

Кинетика и катализ, 2019, T. 60, № 4, стр. 428-439
Развитие идей М.И. Темкина в физической химии
a ФГБУН Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский просп., 31, Россия
* E-mail: tovbinyk@mail.ru
Поступила в редакцию 26.02.2019
После доработки 26.02.2019
Принята к публикации 26.02.2019
Аннотация
Отмечены приоритетные работы М.И. Темкина в области адсорбции, теории элементарных стадий поверхностных процессов и общих вопросов описания конденсированных фаз (на примере процессов на поверхностях в плотных монослоях и для объемной фазы). Кратко рассмотрено последующее развитие его идей в указанных областях. Обсуждены вопросы корректного способа учета кооперативного поведения адсорбированных частиц в равновесии и поверхностных реакциях и принцип самосогласованного описания скоростей стадий и равновесного состояния реагентов для неоднородных поверхностей и неидеальных реакционных систем.
Время начала научной деятельности М.И. Темкина (окончил химфак МГУ в 1932 г.) совпало с периодом становления современной физической химии и химической кинетики. Сформулированные в это время законы квантовой теории позволили по-новому осмыслить статистическую теорию Гиббса, которая стала формироваться в приложениях для физической химии как статистическая термодинамика.
Традиционно с именем Темкина М.И. связывают следующие 4 уравнения: логарифмическая изотерма Темкина [1, 2], уравнение кинетики синтеза аммиака Темкина–Пыжева [3], модель кинетики на неоднородной поверхности Темкина [4] и модель расплава солей сильных электролитов Темкина [5, 6].
Можно выделить следующие направления его работ: статистическая термодинамика идеальных систем, теория абсолютных скоростей реакций на однородных и неоднородных поверхностях, учет неидеальности реакционных систем в приближении среднего поля на однородных поверхностях (включая вопросы самосогласованности описания скоростей кинетических процессов и равновесия в неидеальных и неоднородных системах), теория ионных расплавов, работы по развитию теории кинетики гетерогенных каталитических реакций (включая многостадийные процессы при лимитирующей стадии адсорбции), кинетика сложных многостадийных каталитических процессов и исследование роли внешнего и внутреннего массо- и теплопереноса в гетерогенном катализе.
Из общего числа работ в указанных направлениях обсуждаются основные идеи и пути развития моделей физической химии: адсорбции и растворов (раздел 1) и скоростей элементарных стадий (раздел 2), а также общие принципы описания конденсированных систем (раздел 3).
При этом основное внимание уделяется самим идеям, а не содержанию отдельных работ. Суть этих идей была воплощена в моделях поверхностных процессов, а также для объемных фаз, которые расширили учет физических свойств реагентов, отражаемых в математической записи этих моделей.
1. РАВНОВЕСНЫЕ СИСТЕМЫ
Первой обсуждаемой темой являются равновесные свойства систем. Адсорбционные системы Темкин М.И. рассматривал как аналоги обычных объемных растворов, имеющих специфику, обусловленную присутствием поверхности. Для смесей адсорбированных веществ, как и в объеме, было предложено использовать единую терминологию – в частности идеальный адсорбционный раствор [7]. Такую же общность понятий постоянно развивал Темкин М.И. в химической кинетике (а Хилл Т. – в термодинамике адсорбции на основе представлении об инертном адсорбенте [8]). В работах этого направления общим является использование подходов статистической термодинамики невзаимодействующих частиц для равновесных идеальных систем.
1.1. Обоснование изотермы Лэнгмюра
Здесь следует подчеркнуть, что Темкин М.И. впервые дал статистическое обоснование изотермы Лэнгмюра [9]: aP = θ/(1 – θ), где θ = N/M – степень заполнения поверхности, N – число адсорбированных частиц, занимающих один центр адсорбции, М – число таких центров на единице поверхности, а – коэффициент адсорбции или коэффициент пропорциональности между давлением и степенью заполнения поверхности при малых заполнениях, называемый коэффициентом Генри (в уравнении aP = θ) или Лэнгмюра.
Изотерма Лэнгмюра была получена кинетическим методом путем приравнивания скоростей стадий адсорбции и десорбции [10]. Получение изотермы Лэнгмюра чисто равновесным статистическим методом показало взаимосвязь между константами скоростей адсорбции–десорбции и константой равновесия. Вывод изотермы Лэнгмюра из законов статистического распределения частиц между поверхностью и объемной фазой пара подчеркнул идею о независимости пути процесса для достижения его равновесия. Этот факт сразу поставил вопрос о принципиальном значении самосогласованного описания свойств изучаемой системы исходя из кинетического и строго статистического подхода. Сам Темкин М.И. во всех своих последующих работах последовательно придерживался этой идеи.
Традиционно приоритет статистического обоснования изотермы Лэнгмюра приписывается Фаулеру [11, 12], который является одним из основателей статистической термодинамики [13], но работа Темкина была опубликована на 2 года раньше.
1.2. Учет размера компонентов адсорбционной системы
Близкий круг идей был связан с необходимостью учета размера компонентов адсорбционной системы. В изотерме Ленгмюра каждая частица занимает на поверхности один адсорбционный центр (адцентр). Очевидно, что различные молекулы будут по-разному адсорбироваться на одной и той же поверхности. И, в первую очередь, при этом будет влиять размер молекулы по отношению к размеру адцентра, число которых на единице поверхности зафиксировано. Темкин М.И. впервые вывел изотерму адсорбции димерных молекул на одномерной структуре центров [14]. Димерная молекула занимает два соседних центра, что определяет степень заполнения поверхности как θ = 2N/M, а сама изотерма имеет вид $aP = \frac{{{\text{(2}} - {\theta )\theta }}}{{{\text{4(1}} - {\theta }{{{\text{)}}}^{{\text{2}}}}}}.$ По-видимому, данная изотерма появилась под влиянием работы [15] для растворов в двух- и трехмерных системах, но для адсорбции она явилась своего рода толчком, после чего данное направление по адсорбции многоцентровых частиц стало быстро развиваться.
В то же самое время появились работы [16, 17], а позже работы [18–20] для структур различной размерности, в том числе и для одномерных идеальных [21] и неидеальных адсорбционных систем [22, 23]. Позднее активный интерес был проявлен к более реальным двумерным адсорбционным системам [24–27]. Учет многоцентровости адсорбированных молекул с учетом латеральных взаимодействий был распространен и на кинетические процессы адсорбции–десорбции одномерных (d = 1 [28]) и двумерных систем на открытой поверхности и в порах [29–31].
Одновременно стало ясным, что при статистическом описании больших молекул можно учесть различные ориентации адсорбата. Каждая ориентация молекулы характеризуется своим значением энергии связи с поверхностью, и она может рассматриваться как сорт некоторой частицы, поэтому учет различных ориентаций молекул даже для однокомпонентной системы сводится к теории адсорбции смеси частиц различного размера. Наиболее наглядно роль различных ориентаций молекул иллюстрируется для молекулы в форме “жесткого стержня”. Помимо горизонтального и вертикального положений стержня следует различать различные ориентации адсорбата в плоскости поверхности. Причем по мере увеличения плотности адсорбата более выгодным становится упорядоченное расположение молекул, имеющих одинаковые вертикальные ориентации их длинных осей [32]. Проблема же расчета адсорбции больших молекул (блокирующих более одного адцентра) до настоящего времени является одной из наиболее сложных задач статистической термодинамики.
