - {\kern 1pt} \left[ {\text{O}} \right]{\kern 1pt} --{\kern 1pt} {{{\text{M}}}^{{(n}}}^{{{\text{ }} + {\text{ 1}}) + }} + {\text{A}} \to \\ \to \,\,{{{\text{M}}}^{n}}^{ + }{\kern 1pt} --{\kern 1pt} \left[ {\text{ }} \right]{\kern 1pt} --{\kern 1pt} {{{\text{M}}}^{n}}^{ + } + {\text{AO}}{\text{.}} \\ \end{gathered} $Важной особенностью реакций, протекающих по механизму Марса–ван-Кревелена, или окислительно-восстановительному (ОВ) механизму, является то, что участвующий в нем активный кислород имеет бесконечно большое время жизни даже при отсутствии кислорода в газовой фазе. Т.е. в отличие от процессов окисления, протекающих по механизмам Лэнгмюра–Хиншельвуда или Или–Ридила, которые можно отнести к адсорбционно-десорбционному (АД) типу, активный кислород не находится в равновесии с кислородом газовой фазы и не выделяется в газовую фазу в виде молекул O2. Если же принять во внимание принцип микроскопической обратимости, это означает, что характерное время десорбции много больше времени наблюдения, или что стадия десорбции кислорода является очень медленной – по крайней мере, по сравнению с реакцией (I). С точки зрения описания, для уравнения реакций АД-типа характерны выражения в знаменателе вида [1 + Σ(bi$P_{i}^{n}$)], тогда как в случае уравнений ОВ-типа в простейшем случае в знаменателе присутствует лишь сумма (kвосPA + kок$P_{{{{{\text{O}}}_{2}}}}^{n}$). При анализе экспериментальных данных кинетическим уравнениям для реакций двух типов соответствуют различные линейные анаморфозы.
В качестве практического “теста” на принадлежность реакции к ОВ-типу используется способность соответствующих катализаторов вести процесс в “попеременном” режиме (например, импульсном), т.е. при раздельной подаче реагента-восстановителя и кислорода. При этом скорости превращения окисляемого реагента (или образования продукта) в стадии восстановления должны быть близки к наблюдаемым при стационарном катализе при близких степенях окисления катализатора.
Из сказанного следует, что каталитическая активность оксидов в процессах ОВ-типа напрямую связана с их реакционной способностью – как по отношению к окисляемому веществу в активном (окисленном) состоянии, так и по отношению к окислителю – в восстановленном.
Важно отметить, что с кинетической точки зрения не имеет значения, какова “природа” активного кислорода: является ли он “решеточным” (т.е. занимает определенную позицию в анионной подрешетке кристалла оксида) или сверхстехиометрическим, или находится на поверхности инертного носителя в составе кластеров или слоев нанесенных компонентов или в какой-либо иной хемосорбированной форме. Главным для реализации условий, заложенных в модель, является, как уже было сказано, то, что время его жизни в связанном состоянии (τсв) должно существенно превышать характерное время каталитического процесса (τкат), или реакции (I). В самом грубом приближении можно считать, что время жизни кислорода в связанном (хемосорбированном) состоянии определяется выражением:
Легко показать, что при соблюдении условия τсв$ \gg $ τкат кинетические модели АД-типов становятся неприменимыми, поскольку в этом случае не соблюдается одно из положений, лежащих в их основе, т.е. концентрация непосредственно участвующего в реакции кислорода не определяется условиями равновесия его адсорбированных форм с газовой фазой. При этом, как уже было сказано, вывод уравнения (1) основан на условии равенства скоростей реакций (I) и (II), т.е. стационарности степени окисления катализатора, а не равновесности концентрации адсорбированных реагентов.
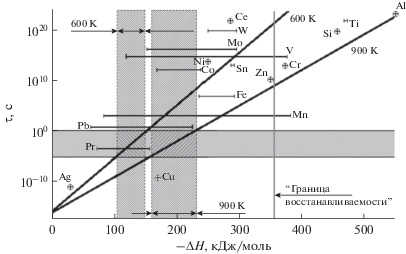
Некоторое представление о возможности протекания реакций каталитического окисления с кинетическими закономерностями ОВ- и АД-типов в присутствии различных оксидов дает рис. 1. Две прямые на нем соответствуют равенству величин τкат и τсв при изменении ΔH для двух температур (600 и 900 К). Если считать, что для каталитических реакций времена τкат лежат в пределах от 10–4 до 10 с, то протеканию реакции по механизму ОВ-типа отвечает область на диаграмме (рис. 1), лежащая правее заштрихованной вертикальной полосы, а по механизму АД-типа – левее нее. При этом в диапазоне величин ΔH, попадающих в эту полосу (например, от ~150 до ~220 кДж/моль при 900 К), такое грубое приближение не дает возможности однозначно определить, к какой группе могут относиться механизмы протекания процесса окисления. Как видно из представленной диаграммы, вероятность наблюдения кинетики ОВ-типа при участии в процессе одних и тех же активных форм кислорода возрастает при понижении температуры, а также если кинетика десорбции более сложная, например:
Рис. 1.
Соотношение между величинами энтальпии восстановления оксидов металлов (положение относительно оси абсцисс произвольное) и областями реализации механизмов АД- и ОВ-типов при 600 и 900 K; для Cr указан только переход Cr2O3 ↔ Cr0, а для оксидов Ce и Pr – переходы M(4+) ↔ M(3+).
– реальный предэкспонент много меньше величины ν, или
– хемосорбция кислорода имеет выраженно активированный характер, или
– наблюдаемая кинетика десорбции существенно отлична от кинетики 1-го порядка.