1.3. Изотермы для неоднородной поверхности
Очень важное направление работ в середине 1930-х гг. было связано с работами по обобщению изотермы Лэнгмюра для однородной поверхности на случай адсорбции на неоднородных поверхностях. Сам Лэнгмюр построил изотерму при рассмотрении адсорбционного равновесия на неоднородных поверхностях [33], используя ненормированные вклады различных участков. Для “кристаллической” поверхности с небольшим числом разных групп участков и “аморфных” поверхностей с большим числом различных участков, характеризующихся непрерывным изменением адсорбционной способности, он выразил степень покрытия как
(1)
${\theta } = \sum\limits_{q = 1}^t {\frac{{{{a}_{q}}P}}{{1 + {{a}_{q}}P}}} \,\,\,\,{\text{и }}\,\,\,\,{\theta } = \int {\frac{{{{a}_{q}}P}}{{1 + {{a}_{q}}P}}} {\text{d}}s,$Величина s связана с q-типом центра через нормированную функцию распределения fq, которая характеризует долю поверхности, занятую узлами q-типа, т.е. ds = fq dq, или ds = f(Q)dQ, если задается правило, определяющее связь между энергией связи адсорбата Q с поверхностью на центре q-типа. Тогда интегральное выражение (1) можно переписать как
(2)
${\theta } = \int\limits_{{{Q}_{{\max }}}}^{{{Q}_{{\min }}}} {f(Q)\frac{{{{a}_{Q}}P}}{{1 + {{a}_{Q}}P}}} {\text{d}}Q.$Уравнение (2) лежит в основе статистики всех процессов на неоднородных поверхностях в отсутствие учета латеральных взаимодействий. В этой связи одной из первых работ по использованию нормированной равномерной функции распределения по энергиям $f(Q)$ для адцентров была работа Темкина в 1934 г. Эти уравнения адсорбции – квазилогарифмическая ${\theta } = \frac{1}{f}\ln \frac{{1 + {{a}_{1}}P}}{{1 + {{a}_{0}}P}}$ для полного диапазона степеней заполнений поверхности и логарифмическая ${\theta } = \frac{1}{f}\ln ({{a}_{1}}P)$ для средних областей заполнений (a1Р $ \gg $ 1 и а0Р $ \ll $ 1) – появились в качестве сноски к статье [34]. Здесь a0 и a1 – коэффициенты адсорбции для наиболее слабосвязанных и сильносвязанных узлов соответственно, $f = {{({{Q}_{{\max }}} - {{Q}_{{\min }}})} \mathord{\left/ {\vphantom {{({{Q}_{{\max }}} - {{Q}_{{\min }}})} {RT}}} \right. \kern-0em} {RT}}$ – параметр линейной функции распределения центров по энергиям. Развернутый вывод изотерм был дан в виде авторской “вставки” внутри статьи [35]. Сам же Темкин М.И. опубликовал свою изотерму в статье под своей фамилией значительно позже [1, 2] (работа [2], по сути, была его докторской диссертацией).
Описание адсорбции с помощью интегральных функций распределения различных типов центров (уравнение (2)) стало самым распространенным подходом в те годы. В качестве примера укажем на опубликованную одновременно работу [36], в которой такое же интегральное описание было использовано для нахождения функции распределения адцентров по энергии, которая отвечает изотерме Френдлиха aP = θn. Возможности использования интегрального описания для объяснения экспериментальных изотерм анализировались также в работе [37].
Для каталитических процессов особенно важна возможность изучения реакций на поверхностях с заданными свойствами. Моделирование структуры и химического состава поверхности позволяет выявить роль каждого фактора. В этом плане очень наглядны эксперименты по влиянию ступеней, изломов ступеней [38, 39] и поверхностного состава сплавов [40–43] на каталитическую активность. Так, наличие ступеней на Pt(III) увеличивает на 2 порядка скорость изотопного обмена водорода, а активность биметаллических катализаторов может превышать активность самого активного компонента [44]. Эксперименты на модельных поверхностях позволили конкретизировать представления, предложенные ранее в теории активных центров Тейлора [45], мультиплетной теории Баландина [46] и теории активных ансамблей Кобозева [47].
Интересно отметить, что такая же “вставка” появилась позже в работе Лэнгмюра [48], который пригласил Тонкса Л. в большой обзор “Монослои на твердом теле”, включая описание адсорбции больших частиц, указанных выше. Вставленная Тонксом Л. часть позже была оформлена как работа [49]. (Эти примеры показывают, что формулировки идей могут быть отнесены к разным ссылкам.)
По физическому смыслу введение функций распределений адцентров для идеальных реакционных систем является примером учета сильного взаимодействия между компонентами адсорбционной системы и поверхностными атомами адсорбента. Условие независимости влияния каждого адцентра позволяет резко упростить задачу и рассматривать каждый адцентр независимо от других (хотя последнее не всегда является удачной аппроксимацией, особенного в случае сильного взаимного влияния адсорбатов). Сегодня, когда интерес в области гетерогенного катализа сильно сместился в сторону процессов, идущих на неметаллических катализаторах (оксиды, сульфиды, цеолиты, металлокомплексы и пр.) и полифункциональных системах, вопрос о функциях распределений адцентров по энергиям также актуален. Эти вопросы начали разрабатываться давно [50–55]. Их суть сводится к переходу от одномерных функций распределений по энергиям (уравнение (2)) на многомерные функции распределений f(Q1, …, Qn), где n – число компонентов, так как различные компоненты смесей по-разному меняют свою энергию связи с поверхностью на адцентрах разного типа.
1.4. Модель ионного расплава
Другим примером использования идеальных моделей для описания сильных взаимодействий служит модель ионного расплава, предложенная Темкиным М.И. в середине 1940-х гг. [5, 6]. Сильные взаимодействия ионов приводят к очень сильной корреляции между положительно и отрицательно заряженным ионами, которым выгоднее формировать структуры наиболее близко к нейтральным соединениям, если они образуют строго упорядоченные структуры. Так, была предложена теория идеальных ионных расплавов [5, 6], которая получила широкое распространение. Данная модель предполагает наличие идеального упорядоченного раствора, в котором катионы и анионы образуют две взаимопроникающие подрешетки без их перемешивания. Это приводит к тому, что во всей системе находятся только ионы и каждый из них окружен ионами только другого сорта. Все ионы одного знака равноценны по размерам и по энергиям взаимодействия с ионами другого знака. Поэтому основную роль играют только величины концентраций ионов в разных подрешетках. Эта модель вошла во все учебники по металлургическим процессам (см. например, [56]). Она была переформулирована и на металлургические шлаки [57], и на ее основе развивается статистическая термодинамика ионных растворов/расплавов для применения к практически важным металлургическим шлакам. Позже идеи Темкина М.И. получили активное развитие в виде моделей регулярных ионных растворов и шлаков.
Сегодня рассматриваются преимущественно более детальные модели типа регулярных растворов или более точные, учитывающие взаимодействия между компонентами расплавов. Например, в работе [58] было изучено влияние вторых соседей в строго регулярной модели расплавов на термодинамические функции раствора для ряда оксидов с целью анализа фазовых диаграмм. Современное развитие техники моделирования фазовых диаграмм привело к созданию пакетов программ Calphad [59], а в работе [60] была разработана теория для расчета поверхностного натяжения на границах раздела фаз в упорядоченных растворах, включая ионные расплавы и шлаки.
2. ЭЛЕМЕНТАРНЫЕ СТАДИИ ПОВЕРХНОСТНЫХ ПРОЦЕССОВ
В настоящее время скорости элементарных поверхностных процессов рассчитываются на основе теории абсолютных скоростей реакций (ТАСР). Эта теория была предложена для газовой фазы Эйрингом [61]. ТАСР использует представление об активированном комплексе (АК), или переходном состоянии, который находится в равновесии с исходными реагентами. Скорость бимолекулярной реакции А + В выражается как
(3)
$\begin{gathered} {{U}_{{{\text{AB}}}}} = {{K}_{{{\text{AB}}}}}{{n}_{{\text{А }}}}{{n}_{{\text{В }}}}, \\ {{K}_{{{\text{AB}}}}} = {\kappa }\frac{{kT}}{h}\frac{{F_{{{\text{AB}}}}^{*}}}{{{{F}_{{\text{A}}}}{{F}_{{\text{B}}}}}}\exp \left( {{{ - {{E}_{{{\text{AB}}}}}} \mathord{\left/ {\vphantom {{ - {{E}_{{{\text{AB}}}}}} {kT}}} \right. \kern-0em} {kT}}} \right), \\ \end{gathered} $ТАСР определяет величину предэкспоненциального фактора в уравнении Аррениуса для гомогенных реакций. Это позволяет уточнить результаты более ранних теорий, по которым этот фактор должен был быть равен числу столкновений для бимолекулярных реакций и частоте колебаний атомов в молекуле для мономолекулярных реакций.
2.1. ТАРС для поверхностных процессов на однородных поверхностях
Для поверхностных процессов ТАСР была разработана Темкиным М.И. применительно к поверхностным процессам на однородных поверхностях [14]. Эта работа является классической и вошла во многие учебники [62–64].
Скорости элементарных стадий для идеальных реакционных систем описываются в виде следующих выражений для моно- (Ui) и бимолекулярных (Uij) реакций:
где Ki и Kij – константы скоростей элементарных процессов (стадий), которые характеризуют удельные скорости элементарных процессов:(5)
$\begin{gathered} {{K}_{{ij}}} = K_{{ij}}^{0}{\text{exp}}\left( {{{ - {{E}_{{ij}}}} \mathord{\left/ {\vphantom {{ - {{E}_{{ij}}}} {{{k}_{{\text{Б }}}}T}}} \right. \kern-0em} {{{k}_{{\text{Б }}}}T}}} \right), \\ K_{{ij}}^{0} = \kappa ({{{{k}_{{\text{Б }}}}T} \mathord{\left/ {\vphantom {{{{k}_{{\text{Б }}}}T} {h\sigma }}} \right. \kern-0em} {h\sigma }})\left( {{{F_{{ij}}^{*}} \mathord{\left/ {\vphantom {{F_{{ij}}^{*}} {{{F}_{i}}{{F}_{j}}}}} \right. \kern-0em} {{{F}_{i}}{{F}_{j}}}}} \right), \\ \end{gathered} $Теория ТАСР позволилa существенно продвинуться в расчетах скоростей адсорбционных процессов по сравнению с моделью столкновений, использованной Лэнгмюром [33]. Статистические суммы исходных реагентов и АК в формуле (5) позволяют учесть вклады внутренних степеней свободы.
Для газовой фазы независимое рассмотрение поступательного, вращательного и колебательного движений реагентов и АК является обоснованным [61, 65]: Fi = $F_{i}^{{{\text{tran}}}}$ $F_{i}^{{{\text{rot}}}}$ $F_{i}^{{{\text{vib}}}}.$ В работе [14] это мультипликативное представление было перенесено на поверхностные процессы в отсутствие латеральных взаимодействий. В настоящее время выражение, аналогичное представлению статистических сумм Fi, переносится на любые состояния реакционной системы. Несмотря на его приближенный характер, оно позволяет учесть фактор вырождения тех или иных степеней свободы (поступательных, колебательных и/или вращательных) в зависимости от энергии связи с поверхностью.
Позже ТАСР для однородных поверхностей Темкина была им же обобщена на неоднородные поверхности и для описания взаимодействия между адсорбированными молекулами при описании адсорбционного равновесия и кинетики гетерогенных процессов [2]. Его изложение учета неоднородности поверхности также стало классической работой [62–64].
2.2. Учет латеральных взаимодействий
При учете латеральных взаимодействий в адсорбции и кинетике на однородных поверхностях [2] Темкин М.И. использовал теорему вириала по аналогии с работой Лэнгмюра по описанию диполь-дипольныого взаимодействия в системе Cs/W [66]. Уравнения Темкина были записаны без конкретизации вида потенциала, чтобы расширить подход на более широкий диапазон степеней заполнений по сравнению с работой Лэнгмюра, обоснованной для относительно малых степеней заполнений. В итоге были построены уравнения в приближении среднего поля, в котором отсутствуют эффекты корреляций между взаимодействующими частицами.
Следует отметить, что к тому времени уже существовали уравнения для равновесия и кинетики адсорбции и десорбции в рамках модели столкновений [67]. В модели столкновений состояние адсорбированных частиц никак практически не связано с состоянием молекул газовой фазы, поэтому в такой модели отсутствует переходное состояние, являющее необходимым понятием ТАСР. Учет свойств АК долгое время не был выполнен последовательным образом, что было определенным вызовом теории. Многочисленные попытки преодолеть данную проблему подробно изложены в обзоре [68].
2.3. Кластерный подход
К 1972 г. сложилась такая ситуация, когда для решения задачи учета свойств АК стал привлекаться метод теории корреляционных функций [69, 70], который более гибко, чем метод статистической суммы позволял отразить локальные корреляции взаимодействующих частиц. Привлечение этих методов было реализовано в рамках примесного подхода [71, 72] к задачам в модели Изинга. Предложили использовать примесный подход к расчету скорости элементарного процесса, полагая, что АК представляет собой примесь к реагентам в реакционной системы [73]. Данный подход учитывал статистический характер распределения примеси и весь спектр корреляций между адсорбированными частицами, но ассоциация АК с примесью исключала динамический характер элементарного процесса, который предполагался в ТАСР [2, 14, 61, 65].