Если считать, что в реакции (I) происходит переход между двумя стабильными оксидами переходного металла:
Наконец, следует принять во внимание то, что при восстановлении оксидов различными реагентами происходит выделение энергии за счет образования продуктов (например, воды и CO2 при восстановлении водородом и CO соответственно). Это позволяет определить некоторую границу, за пределами которой в области высоких ΔH некоторые оксиды становится практически невосстанавливаемыми в диапазоне типичных температур катализа. На рис. 1 эта граница обозначена вертикальной чертой при ~360 кДж/моль. При всей приблизительности проведенного анализа его результат соответствует тому, что наблюдается экспериментально: оксиды щелочных, щелочноземельных, редкоземельных (в степени окисления 3+) металлов, титана, кремния, алюминия, а также низшие оксиды хрома и марганца можно считать практически невосстанавливаемыми. Оксид цинка лежит близко к границе, что также находит подтверждение в эксперименте: при достаточно жестких условиях его удается восстановить, например, водородом.
Проведенный анализ при его наглядности имеет, однако, ограниченную ценность для реальных катализаторов, которые представляют собой, как правило, многокомпонентные оксидные системы. Входящие в них компоненты образуют многочисленные стехиометрические и нестехиометрические соединения и фазы, обладающие к тому же дефектностью различного рода. В таких сложных системах, как правило, присутствуют формы кислорода, которые весьма далеки по свойствам от анионов кислорода в индивидуальных стехиометрических оксидах металлов. Окислительно-восстановительные процессы в этих системах могут протекать в пределах фазовой гомогенности лишь с изменением степени окисления некоторых катионов, входящих в их состав, но могут и сопровождаться образованием других фаз. Характерные времена перестроек могут быть близки к характерному времени каталитического процесса или даже превышать их, а, значит, стационарное состояние оксидной системы – весьма далеким от того, что наблюдается в двух “крайних” состояниях (окисленном и восстановленном). Это существенно затрудняет кинетический анализ и делает невозможным использование величин ΔH, получаемых из табличных термодинамических данных.
Предложены различные косвенные методы определения энергии связи активного кислорода, участвующего в реакциях окислительно-восстановительного катализа (статическая калориметрия, кинетические методы [6]). По-видимому, более достоверная информация о формах активного кислорода, их концентрациях, реакционной способности и энергетике, а также о динамике изменений, происходящих в оксидных системах в условиях ОВ-превращений, может быть получена из данных реакционной (in situ) динамической калориметрии [7].
Имеются многочисленные примеры реализации механизмов ОВ-типа в реакциях парциального окисления органических соединений, в первую очередь – в присутствии оксидных катализаторов, содержащих ионы переходных металлов, которые способны менять не только степень окисления, но и координацию в пределах близких по строению локальных структур. Речь, прежде всего, идет о молибдене, вольфраме и переходных металлах III периода от ванадия до меди. Массивные и нанесенные индивидуальные и смешанные оксиды этих металлов (включая молибдаты, вольфраматы и ванадаты) являются основой большого числа типичных (в том числе – промышленных) катализаторов селективного парциального окисления олефинов, ароматических углеводородов и функциональных органических соединений. Для многих из них доказано инструментальными методами и кинетически (в частности, упомянутым выше методом сравнения скоростей стационарного катализа и раздельных процессов восстановления–реокисления [6, 8], а также с использованием меченного изотопом 18O кислорода (см., например, [9–11])), что они протекают по ОВ-механизму с участием именно “решеточных” форм кислорода оксидов.
Особо следует отметить в этом ряду использование метода так называемого “реактора с анализом продуктов с высоким временным разрешением” – Temporal Analysis of Products, или TAP-реактора [12, 13]. В настоящее время он является одним из наиболее мощных инструментов для определения концентрации, реакционной способности и кинетических характеристик различных интермедиатов, включая активные формы кислорода, а также динамики оксидных систем.
Имеются примеры применения способности ряда оксидных систем запасать “активный” кислород и поставлять его к центрам взаимодействия с окисляемыми субстратами при создании промышленных процессов с пространственно разделенными зонами восстановления и реокисления, например, при получении малеинового ангидрида окислением бутана [14]. Оксиды с высокой емкостью по “активному кислороду” (например, CeO2) используются также при создании сложных каталитических композитов, применяемых для очистки газовых выбросов, в частности для решения так называемой “проблемы холодного старта” нейтрализаторов выхлопных газов автотранспорта.
ОКИСЛИТЕЛЬНЫЕ ПРЕВРАЩЕНИЯ ЛЕГКИХ АЛКАНОВ
В течение последних десятилетий процессы прямого превращения легких алканов в продукты с добавленной стоимостью вызывают большой интерес исследователей как потенциальная альтернатива процессам получения низших олефинов из более тяжелого сырья методами термического крекинга и дегидрирования. К этой группе процессов относятся окислительная конденсация метана (ОКМ), окислительное дегидрирование (ОД) C2+- и окислительный крекинг (ОК) C3+-алканов. Наибольшее внимание в эти годы было сосредоточено на разработке эффективных катализаторов и исследовании особенностей механизма процессов, таких как выяснение природы поверхностных активных центров, активации молекул алкана и путей их превращения в конечные продукты, роли свободных радикалов как промежуточных продуктов, а также на исследовании кинетики. Ввиду родственности исходных соединений все три процесса рассматриваемой группы имеют много общих черт, к которым можно отнести следующие (см., например, [15, 16]):
1) процессы ОКМ, ОД и ОК эффективно протекают при относительно высоких температурах (>600–700°С) в условиях большого избытка углеводорода (УВ) против стехиометрии (мольное соотношение УВ : O2 > 2);
2) наиболее эффективными катализаторами этих процессов являются индивидуальные и смешанные оксиды (массивные и нанесенные), не содержащие благородных и переходных металлов; исключением из этого правила являются некоторые Mn-, Ce- и Pr-содержащие системы, о которых подробно пойдет речь ниже;
3) на первой стадии каталитического превращения исходного алкана происходит гомолитический разрыв C–H-связи с отрывом атома водорода и образованием алкильного радикала и OH-группы с участием атома кислорода поверхности;
4) молекулярные продукты реакции (целевые и побочные) образуются в сложной цепочке превращений промежуточных частиц, в первую очередь – свободных радикалов, как в газовой фазе, так и с участием активных центров поверхности.