В работе [73] концентрация АК выражалась как Cm = exp(–βεАК) 〈Um〉, где εАК – величина энергии активации появления АК, β = 1/(kБT), средняя величина $\left\langle {{{U}_{m}}(f)} \right\rangle = 1 - {{{\theta }}_{f}} + J_{m}^{'}\sum\nolimits_{k = 0}^z {\frac{{{{x}^{k}}}}{{k!}}} {{F}_{k}}(f)$ получалась из усреднения по всем локальным конфигурациям соседей вокруг разных состояний центрального узла, считавшегося примесным, $J_{m}^{'}$ – отношение статистических сумм АК и исходного “реагента” (частицы при десорбции или вакансии при адсорбции), x = exp[β(ε* – ε)] – 1, ε – энергия взаимодействия двух соседних адсорбированных частиц, ε* – энергия взаимодействия АК с соседней частицей. Функции ${{F}_{k}}$ отражают спектр вероятностей различных конфигураций соседей частицы, участвующей в элементарном процессе.
Этот вариант теории был доложен 23 апреля 1973 г. на встрече специалистов в Москве на семинаре “Теоретические проблемы диффузионной кинетики” и 31 октября 1973 г. на Ленинградском семинаре “Теория поверхностных явлений” (Федянин В.К., Товбин Ю.К. “Кинетика мономолекулярной адсорбции и десорбции на однородной поверхности”). Однако на лабораторном семинаре (в то время Федянин В.К. был сотрудником лаборатории Темкина, а Товбин Ю.К. – сотрудником лаборатории математического моделирования) Темкин М.И. сделал акцент на необходимости отражения динамического характера присутствия АК в реакционной системе, а не как стабильной частицы малой концентрации. Поэтому в работе [74] эта модель была представлена как сопоставление вкладов двух слагаемых суммы функции U: (1 – θf) и $J_{m}^{'}\sum\nolimits_{k = 0}^z {\frac{{{{x}^{k}}}}{{k!}}} {{F}_{k}}(f).$ Вклад первого слагаемого мог изменяться от нуля до единицы. Вклад второго слагаемого, содержащего известный в ТАСР сомножитель – частотный фактор, входящий в $J_{m}^{'}$ и пропорциональный величине kБT/h – много больше единицы. А поэтому вкладом первого слагаемого можно пренебречь. По этой упрощенной формуле были проведены все расчеты и описан эксперимент по десорбции атомов калия с поверхности вольфрама [75], отражающий влияние латеральных взаимодействий.
Таким образом, сложилась дилемма: либо модель Темкина с отсутствием эффектов корреляции, но с динамическим характером АК в системе латерально взаимодействующих частиц, либо примесный подход на основе корреляционных функций с полным спектром корреляций. Требовалось иное построений теории. Полное согласие между соавторами было достигнуто спустя 5 лет, когда удалось переформулировать примесную модель так, чтобы она отражала динамический характер АК [76, 77]. Во второй версии Cm = exp(–βεАК) 〈nfUm〉, где nf – число заполнений узла f адсорбированными частицами (nf = 1 для частиц А, и nf = 0 для вакансии). Тогда привязка процесса к исходной частице в узле f автоматически отражала состояние занятости этого узла и единственно возможный ход процесса (либо адсорбция, либо десорбция). В итоге удалось сохранить всю технику расчета вероятностей из работы Тябликова–Федянина [72], а физический смысл был сохранен как у Эйринга и Темкина.
На этой основе был разработан “кластерный подход” к атомно-молекулярным процессам в любых конденсированных фазах [78, 79], и он был расширен на любое число компонентов смеси. Суть данного альтернативного подхода [78, 79] к методу Боголюбова для однородных систем [69] заключалась в изначальном рассмотрении локально неоднородных систем, когда для каждого локального кластера размером, определяемом радиусом потенциала взаимодействия, строилась точная система уравнений на полный спектр локальных корреляций, и затем расчет всех кластерных распределений компонентов системы согласовывался для всего объема в целом. Это требовало использования приближенных методов расчета локальных распределений молекул, но способ описания был единым для всей системы.
2.4. Современное состояние теории
В настоящее время кинетическая теория атомно-молекулярных процессов в конденсированных фазах на основе кластерного подхода отражает следующие физические факторы: 1) неоднородность поверхности или объема твердых тел, 2) взаимное влияния ад- и абсорбированных частиц или компонентов раствора, 3) разные размеры частиц и блокировка реагентами нескольких адсорбционных центров, 4) ограниченные скорости подвижности реагентов, 5) перестройку поверхности и/или объема твердых тел, 6) внешние поля и 7) большие давления в газовой фазе. В том числе идея об упорядочении компонентов в растворах (как в ионных расплавах) была перенесена на реакционные системы [80] и использована в поверхностных процессах [81, 82]. Важно отметить, что выражения для расчета скоростей различных типов стадий согласованы между собой и выражение для скорости каждой стадии учитывает все перечисленные выше реальные свойства системы во всем диапазоне плотностей и температур. Более детально эти вопросы отражены в монографии [79] и публикациях [83–85].
Следует упомянуть модель двумерного электронного газа, которую Темкин М.И. предложил в работе [86]. Она дает такие же концентрационные зависимости для изотермы и теплоты адсорбции, как и модель учета латеральных взаимодействий в приближении среднего поля или учета неоднородности поверхности в области средних заполнений, к которой он возвращался вплоть до обзорной статьи [87]. Основной идей была линейная зависимость кинетической энергии электронного газа по мере заполнения изолированной зоны приповерхностных состояний. Однако последующие более детальные исследования не подтвердили ее обоснованность. Это касается учета эффектов экранирования электронов двумерной системы [88–90] и многочисленных последующих работ по квантово- химическим расчетам электронных состояний поверхности. Модель [86] допускает простое обобщение на кинетику поверхностных процессов, но, по сути, представляет феноменологическое описание эффектов, отличных от прямых корреляционных влияний ближайших соседей.
3. ОБЩИЕ ВОПРОСЫ ОПИСАНИЯ КОНДЕНСИРОВАННЫХ ФАЗ
Следует выделить круг вопросов, связанных с общими проблемами описания конденсированных фаз на примерах поверхностных процессов в плотных монослоях и в объемном газе. К ним относятся, во-первых, работа по описанию каталитического процесса синтеза аммиака при высоких давлениях [91] (3.1.), во-вторых, отличие трактовок учета неидеальности реакционной среды в работах Темкина М.И. [2, 91] и школы Эйринга [65], а также требование к теориям, чтобы они удовлетворяли концепции самосогласованности описания скоростей элементарных стадий (3.2.), и, в-третьих, состояния равновесия реакционной системы [2, 91] (3.3.).
3.1. Синтеза аммиака при высоких давлениях
При описании процесса синтеза аммиака при высоких давлениях было введено качественно новое понятие о сжимаемости адсорбционного слоя [91]. В этих условиях понятие давления, относящееся к газовой фазе, должно быть заменено на летучесть, а константы скоростей в выражениях скоростей элементарных реакций должны быть модифицированы за счет учета влияния смещения адсорбционного равновесия на поверхностях катализатора. В работе [91] были учтены неидеальность газовой фазы, неидеальность системы адсорбированных частиц, влияние неидеальности адсорбированных частиц на кинетику элементарных стадий и неоднородность поверхности катализатора. Включение этих факторов позволило [91] получить конечное выражение для скорости процесса синтеза аммиака, в котором удовлетворялось самосогласованность описания скоростей элементарных стадий и состояния равновесия реакционной системы.
3.2. Учет неидеальности реакционной среды
Следует напомнить, что в теории Эйринга при переходе к неидеальным системам было рекомендовано ввести коэффициенты активности как для реагентов, так и для АК рассматриваемой стадии. Скорость реакции записывается в виде закона действующих масс UAB= $K_{{{\text{AB}}}}^{*}$nАnВ (3), но для неидеальных реакционных систем константы скорости элементарных реакций записываются как $K_{{ij}}^{*}$ = Kijαiαj/$\alpha _{{ij}}^{*}$exp(–Eij/kБT), где αi = ai/ni – коэффициент активности реагента i, $\alpha _{{ij}}^{*}$ – коэффициент активности АК, Kij – константа скорости реакции в идеальной системе.
По определению активности в термодинамике величина ai зависит от всех концентраций компонентов раствора и от всех их молекулярных свойств, в том числе от энергий межмолекулярного взаимодействия [92]. Поэтому через коэффициенты активности константа скорости “зацепляется” за все физико-химические свойства реакционной системы [93, 94]. Дополнительные противоречия связаны с тем, что в случае отсутствия равновесия не существует само понятие “химический потенциал” μi, а также величина коэффициента активности АК ($\alpha _{{ij}}^{*}$) не всегда может быть определена и обоснована. Для ее построения требуется усреднение по всем возможным состояниям окружения (по всем конфигурациям и сортам соседних молекул), тогда как смена соседа меняет условия протекания реакции. Выход из этой ситуации возможен только через прямой учет молекулярных взаимодействий и отказ от термодинамических связей.
В этом отношении работы Темкина М.И. [2, 91] были ориентированы на включение прямых модельных построений, отражающих взаимодействия АК с соседями, чтобы избежать использования термодинамических соотношений. Как указано выше, проблема корректного учета неидеальности реакционных систем во всем диапазоне плотностей долгое время оставалась нерешенной. В качестве иллюстрации противопоставления концепций Темкина (отсутствие коэффициента активности АК) и Эйринга (присутствие коэффициента активности АК) приведем два примера по этим двум способам учета влияния среды для изотермического и неизотермического процесса десорбции. Для простейшей однокомпонентной системы “среду” представляют другие молекулы А, и релаксация (установление равновесия) состоит в перераспределении молекул вокруг АК при их миграции.
На рис. 1 показаны рассчитанные скорости быстрой (б) и медленной (м) мономолекулярной реакции и величина –lnηА, где lnηА = –ln(UА(м)/UА(б)) = = β [$\Delta E_{{\text{A}}}^{{{\text{э ф }}}}$(м) – $\Delta E_{{\text{A}}}^{{{\text{э ф }}}}$(б)], β$\Delta E_{{\text{A}}}^{{{\text{э ф }}}}$ = –ln(UА/$U_{{\text{A}}}^{{{\text{и д }}}}$) для всего диапазона изменения плотности реагента А [95]. Данные расчеты отвечают скорости недиссоциативной десорбции на квадратной решетке (z = 4). Здесь эффективные энергии активации характеризуют степень отклонения скорости реакции в неидеальной реакционной системе по сравнению с идеальной. На вставке рис. 1 показаны концентрационные зависимости отношений TAA = $t_{{{\text{AA}}}}^{*}$/tAA (где tAA и $t_{{{\text{AA}}}}^{*}$ – вероятности нахождения частицы А рядом с другой частицей А и рядом с АК соответственно), которые характеризуют локальные изменения распределений компонентов А за счет их миграции под влиянием АК для медленной реакции. Это отношение стремится к единице при θ ≥ 1, а при малых θ проявляется максимальное влияние АК: TAA = θexp(βδεAA). Для быстрой реакции TAA = 1 при всех θ.
Рис. 1.
Эффективные энергии активации для скоростей медленной (1, 4, 7) и быстрой (2, 5, 8) мономолекулярных реакций, их разности β[$\Delta E_{{\text{A}}}^{{{\text{э ф }}}}$(м) – $\Delta E_{{\text{A}}}^{{{\text{э ф }}}}$(б)] (3, 6, 9) и функции TAA = $t_{{{\text{AA}}}}^{*}$/tAA (10–12) (вставка) при s = 2, βεАА = 1 и различных отношениях $\varepsilon _{{{\text{AA}}}}^{*}$/εAA: (1–3, 10), 0.5 (4–6, 11) и 1.5 (7–9, 12).
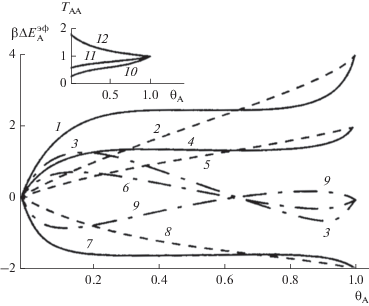
Рисунок 1 показывает качественное отличие концентрационных зависимостей скоростей реакций для разных релаксаций среды: в отсутствие релаксации lnUA(б) меняется практически линейно с увеличением θ, а при равновесной релаксации среды lnUA(м) резко меняется в областях θ < 0.2 и θ > 0.8 и остается практически постоянным при 0.2 < θ < 0.8. Это общее свойство влияния характера релаксации среды сохраняется и для других ситуаций: моно- и бимолекулярные стадии в растворах, которые были исследованы в работах [95]. Оно составляет основу при анализе экспериментальных концентрационных зависимостей логарифмов скоростей реакций в широком диапазоне концентраций.
Термодесорбционные кривые на рис. 2 приведены для медленной (а) и быстрой (б) релаксации окружения [96]. Номера кривых относятся к различным начальным степеням заполнений поверхности: 0.99 (1), 0.7 (2), 0.5 (3) и 0.3 (4). Термодесорбционные кривые получаются при линейном нагреве поверхности по закону Т = Т0+ bt, где b – скорость нагрева (град/с), Т0 – начальная температура, для которой в начальный момент времени считается заданной начальная степень заполнения поверхности θ0. Решается дифференциальное уравнение dθ/dT = –Uд, где Uд – скорость десорбции при заданном начальном заполнении θt= 0 = θ0.
Рис. 2.