Отдельного обсуждения требует вопрос о наблюдаемых кинетических закономерностях. Если для анализа распределений продуктов приходится привлекать сложные многостадийные гетерогенно-гомогенные схемы (см. выше пп. 3 и 4), то для описания наблюдаемых скоростей превращения зачастую бывает достаточно простых кинетических соотношений, вполне традиционных для гетерогенного катализа. Проведенный анализ показал [16], что наиболее применимыми для описания процессов ОКМ и ОД этана являются уравнения ОВ-типа – уравнение Марса–ван-Кревелена (1) и родственное ему уравнение, учитывающее участие в процессе двух активных центров поверхности [17]:
Поскольку это утверждение справедливо практически для всех катализаторов, для которых оно проверялось, в том числе – для систем, содержащих оксиды, отнесенные выше к категории “невосстанавливаемых”, оно вступает в видимое противоречие со сказанным относительно возможности реализации процессов ОВ-типа в присутствии различных оксидов (рис. 1 и комментарии к нему). Это противоречие не может быть разрешено в рамках рассмотрения стадийной схемы, основанной на реакциях (I)–(III). Действительно, если мы запишем, например, брутто-уравнение реакции образования этана – первичного продукта ОКМ:
(IV)
${\text{2C}}{{{\text{H}}}_{{\text{4}}}} + {\text{1/2}}{{{\text{O}}}_{{\text{2}}}} \to {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{6}}}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}$(V)
$[{\text{O}}] + {\text{C}}{{{\text{H}}}_{{\text{4}}}} \to [{\text{OH}}] + {\text{CH}}_{3}^{\centerdot },$а) кислород входит только в один продукт реакции – воду, которая не может образоваться непосредственно в стадии восстановления активного центра;
б) ввиду высокой энергии связи кислорода с решеткой оксида – энергии его отрыва (E[O]), линейно связанной с величиной ΔH, – образование кислородных вакансий и, следовательно, протекание реокисления катализатора по реакции (II) весьма маловероятно.
Первое противоречие может быть формально преодолено с учетом того, что кинетически значимой (в смысле вхождения соответствующих параметров в стационарное уравнение Марса–ван-Кревелена) не обязательно должна быть именно реакция (V). Это предположение, однако, не согласуется с наблюдаемой кинетикой процесса ОКМ, всегда показывающей близкий к 1 положительный порядок реакции по метану. Второе противоречие не может быть устранено никакими формальными соображениями в рамках рассмотрения традиционной схемы процесса ОВ-типа – совокупности реакций (I) и (II). Можно предположить, что за стадией (V) следует быстрый процесс дегидроксилирования поверхности:
(VI)
${\text{2}}[{\text{OH}}] \to [{\text{O}}] + [{\text{ }}] + {{{\text{H}}}_{{\text{2}}}}{\text{O}},$Однако, как показано ниже, такое предположение далеко не всегда может быть правомерным.
Таким образом, простота описания наблюдаемой кинетики по схеме и уравнению Марса–ван-Кревелена является кажущейся в том смысле, что реальные превращения (промежуточные частицы, состояния активного центра катализатора и стадии процесса) с очевидностью отличны от рассматриваемых в рамках “традиционных” представлений.
РЕАЛИЗАЦИЯ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОГО МЕХАНИЗМА В ПРИСУТСТВИИ НЕВОССТАНАВЛИВАЕМЫХ ОКСИДОВ
Невозможность принять применимость уравнения (1) как однозначное указание на протекание процесса по “традиционной” схеме Марса–ван-Кревелена подтверждается следующими наблюдениями [7, 18, 19]. При подаче на окисленные катализаторы ОКМ при типичных температурах катализа импульсов метана или этана наблюдается быстрое выделение С-содержащего продукта (этана и этилена соответственно). Как сказано выше, это свидетельствует о том, что в катализаторе присутствуют долгоживущие центры активации углеводорода. Характерное снижение скорости превращения реагента-восстановителя и образования продукта от импульса к импульсу показывает, что число таких центров в катализаторе ограничено и что они “срабатываются” по мере протекания реакции с исходным углеводородом. Однако в ряде случаев (например, в случае Li/MgO – типичного катализатора ОКМ) при этом не выделяется вода; характерное время ее образования может более чем на 4 порядка превышать характерное время образования С-содержащего продукта. При этом катализатор не реокисляется при подаче N2O, что подтверждает отсутствие в нем достаточно высоких концентраций кислородных вакансий. Однако при подаче импульсов кислорода происходит быстрое (за время прохождения импульса менее 1 с) выделение воды и реокисление катализатора. Полученные результаты показывают, что:
– процесс в целом является окислительно-восстановительным;
– в восстановленном состоянии катализатор содержит водород, по-видимому, в форме OH-групп, образующихся по реакции (V);
– образование кислородных вакансий по реакции (VI) затруднено, а регенерация активных центров (реокисление) осуществляется через взаимодействие молекулярного кислорода с восстановленной формой активного центра в форме OH-группы.
Упомянутые опыты по импульсному восстановлению–реокислению катализаторов ОКМ проводились в реакторе, помещенном в измерительную ячейку калориметра, с хроматографическим анализом потока реакционной смеси, что позволяло одновременно регистрировать как поглощение исходного реагента и образование продуктов, так и тепловые эффекты протекающих при этом процессов. К сказанному выше следует добавить, что при подаче на окисленный катализатор Li/MgO водорода наблюдалось его поглощение и выделение тепла, но вода, как и в случае экспериментов с метаном и этаном, образовывалась только при подаче импульсов кислорода [18, 19]. Только при длительной выдержке восстановленного образца в токе инертного газа при 700°С удавалось зафиксировать частичное реокисление импульсами N2O с выделением молекулярного азота.