Термодесорбционные кривые системы CO–Pt для быстрого (а) и медленного (б) процесса мономолекулярной десорбции при различных начальных заполнениях θ0: 0.99 (1), 0.7 (2), 0.5 (3) и 0.3 (4) [96]. в – Концентрационная зависимость эффективной энергии активации $\Delta E_{{\text{A}}}^{{{\text{э ф }}}}$ за счет влияния латеральных взаимодействий, рассчитанной для мономолекулярной десорбции при Т = 300 К в случае быстрого (1) и медленного (2) процессов.
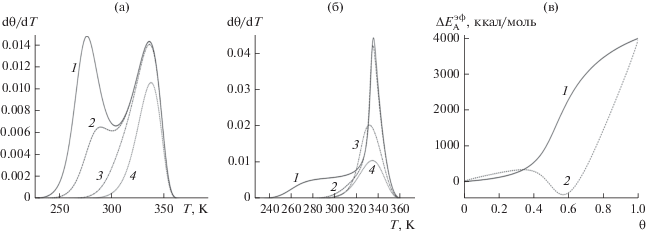
Параметры кривых на рис. 2а получены из экспериментальных данных для системы CO–Pt [68, 79] по модели [76, 77]. Те же параметры по концепции Эйринга дают кривые на рис. 2б. Сопоставление эффективной энергии активации процесса на рис. 2в, где кривая 1 отвечает рис. 2а, а кривая 2 – рис. 2б, показывает, что сильное отталкивание молекул СО приводит к расщеплению термодесорбционной кривой. По схеме Эйринга получается характерная кривая для фазового перехода первого рода, соответствующая случаю сильного притяжения частиц системы (что качественно искажает физический смысл параметров межчастичного взаимодействия). Приведенные примеры показывают, что концепция Темкина является физически корректной в отличие от концепции Эйринга.
3.3. Концепция самосогласованности описания скоростей элементарных стадий
Концепция самосогласованного описания скоростей кинетических процессов и равновесия в неидеальных и неоднородных системах, использованная в работах [2, 91], является ключевой для взаимосвязи равновесия и кинетики, как предельного перехода по времени. Она означает, что из условия равенства скоростей в прямом и обратном направлениях элементарной реакции должны вытекать уравнения равновесного распределения молекул в реакционной системе. В кинетической теории газов такая проверка является обязательной для всех новых предлагаемых подходов. В конденсированных фазах она практически не проводилась по многим причинам (что, в первую очередь, связано с отсутствием уравнения состояния в плотных жидких, адсорбционных и твердых фазах).
Выше было отмечено, что грубое описание эффектов корреляции не позволяет построить корректные выражения для скоростей элементарных стадий. В работах [97–102] были проанализированы различные версии учета межчастичных взаимодействий на однородных и неоднородных поверхностях. Доказательство выполнения условия самосогласования для общего случая учета неоднородности поверхности и латеральных взаимодействий в квазихимическом приближении было дано в работе [68] (см. также работы [103–105]). Было доказано, что теория обеспечивает самосогласованное описание динамики и равновесия на всех пространственных масштабах для любых плотностей, температур, интенсивностей латеральных взаимодействий и внешних полей только в случае учета эффектов корреляции как минимум для ближнего порядка. В противном случае условие самосогласованности нарушается, поэтому приближение молекулярного/среднего поля, хаотическое приближение и теория функционала плотности не могут быть использованы для описания динамики в плотных фазах.
Однако необходимо указать, что концепция самосогласованности является необходимой, но не достаточной. Наличие самосогласованности не означает автоматически корректности уравнений для скорости стадий, так как одновременно могут быть некорректными и уравнения для изотерм. В качестве примера укажем на приближения среднего поля, хаотическое и функционала плотности для любых одно- и двухузельных стадий на однородной поверхности, а также для одноузельных стадий на неоднородной поверхности в приближении среднего поля, так как оно нивелирует все неоднородности системы [106]. То же самое относится и к уравнениям Темкина 1941 и 1950, использующим это же приближение. Для однородной поверхности уравнения [2] не дают расщепления термодесорбционных кривых, на которые указывают эксперименты [68, 79, 107]. Этот недостаток является следствием отсутствия учета эффектов корреляции. Наконец, это в такой же мере относится ко всем случаям использования уравнения закона действующих масс, когда он не отражает физического состояния системы, обусловленного взаимодействиями между компонентами реакционной системы.
Отметим, что в обсуждаемых процессах на атомно-молекулярном уровне скорости стадий относятся к любым степеням отклонений от состояния равновесия, а не только к малым отклонениям, как это предполагается в подходе Онсагера [108, 109]. Концепция самосогласованности налагает ограничение на способ построения уравнений для скоростей стадий на данном микроскопическом уровне (хотя формально она полностью переносится на более детальную поуровневую кинетику в уравнениях типа уравнений Паули [110, 111]).
В развитие идей Темкина о динамическом характере элементарного акта любого взаимодействия частиц и Боголюбова о едином способе расчета распределения частиц [69] был разработан “кластерный подход” для неоднородных систем любой плотности (любых фаз) как в равновесии, так и в кинетике при любых степенях отклонений от равновесия [79, 112, 113]. Основное кинетическое уравнение (Master Equation) для эволюции полной функции распределения системы P({I},τ), находящейся в момент времени τ в состоянии {I}, {I} ≡ $\left\{ {\gamma _{f}^{i}} \right\}$ ≡ $\gamma _{1}^{i},$ …, $\gamma _{{\text{М }}}^{i}$ (где символ $\gamma _{f}^{i}$ означает состояние занятости i узла с номером f, а фигурные скобки – полный список узлов системы М) за счет реализации элементарных процессов α имеет вид:
(6)
$\begin{gathered} \frac{{\text{d}}}{{{d\tau }}}P(\{ {\text{I}}\} ,{\tau }) = \sum\limits_{{\alpha },\{ I\} } {[{{W}_{{\alpha }}}(\left\{ {{\text{II}}} \right\} \to \left\{ {\text{I}} \right\})P(\left\{ {{\text{II}}} \right\},{\tau })} - \\ - \,\,{{W}_{{\alpha }}}(\left\{ {\text{I}} \right\} \to \left\{ {{\text{II}}} \right\})P(\left\{ {\text{I}} \right\},{\tau })], \\ \end{gathered} $Вероятности Wα переходов α подчиняются условию детального равновесия
(7)
$\begin{gathered} {{W}_{\alpha }}(\{ {\text{I}}\} \to \{ {\text{II}}\} ){\text{е х р }}(--\beta H(\{ {\text{I}}\} )) = \\ = {{W}_{\alpha }}(\{ {\text{II}}\} \to \{ {\text{I}}\} ){\text{е х р }}(--\beta H(\{ {\text{II}}\} )),\quad \\ \end{gathered} $Следует обратить внимание, что к началу 1980-х гг. в кинетических уравнениях Глаубера (Master Equation), задействованных для описания спиновых систем и процессов роста кристаллов, как правило, использовали феноменологические вероятности переходов Wα между различными состояниями спинов, молекул или системы [68, 114]. Только в 1982 г. впервые было сформулировано требование к вероятностям переходов Wα в уравнениях Глаубера и/или Паули на базе Master Equation, чтобы они удовлетворяли теории химических реакций и/или рассеяния [112, 113] и условию самосогласованности, которое так активно использовал М.И. Темкин. В настоящее время это является общепризнанным положением.
Этот принцип согласовал три уровня: квантовый для расчета энергетики, динамический для реализации элементарного акта и статистический для расчета вероятностей конкретных конфигураций соседей, определяющий первые два уровня.
В настоящее время кластерный подход имеет такую же общность областей применения, как и термодинамика [115]: можно самосогласованно описывать равновесные и неравновесные процессы в трехагрегатных состояниях и на границах раздела фаз.
В современной теории ключевую роль играют эффекты корреляции взаимодействующих адсорбированных частиц на поверхности или атомов внутри твердого катализатора или адсорбента при их пространственном распределении. В последнем случае эффекты корреляции формируют адсорбционные и каталитические центры, и их описание является необходимым для описания процессов на сложных неметаллических катализаторах [116, 117]. Кинетические уравнения [79, 112, 113] создают основу для моделирования формирования различных типов катализаторов и позволяют дать методы построения многомерных функций распределений адцентров для многокомпонентных реакционных систем f(Q1, …, Qn) [118, 119]. Таким образом, идея о важной роли сильных корреляций взаимодействующих компонентов твердых тел проходит от модели ионных расплавов до моделей формирования современных каталитических и полифункциональных систем.
ЗАКЛЮЧЕНИЕ
Статистическая термодинамика идеальных систем позволила получить М.И. Темкину его классические результаты в начале формирования физической химии в 30–40-х гг. прошлого века. Такие же строгие методы статистической термодинамики необходимы для дальнейшего развития современной физической химии, теории кинетических процессов и катализа.
Теория абсолютных скоростей реакций для всех однородных и неоднородных поверхностей (в отсутствие латеральных взаимодействий) должна носить имя Темкина М.И. как ее реального создателя.
Как упомянуто выше, сегодня в области гетерогенного катализа интерес сильно сместился в сторону процессов, идущих на неметаллических катализаторах и полифункциональных системах. Их детальное кинетическое описание чрезвычайно сложно в силу проблем определения природы активационных центров. Тем не менее развитые ранее подходы могут быть применены для описания сложных систем путем обобщения функций распределений адцентров для различных компонентов реакционных смесей. Такие модели, естественно, могут быть применены ко всем многостадийным процессам гетерогенных каталитических реакций, а также при моделировании процессов внешнего и внутреннего массо- и теплопереноса в гетерогенном катализе. Современное состояние теории адсорбции и кинетики поверхностных процессов позволяет одновременно учесть различные физико-химические факторы реакционных систем.
Использованная М.И. Темкиным концепция самосогласованного описания скоростей реакций и равновесия является ключевой в любых сложных ситуациях и вне применимости моделей идеальных реакционных систем. Применение методов конденсированных фаз позволило получить решение задачи о самосогласованном описании во времени и в равновесном состоянии распределения взаимодействующих частиц по узлам однородных и неоднородных решеток, а это дает возможность решения большого числа задач по описанию процессов на границе газ–твердое тело. Исследованные примеры различных процессов (адсорбции, абсорбции, диффузии частиц и поверхностных реакций) указывают на принципиальную роль кооперативных эффектов, обусловленных взаимодействием между компонентами реакционной системы, в кинетике этих процессов. Современная теория может быть использована для практически важных каталитических процессов, так как на их основе можно исследовать такие вопросы изменения активности катализаторов, как термическая устойчивость, спекание, отравление и т.д.). Близкие задачи возникают при исследовании вопросов о характере поверхностной сегрегации сплавов в вакууме и газовой фазе, при росте кристаллов из газовой фазы и их сублимации, при формировании и снятии защитных пленок, при травлении поверхности и воздействии на нее потоками частиц различной энергии и т.д.
Сформулированные общие условия реализации самосогласованности указывают, как достичь ее выполнения во всех конденсированных фазах.
Список литературы
Темкин М.И. // ЖФХ. 1940. Т. 14. С. 1153.
Темкин М.И. // ЖФХ. 1941. Т. 15. № 3. С. 296.
Темкин М.И., Пыжев В.М. // ЖФХ. 1939. Т. 13. С. 851.
Темкин М.И. // ДАН СССР. 1965. Т. 161. № 1. С. 160.
Temkin M.I. // Acta Physicochimica URSS. 1945. V. 20. P. 411.
Темкин М.И. // ЖФХ. 1946. Т. 20. № 1. С. 105.
Темкин М.И. Гетерогенный катализ в химической промышленности. Сб. М.: Госхимиздат, 1955. С. 256.
Хилл Т. Катализ, вопросы теории и методы исследований. Сб. ст. М.: Изд-во иностр. лит., 1955. С. 276.
Темкин М.И. // ЖФХ. 1933. Т. 4. С. 573.
Langmuir I. // J. Amer. Chem. Soc. 1916. V. 38. P. 2217.
Fowler R.H. // Proc. Cambridge Phil. Soc. 1935. V. 31. P. 260.
Fowler R.H. Statistical Mechanics. Cambridge: Cambridge Univer. Press, 1936.
Fowler R.H., Guggenheim E.A. Statistical Thermodynamics. Cambridge: Cambridge Univer. Press, 1939.
Темкин М.И. // ЖФХ. 1938. Т. 11. С. 169.
Fowler R.H., Rushbrooke G.S. // Trans. Faraday Soc. 1937. V. 33. P. 1272.
Roberts I.K. // Proc. Cambrridge Phil. Soc. 1938. V. 34. P. 577.
Rushbrooke G.S. // Proc. Roy. Soc. London. A. 1938. V. 166. P. 296.
Chang T.S. // Proc. Roy. Soc. A. 1939. V. 169. P. 512.
Chang T.S. // Proc. Cambrridge Phil. Soc. 1939. V. 35. P. 265.
Roberts I.K., Miller A.K. // Proc. Cambrridge Phil. Soc. 1939. V. 35. P. 293.
Ходаков Ю.С., Берлин А.А., Каляев Г.И., Миначев Х.М. // Теорет. и эксперим. химия. 1969. Т. 5. № 5. С. 631.
Товбин Ю.К. // ЖФХ. 1974. Т. 48. № 5. С. 1239.
Товбин Ю.К. // Теорет. и эксперим. химия. 1974. Т. 10. № 2. С. 258.
Островский В.Е., Темкин М.И. // Кинетика и катализ. 1969. Т. 10. С. 118.
Островский В.Е., Темкин М.И. // Кинетика и катализ. 1969. Т. 12. С. 1070.
Снаговский Ю.С. // ЖФХ. 1972. Т. 46. № 6. С. 2367.
Снаговский Ю.С., Островский Г.М. Моделирование кинетики гетерогенных каталитических процессов. М.: Химия, 1976. 248 с.
Товбин Ю.К. // Теорет. и эксперим. химия. 1981. Т. 17. № 1. С. 28.