Полученные данные позволили определить величины термохимических параметров ряда модельных катализаторов окисления – величину энергии связи кислорода с решеткой (E[O]) и прочности O–H-связи в поверхностной OH-группе, образующейся в реакции (5) (DO–H).
Была предложена [19] альтернативная схема реокисления активных центров, которая реализуется без образования кислородных вакансий. Она включает в себя четыре стадии с участием свободных радикалов:
(VII)
$\left[ {{\text{OH}}} \right] + {{{\text{O}}}_{{\text{2}}}} \to \left[ {\text{O}} \right] + {\text{HO}}_{2}^{\centerdot },$(VIII)
$\left[ {{\text{OH}}} \right] + {\text{ HO}}_{2}^{\centerdot } \to \left[ {\text{O}} \right] + {{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{2}}}},$(IX)
${{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{2}}}} \to {\text{2O}}{{{\text{H}}}^{\centerdot }},$(X)
$\left[ {{\text{OH}}} \right] + {\text{O}}{{{\text{H}}}^{\centerdot }} \to \left[ {\text{O}} \right] + {{{\text{H}}}_{{\text{2}}}}{\text{O}}.$Суммарно в результате реакции одной молекулы кислорода с четырьмя поверхностными OH-группами образуется две молекулы воды и регенерируется четыре активных центра в окисленном состоянии.
Кинетические параметры стадий (VII), (VIII) и (X) были определены в рамках предложенной автором модели протекания гетерогенно-гомогенных процессов окисления с участием оксидных катализаторов [20–22], в основу которой положено предположение об аналогии между гетерогенными реакциями свободных радикалов и их реакциями тех же типов в газовой фазе. Согласно этой модели, величины энергий активации процессов с участием активных центров определяются двумя термохимическими параметрами – E[O] и DO–H.
При типичных температурах процесса ОКМ реакция гомогенного распада перекиси водорода (IX) идет достаточно быстро. Из трех реакций с участием активных центров катализатора наиболее медленной является реакция (VII), поскольку радикалы ${\text{HO}}_{2}^{\centerdot }$ и, тем более, OH• имеют гораздо более высокое сродство к атому водорода, чем молекула кислорода. Соответственно, при близких величинах предэкспоненциальных множителей константы скоростей реакций (VIII) и (X) должны быть намного выше, чем для реакции (VII), которая, таким образом, будет первой и лимитирующей в цепочке превращений молекул кислорода и образования воды, необходимых для “замыкания” каталитического цикла. С этим, очевидно, связано то, что экспериментально наблюдаемые величины порядка реакции по кислороду (n) в уравнении типа Марса–ван-Кревелена для катализаторов ОКМ всегда равны 1 [16].
Важно подчеркнуть, что механизмы реокисления по “традиционной” схеме (через предварительное дегидроксилирование и присоединение кислорода к кислородным вакансиям) и по реакциям (VII)–(X) (условно названной схемой окислительного дегидрирования OH-групп) реализуются параллельно. Вклад каждого из них в суммарное реокисление определяется термохимией активных центров (в основном – параметром E[O]) и условиями реакции – температурой и парциальным давлением воды в реакционной смеси. Чем выше энергия связи кислорода, тем меньше вклад “традиционного” маршрута через стадии (V) и (II). Однако даже при достаточно низких величинах E[O] вклад этого маршрута снижается с ростом концентрации воды в реакционной среде. Качественное представление о том, как термохимические параметры влияют на соотношение между маршрутами реокисления, дает табл. 1.
Таблица 1.
Термохимические параметры активных центров модельных катализаторов ОКМ и возможность осуществления их реокисления по маршрутам “дегидроксилирования” (маршрут A) и “окислительного дегидрирования” (маршрут B) в режиме попеременной подачи восстановителя (метан, водород) и кислорода (по данным [5])
| Параметр или маршрут реокисления |
Катализатор | ||
|---|---|---|---|
| Li/MgO | K/Al2O3 | Pb/Al2O3 | |
| E[O], кДж/моль | 535 | ~450 | 407 |
| DO–H, кДж/моль | 320 | 270 | 250 |
| Маршрут A | (–) | (+/–) | (+) |
| Маршрут B | (+) | (+/–) | (–) |
В качестве активных центров катализаторов ОКМ и родственных процессов могут выступать различные формы кислорода. Одним из первых типов центров такого рода, для которых были доказаны их присутствие в оксидных системах и участие в активации молекул алканов, были ион-радикальные центры [O–], которые образуются, например, при рентгеновском или γ-облучении MgO или при изоморфном замещении ионов Mg2+ ионами Li+ в решетке MgO (см., например, [23–25]). В системах, содержащих крупные ионы щелочных, щелочноземельных и, возможно, редкоземельных элементов, активными центрами могут быть пероксид-ионы ${\text{O}}_{2}^{{2 - }}$ [26–28]. Не исключено также участие в катализе ионов кислорода с пониженной координацией, которые также могут обладать высоким сродством к атому водорода и участвовать в реакции типа (V) в сильно разупорядоченных оксидах [29, 30], принадлежащих к группе “невосстанавливаемых”.
Таким образом, за наблюдаемой кинетикой, формально соответствующей уравнениям типа (1), может стоять последовательность процессов, которые, оставаясь окислительно-восстановительными по сути, протекают через промежуточные частицы и состояния активных центров, кардинально отличные от присутствующих в “классической” схеме Марса–ван-Кревелена.
ХИМИЧЕСКИЕ И ФАЗОВЫЕ ПРЕВРАЩЕНИЯ В СИСТЕМЕ NaWMn/SiO2
Как отмечалось выше, имеется одна группа катализаторов ОКМ и родственных процессов, в состав которых входят ионы переходного металла, а именно – марганец-содержащие (и родственные им Pr- и Ce-содержащие) системы. Интересно отметить, что первое поколение катализаторов этого типа предлагалось использовать, в том числе, в качестве переносчиков кислорода, т.е. для осуществления процесса ОКМ в попеременном окислительно-восстановительном режиме [31, 32]. Это можно считать однозначным указанием на участие в активации метана анионов кислорода, входящих в состав оксидных соединений марганца. При этом энергия связи кислорода в этих соединениях оптимальна. С одной стороны, переходы между Mn(2+) и состояниями с более высокими степенями окисления позволяют им участвовать в окислительно-восстановительных процессах при типичных температурах ОКМ и родственных реакций (рис. 1). С другой стороны, по обсуждавшимся выше критериям MnO однозначно является невосстанавливаемым оксидом (в отличие, например, от FeO, CoO, NiO), и даже при максимально жестком восстановлении оксидной системы в ней не появляется металлического компонента, что неизбежно привело бы к резкому падению селективности по целевым продуктам (олефинам). Вскоре было показано, что Pr-содержащие системы проявляют аналогичные свойства [33]. Попутно отметим, что также как в случае оксидов марганца существует предельная степень восстановления оксидов празеодима до Pr2O3.
К группе нанесенных Mn-содержащих оксидных систем относится и наиболее эффективный из известных катализатор ОКМ – NaWMn/SiO2, впервые предложенный в работах сотрудников Института химической физики (г. Ланьчжоу, КНР) [34]. Эта система интенсивно изучалась многими исследователями – как с точки зрения поиска возможности дальнейшего повышения ее эффективности и долговечности, так и с позиций раскрытия механизма ее каталитического действия [35]. В состав этой системы помимо марганца (возможна его замена на церий без потери эффективности) входит еще один элемент, образующий оксидные соединения в разных степенях окисления, – вольфрам. По этой причине в литературе высказывались различные мнения о том, какой именно кислород – принадлежащий связям Mn–O, W–O или смешанным структурам типа Mn–O–W – принимает непосредственное участие в каталитическом процессе.
Было также показано, что для достижения высокой эффективности катализаторы этого типа должны непременно иметь в своем составе марганец (или церий) и вольфрам, замена которого на родственные элементы (например, молибден или ванадий) приводит к полной потере селективности по олефинам. Справедливости ради следует отметить, что допустимой заменой вольфрама могут быть некоторые другие анионобразующие элементы, например, фосфор: как уже указывалось, открытию системы NaWMn/SiO2 предшествовала серия работ, в которых была показана эффективность фосфатсодержащих систем (см., например, [32]). Однако и в этих случаях – как и в вольфраматсодержащих системах – присутствие щелочного (возможно – щелочноземельного или редкоземельного) металла является непременным условием высокой активности и селективности катализатора [31–33, 36, 37].
В наших недавних работах было показано, что:
– системы NaWMn/SiO2 содержат два типа активного кислорода, способного вступать во взаимодействие с молекулами алканов и обратимо удаляться из оксида; они обе ведут процесс ОКМ в режиме попеременного восстановления–реокисления, но существенно отличаются по реакционной способности и времени жизни: относительно короткоживущая форма гораздо более активна и несколько менее селективна в отношении образования продуктов ОКМ при восстановлении метаном [38];
– энергия связи относительно короткоживущей формы кислорода с оксидной матрицей позволяет однозначно отнести ее обратимое удаление из системы к окислительно-восстановительному переходу между ионами Mn(3+) и Mn(2+) [39].
Было также показано, что время жизни этой формы кислорода существенно превышает характерное время стационарной каталитической реакции и что с большой вероятностью именно она ответственна за протекание каталитического процесса. Вторая, существенно более сильно связанная форма кислорода, удаление которой из системы приводит к восстановлению вольфрама, в стационарном каталитическом процессе не участвует, т.е. в ходе него вольфрам неизменно остается в высшей степени окисления в виде вольфрамат-ионов.
Функции, которые в системе выполняют вольфрам, не претерпевающий в ходе катализа оксилительно-восстановительных превращений, а также ионы щелочных металлов, долгое время оставались невыясненными. Неясна была причина, по которой именно оксид кремния является оптимальным носителем для смешанного NaWMn-оксидного катализатора ОКМ – применение других носителей с близкими параметрами, например, α-Al2O3, приводит к снижению как активности, так и селективности. Также имелось противоречие между достаточно высокой температурой выделения из системы в режиме термодесорбции кислорода, связанного с марганцем (не ниже 600°С), и весьма слабой зависимостью скорости его выделения от температуры выше некого порогового ее значения.
Ответ на эти вопросы оказался в значительной мере единым. Он был получен с использованием комплекса физико-химических методов и кинетического анализа [40, 41]. Исходным для этого оказался факт, установленный методом рентгеновской фотоэлектронной спектроскопии ex situ: было обнаружено резкое увеличение наблюдаемых поверхностных концентраций ионов нанесенных компонентов в системе после катализа по сравнению со свежеприготовленным образцом катализатора. Аналогичные изменения наблюдаются при прогреве катализатора в вакууме или в потоке инертного газа до типичных температур катализа (>800°С), в ходе которого происходит выделение (термодесорбция) кислорода – той его формы, которая выше охарактеризована как менее долгоживущая и принимающая участие в каталитическом окислительно-восстановительном цикле. В наибольшей мере возрастает наблюдаемая концентрация марганца – элемента, который претерпевает окислительно-восстановительные превращения. Такое изменение однозначно указывало на то, что окислительно-восстановительный переход связан с редиспергированием нанесенных компонентов. Выяснилось также, что в свежеприготовленном катализаторе присутствуют фазы вольфрамата натрия и оксида марганца (Mn2O3). Эти же фазы обнаружены в образце, который был резко охлажден до комнатной температуры в ходе стационарной реакции ОКМ при наличии остаточного кислорода в реакционной смеси. Если же реакция велась при почти полной конверсии кислорода, либо образец был прогрет в инертном газе, то в нем методом рентгенофазового анализа (РФА) фиксируется лишь наличие фазы вольфрамата марганца MnWO4. Ключевыми оказались результаты исследования образцов, подвергнутых различным предварительным обработкам, методом сканирующей электронной микроскопии с элементным анализом: наряду с кристаллической фазой MnWO4 в восстановленной системе была обнаружена аморфная фаза, в состав которой входят натрий и марганец (рис. 2б). Эта фаза была условно названа “клееподобной”, поскольку в охлажденном образце она обволакивает сплошным слоем глобулы носителя (SiO2 в кристаллической модификации кристобалит). После прогрева восстановленного образца в кислороде в нем вновь возникают фазы Na2WO4 и Mn2O3, однако по сравнению со свежеприготовленным катализатором (рис. 2а) их дисперсность изменяется: наряду с относительно крупными кристаллами (~1 мкм) появляются многочисленные кристаллы гораздо меньшего размера. Они оказываются локализованными в областях, где в восстановленном образце присутствовала “клееподобная” аморфная фаза – в основном на границах глобул кристобалита (рис. 2в).
Рис. 2.
Микрофотографии образца NaWMn/SiO2, полученные методом электронной сканирующей микроскопии [40]: свежеприготовленный (а); после термодесорбции кислорода (б); после реокисленния (в).

Наконец, исследование системы методом высокотемпературного РФА показало, что выделению кислорода из системы NaWMn/SiO2 при термодесорбции предшествует постепенное снижение интенсивности и полное исчезновение рефлексов, соответствующих фазе Na2WO4, происходящее на 50–100°С ниже стандартной температуры плавления (698°С) [41]. Этот эффект связан с достаточно высокой адгезией расплава вольфрамата натрия к поверхности кристобалита; в результате понижения энтальпии плавления на величину энергии взаимодействия между ними происходит снижение температуры плавления, поскольку
Полученные данные позволяют описать происходящие в системе процессы следующим образом. При типичных температурах ОКМ (>700°С) происходит плавление фазы Na2WO4 и создаются условия протекания процесса
(XI)
$\begin{gathered} {\text{M}}{{{\text{n}}}_{{\text{2}}}}{{{\text{O}}}_{3}} + 4{\text{N}}{{{\text{a}}}^{ + }} + 2{\text{WO}}_{4}^{{2 - }} \rightleftarrows 2{\text{MnW}}{{{\text{O}}}_{{\text{4}}}} + \\ + \,\,\left\{ {2{\text{N}}{{{\text{a}}}_{{\text{2}}}}{\text{O}}} \right\} + 0.5{{{\text{O}}}_{2}}. \\ \end{gathered} $Здесь формулой {Na2O} условно обозначен оксид натрия, высвобождающийся при связывании вольфрамат-аниона с ионами Mn(2+) в кристаллическом MnWO4. Щелочной оксид должен иметь высокую адгезию к поверхности SiO2, с чем, очевидно, связано образование им сплошной аморфной (возможно – жидкой при температурах катализа) пленки на поверхности глобул кристобалита, в которой присутствуют также ионы марганца, не связанные в вольфрамат. По-видимому, сама по себе скорость реакции (XI) слабо зависит от температуры, но условием ее протекания является наличие расплава Na2WO4, чем может объясняться низкая энергия активации термодесорбции кислорода при наличии температурного порога.
Присутствие в системе жидкой фазы солевого (оксидного) расплава должно повышать скорость химических превращений, в т.ч. окислительно-восстановительного типа, поскольку все компоненты системы в этом случае гомогенно перемешаны и скорости переноса (например, поверхностной и объемной диффузии в кристаллах) не оказывают влияния на суммарную скорость процесса. Роль отдельных компонентов нанесенной фазы также становится понятной: марганец обеспечивает функционирование оптимальной редокс-пары, вольфрамат натрия – солевого расплава с оптимальными свойствами как в окисленном, так и в восстановленном состоянии системы. По сравнению с другими солями натрия, плавящимися в этом же температурном интервале, вольфрамат более устойчив – как термически, так и с точки зрения возможных окислительно-восстановительных превращений. Так, при замене вольфрамата натрия на ванадат или молибдат из-за более низкой энергии связи кислорода с оксидами ванадия и молибдена полученные катализаторы имеют гораздо более низкую селективность в процессе ОКМ ввиду протекания побочных процессов глубокого окисления.
Интересно и важно отметить, что в системе NaWMn/α-Al2O3 эффекта снижения температуры плавления Na2WO4 не наблюдается [41]. По-видимому, в этом случае из-за более слабого взаимодействия между носителем и вольфраматом натрия не создается условий для образования пленки его расплава и всех физико-химических процессов, обусловленных ее появлением.
В связи со сказанным важно отметить, что в катализаторах, не содержащих вольфрамата, отмечено образование свободного оксида MnO [37], а также связывание марганца в силикаты и алюминаты [31, 32], что может приводить к затруднению стадии реокисления и выводить часть марганца из участия в окислительно-восстановительном цикле.
Возвращаясь к кинетическому описанию стационарного каталитического процесса, мы должны констатировать следующее: его простота в данном случае является кажущейся, т.е. соответствие экспериментальных данных уравнению Марса–ван-Кревелена отражает лишь тот факт, что протекание многочисленных химических и фазовых превращений в системе оптимизировано. В результате стационарная скорость катализа не столь сильно зависит от их скоростей, чтобы это могло отразиться на форме кинетического уравнения (1), хотя, совершенно очевидно, его параметры каким-то сложным образом зависят от скоростей этих “скрытых” превращений.
Однако еще большее влияние на величины параметров уравнения (1) должны оказывать факторы, кратко рассмотренные в следующем разделе. Здесь же еще раз отметим следующее: концепция “решеточного” кислорода и кислородных вакансий, с которой часто связывают кинетическую модель Марса–ван-Кревелена, в случае системы NaWMn/SiO2 не отражает существа процесса, хотя обратимое окисление-восстановление одного из компонентов каталитической системы составляет важную часть каталитического цикла.
СВОБОДНОРАДИКАЛЬНЫЕ ПРОЦЕССЫ
Выше уже говорилось о роли свободных радикалов в каталитических процессах окисления легких алканов. Эта тема является весьма обширной и требует отдельного детального рассмотрения (см., например, [22, 43, 43 ]). Здесь же кратко отметим только тот ее аспект, который связан с кинетическим описанием стационарной каталитической реакции.
После образования первичных радикалов – например, по реакции (V) в случае метана – они претерпевают многочисленные последовательные превращения, приводящие к образованию конечных продуктов. Эти превращения протекают как в газовой фазе, так и с участием активных центров катализатора – в какой бы форме они не существовали. В этих промежуточных превращениях образуются вторичные радикалы, которые являются зачастую гораздо более активными восстановителями (алкильные радикалы, атомы водорода) и окислителями (гидроксильные, алкоксильные, пероксидные радикалы, атомы кислорода), чем исходные молекулы алканов и кислорода соответственно. Все они могут участвовать, и, безусловно, участвуют как в активации исходных реагентов, так и в процессах восстановления и реокисления активных центров катализатора. Соответственно, представление измеряемой в эксперименте скорости реакции в виде зависимости исключительно от концентраций исходных реагентов не может в явном виде отражать всей совокупности этих процессов.
Что же скрывается тогда за столь относительно простой формой описания? Ответ на этот вопрос, по-видимому, состоит в том, что все стадии активации исходной молекулы алкана протекают однотипно – через гомолитический отрыв атома водорода:
Выше был рассмотрен относительно простой случай, когда процессы с участием радикалов OH• и ${\text{HO}}_{2}^{\centerdot }$ определяют скорость стадии реоксиления активных центров. В условиях реального каталитического процесса вклад свободнорадикальных стадий в активацию реагентов и “оборот” активных центров зависит от многих факторов – состава реакционной смеси, температуры, активности катализатора. При этом приписать определенный физический смысл величинам параметров уравнения (1), получаемым из кинетического эксперимента, зачастую не представляется возможным.
Косвенным образом относительный вклад разных активных кислородсодержащих частиц в суммарный стационарный каталитический процесс можно оценить путем сравнения кинетических параметров уравнения (1) и аналогичных ему, определяемых по данным экспериментов по окислению разных углеводородов – например, метана и этана. Действительно, если вклад гетерогенного окисления является превалирующим, эмпирические значения величин предэкспоненциального множителя и энергии активации для стадии реокисления, полученные из двух серий экспериментов, должны совпадать. Такое наблюдается (с приемлемой точностью), например, для катализатора Pb/Al2O3 [17, 44]. Однако при исследовании кинетики окисления этана (в отличие от метана) в присутствии La/MgO и NaWMn/SiO2 в диапазоне температур 680–800°С формально определяемая по (1) скорость реокисления была столь велика, что ее кинетические параметры не удалось установить [45].
Как показывают результаты моделирования [46], при окислении этана вклад гомогенных процессов в суммарную кинетику определяется более высокой скоростью разветвления и развития цепей. В связи с этим при относительно низкой скорости гетерогенной реакции (V) и аналогичной ей с участием этана относительный вклад гомогенных процессов может становиться основным, что влечет за собой, в том числе, несовпадение величин кинетических параметров стадии реокисления, определенных из серий экспериментов по окислению метана и этана.
ЗАКЛЮЧЕНИЕ
Главной целью при написании этой статьи было – далеко не в первый раз! – обратить внимание на сложность и неоднозначность соотношения между наблюдаемыми кинетическими закономерностями и механизмом химического превращения, что, безусловно, имеет место не только в гетерогенном окислительном катализе. Наглядный пример такого рода дает группа процессов высокотемпературного окисления метана и его ближайших гомологов в присутствии оксидных катализаторов. Рассмотрение детальных механизмов эволюции окислительных активных центров катализаторов в этих процессах показывает, что за относительно простыми кинетическими закономерностями, наблюдаемыми в стационарном кинетическом эксперименте, скрываются весьма сложные явления. В частности, формальное соответствие экспериментальных данных простым уравнениям, вытекающим из окислительно-восстановительной стадийной схемы Марса–ван-Кревелена и аналогичных ей, не может служить однозначным указанием на то, что процесс протекает через образование кислородных вакансий в поверхностном слое оксидного катализатора и их восполнение при реакции с молекулой окислителя (например, O2).
Тем не менее, определяемые из эксперимента величины параметров уравнения типа уравнения Марса–ван-Кревелена в сочетании с данными о химических и структурных превращениях в работающем катализаторе, а также с результатами моделирования кинетики гетерогенно-гомогенных процессов с участием свободных радикалов дают дополнительную важную информацию о факторах, определяющих эффективность сложных каталитических систем.
БЛАГОДАРНОСТИ
В течение долгого времени взгляды на механизм окисления метана и его ближайших гомологов, изложенные в этой работе, были предметом обсуждения автора с проф. О.В. Крыловым и проф. Л.Я. Марголис. Однако значительная часть представленных конкретных результатов и основанные на них выводы получены в недавнее время и уже не могли войти в наши совместные публикации. С большим теплом и благодарностью автор вспоминает акад. А.Б. Налбандяна и д. х. н. Т.А. Гарибян – пионеров в области экспериментального изучения роли реакций свободных радикалов в гетерогенном катализе – и работу с ними и их сотрудниками в Институте химической физики в Ереване. На этапе подготовки рукописи этой статьи весьма плодотворным было обсуждение ее положений с проф. Д.Ю. Мурзиным (Университет Або Академи, г. Турку, Финляндия), которому автор выражает глубокую признательность.
Список литературы
Темкин М., Пыжев В. // Журн. физ. химии. 1939. Т. 13. С. 851.
Mars P., van Krevelen D.W. // Chem. Eng. Sci. 1954. V. 3. P. 41.
Темкин М.И. // ДАН СССР. 1965. Т. 161. С. 160.
Vannice M.A. // Catal. Today. 2007. V. 123. P. 18.
Темкин О.Н. Гомогенный металлокомплексный катализ: кинетические аспекты. М.: Академкнига, 2008. 918 с.
Боресков Г.К. Гетерогенный катализ. М.: Наука, 1986. 304 с.
Синев М.Ю., Бычков В.Ю. // Кинетика и катализ. 1999. Т. 40. С. 906.
Боресков Г.К., Веньяминов С.А., Сазонова Н.Н., Панкратьев Ю.Д., Питаева А.Н. // Кинетика и катализ. 1975. Т. 15. С. 1442.
Sakita K., Nakamura T., Misono M., Yoneda Y. // Chem. Lett. 1979. P. 273.
Уэда В., Мороока Й., Икава Ц. // Сёкубай. 1980. V. 22. P. 211.
Мороока Й. // Хёмэн. 1981. V. 19. P. 421.
Gleaves J.T., Ebner J.R., Kuechler T.C. // Catal. Rev. Sci. Eng. 1988. V. 30. P. 49.
Morgan K., Maguire N., Fushimi R., Gleaves J.T., Goguet A., Harold M.P., Kondratenko E.V., Menon U., Schuurman Y., Yablonsky G.S. // Catal. Sci. Technol. 2017. V. 7. P. 2416.
Patience G.S., Bockrath R.E. // Appl. Catal. A: General. 2010. V. 376. P. 4.
Sinev M.Yu. // J. Catal. 2003. V. 216. P. 468.
Ломоносов В.И., Синев М.Ю. // Кинетика и катализ. 2016. Т. 57. C. 647.
Синев М.Ю., Корчак В.Н., Крылов О.В. // Кинетика и катализ. 1989. V. 30. P. 860.
Sinev M.Yu., Bychkov V.Yu., Korchak V.N., Krylov O.V. // Catal. Today. 1990. V. 6. P. 543.
Синев М.Ю., Бычков В.Ю. // Кинетика и катализ. 1993. Т. 34. С. 309.
Sinev M.Yu. // Catal. Today. 1992. V. 13. P. 561.
Sinev M.Yu. // Catal. Today. 1995. V. 24. P. 389.
Sinev M.Yu. // Russ. J. Phys. Chem. B. 2007. V. 1. P. 329.
Aika K.-I., Lunsford J.H. // J. Phys. Chem. 1978. V. 82. P. 1794.
Takita Y., Lunsford J.H. // J. Phys. Chem. 1979. V. 83. P. 683.
Wang J.X., Lunsford J.H. // J. Phys. Chem. 1986. V. 90. P. 3890.
Синев М.Ю., Корчак В.Н., Крылов О.В. // Кинетика и катализ. 1986. Т. 27. С. 1274.
Otsuka K., Murakami Y., Wada Y., Said A.A., Morikawa A. // J. Catal. 1990. V. 121. P. 122.
Mestl G., Knozinger H., Lunsford J.H. // Berichte der Bunsengesellschaft für Physikalische Chemie. 1993. V. 97. P. 319.
Burrows A., Kiely Ch.J., Hutchings G.J., Joyner R.W., Sinev M.Yu. // J. Catal. 1997. V. 167. P. 77.
Burrows A., Kiely Ch.J., Hargreaves J.S.J., Joyner R.W., Hutchings G.J., Sinev M.Yu., Tulenin Yu.P. // J. Catal. 1998. V. 173. P. 383.
Sofranko J.A., Leonard J.J., Jones C.A. // J. Catal. 1987. V. 103. P. 302.
Jones C.A., Leonard J.J., Sofranko J.A. // J. Catal. 1987. V. 103. P. 311.
Gaffney A.M., Jones C.A., Leonard J.J., Sofranko J.A. // J. Catal. 1988. V. 114. P. 422.
Fang X.P., Li S.B., Lin J.Z., Chu Y.L. // China J. Molec. Catal. 1992. V. 6. P. 255.
Arndt S., Otremba T., Simon U., Yildiz M., Schubert H., Schomäcker R. // Appl. Catal. A: General. 2012. V. 425–426. P. 53.
Minachev Kh.M., Usachev N.Ya., Belanova E.P., Shirokova G.A. // Catal. Today. 1992. V. 13. P. 565.
Крылов О.В., Мамедов А.Х. // Успехи химии. 1995. Т. 64. С. 935.
Gordienko Y., Usmanov T., Bychkov V., Lomonosov V., Fattakhova Z., Tulenin Y., Shashkin D., Sinev M. // Catal. Today. 2016. V. 278. P. 127.
Ломоносов В.И., Гордиенко Ю.А., Синев М.Ю., Рогов В.А., Садыков В.А. // Журн. физ. химии. 2018. Т. 92. С. 386.
Sinev M., Ponomareva E., Sinev I., Lomonosov V., Gordienko Yu., Fattakhova Z., Shashkin D. // Catal. Today. 2019. DOI: 10.1016/j.cattod.2018.06.028.
Гордиенко Ю.А., Ломоносов В.И., Пономарева Е.А., Синев М.Ю., Бухтияров А.В., Винокуров З.С. // Журн. физ. химии. 2019. Т. 93. № 3. С. 347.
Синев М.Ю., Марголис Л.Я., Корчак В.Н. // Успехи химии. 1995. Т. 64. С. 373.
Sinev M., Arutyunov V., Romanets A. // Adv. Chem. Eng. 2007. V. 32. P. 167.
Синев М.Ю., Корчак В.Н., Крылов О.В. // Кинетика и катализ. 1989. Т. 30. С. 855.
Ломоносов В.И., Гордиенко Ю.А., Синев М.Ю. // Кинетика и катализ. 2013. Т. 54. С. 474.
Lomonosov, V., Gordienko Yu., Ponomareva E., Sinev M. // Chem. Eng. J. 2019. DOI: 370. 10.1016/j.cej.2019.04.006.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