Товбин Ю.К. // Хим. физика. 2005. Т. 24. № 9. С. 91.
Товбин Ю.К. // ЖФХ. 2006. Т. 80. № 6. С. 1134.
Товбин Ю.К. Молекулярная теория адсорбции в пористых телах. М.: Физматлит, 2012. 624 с.
Boehm B.E., Martire D.E. // J. Chem. Phys. 1977. V. 67. P. 1961.
Langmuir I. // J. Am. Chem. Soc. 1918. V. 40. P. 1361, 1403.
Фрумкин А.Н., Шлыгин А.И. // ДАН СССР. 1934. Т. 2. С. 173.
Shlygin A.I., Frumkin A.N. // Acta Physicochimica URSS. 1935. V. 1. S. 791.
Zeldowitsch Ya.B. // Acta Physicochimica URSS. 1935. V. 1. S. 961.
Темкин М.И., Левич В.Г. // ЖФХ. 1946. Т. 20. С. 1441.
Somorjai G.A. Chemistry in Two-Dimension Surface. N.Y., Ithaca: Cornell Univ. Press L., 1981.
Somorjai G.A. // Catal. Rev. 1972. V. 7. P. 87.
Ponec V. // Catal. Rev. 1975. V. 11. P. 1.
Clarke K.A. // Chem. Rev. 1975. V. 76. P. 291.
Sachtler W.M.H. // Catal. Rev. 1976. V. 14. P. 193.
Madix R.J. // Catal. Rev. 1977. V. 15. P. 293.
Sinfelt J.H. // Adv. Catal. 1973. V. 28. P. 91.
Taylor H.S. // Proc. Roy. Soc. 1925. V. A108. P. 105.
Баландин А.А. Мультиплетная теория катализа. М.: Изд-во МГУ, 1963. ч. 1, 1964. ч. 2.
Кобозев Н.И. // ЖФХ. 1939. Т. 18. С. 1; Успехи химии. 1956. Т. 25. С. 545.
Langmuir I. // J. Chem. Soc. 1940. April–May. P. 513.
Tonks L. // J. Chem. Phys. 1940. V. 8. P. 477.
Рогинский С.З., Тодес О.М. // Асta Physicjchimica USSR. 1945. V. 20. P. 307.
Рогинский С.З., Тодес О.М. //Асta Physicjchimica USSR. 1945. V. 20. P. 696.
Рогинский С.З., Тодес О.М. // Проблемы кинетики и катализа. 1949. V. 8. P. 339.
Рогинский С.З. Адсорбция и катализ на неоднородных поверхностях. М.–Л.: Изд-во АН CCСР, 1948.
Рогинский С.3., Тодес О.М. Проблемы кинетики и катализа. Сб. М.: Изд-во АН СССР. 1949. Т. 8. С. 336.
Tovbin Yu.K. Dynamics of Gas Adsorption on Heterogeneous Solid Surfaces / Eds. Rudzinski W., Steele W.A., Zgrablich G. Amsterdam: Elsevier, 1996. P. 105.
Рыжонков Д.И., Арсентьев П.П., Яковлев В.В., Крашенинников М.Г., Пронин Л.А., Филиппов Е.С. Теория металлургических процессов. Учеб. для вузов. М.: Металлургия, 1989. 392 с.
Кожеуров В.А. Термодинамика металлургических шлаков. Свердловск: ГНТИ СО, 1955. 164 с.
Пак В.М., Серов Г.В., Товбин Ю.К. // Расплавы 1988. Т. 2. № 5. С. 21.
Sundman B., Kattner U.R., Palumbo M., Fries S.G. // Integrating Materials and Manufacturing Innovation. 2015. V. 4. P. 1.
Товбин Ю.К. // ЖФХ. 1992. Т. 66. № 5. С. 1395.
Eyring H. // J. Chem. Phys. 1935. V. 3. P. 107.
Киперман С.Л. Введение в кинетику гетерогенных каталитических реакций. М.: Наука, 1964. 60 с.
Киперман С.Л. Основы химической кинетики в гетерогенном катализе. М.: Химия, 1978. 350 с.
Еремин Е.Я. Основы химической кинетики. М.: Высш. шк., 1976. 374 с.
Глесстон С., Лейдлер К., Эйринг Г. Теория абсолютных скоростей реакций. М.: Изд-во иностр. лит., 1948. 583 с.
Лэнгмюр И. // Успехи химии. 1933. Т. 2. С. 649.
Roberts I.K. // Proc. Royal Soc. A. 1937. V. 161. P. 141.
Tovbin Yu.K. // Progress in Surface Science. 1990. V. 34. № 1–4. P. 1.
Боголюбов Н.Н. Проблемы динамической теории в статистической физике. М.: Гостехиздат, 1946.
Тябликов С.В. Методы квантовой теории магнетизма. М.: Наука, 1965. 400 с.
Тябликов С.В., Федянин В.К. // Физика металлов и металловедение. 1967. Т. 23. № 2. С. 193.
Тябликов С.В., Федянин В.К. // Физика металлов и металловедение. 1968. Т. 26. № 4. С. 589.
Fedjanin V.K. // Chem. Phys. Lett. 1973. V. 22. P. 99.
Товбин Ю.К. Дисс. … канд. физ.-мат. наук. М.: НИФХИ им. Л.Я. Карпова, 1974.
Товбин Ю.К., Федянин В.К. // ФТТ. 1975. Т. 17. С. 1511.
Товбин Ю.К., Федянин В.К. // Кинетика и катализ. 1978. Т. 19. № 4. С. 989.
Товбин Ю.К., Федянин В.К. // Кинетика и катализ. 1978. Т. 19. № 5. С. 1202.
Товбин Ю.К. Дисс. … д-ра физ.-мат. наук. М.: НИФХИ им. Л.Я. Карпова, 1985.
Товбин Ю.К. Теория физико-химических процессов на границе газ–твердое тело. M.: Наука, 1990. 288 p.
Товбин Ю.К. // ДАН СССР. 1984. Т. 277. № 4. С. 917.
Товбин Ю.К., Вотяков Е.В. // ЖФХ. 1993. Т. 67. № 1. С. 136.
Tovbin Yu.K., Votyakov E.V. // Langmuir. 1999. V. 15. P. 6070.
Товбин Ю.К. // Электрохимия. 2009. Т. 45. № 9. С. 1030.
Tovbin Yu.K. Dynamics of Gas Adsorption on Heterogeneous Solid Surfaces / Eds. Rudzinski W., Steele W.A., Zgrablich G. Amsterdam: Elsevier, 1996. P. 240.
Tovbin Yu.K. Thin Films and Nanostructures. V. 34. / Eds. Trakhtenberg L.I., Lin S.H., Ilegbusi O.J. Amsterdam: Elsevier, 2007. P. 347.
Темкин М.И. Вопросы химической кинетики, катализа и реакционной способности. М.: Изд-во АН СССР, 1955. С. 484.
Temkin M.I. // Adv. Catal. 1979. V. 28. P. 173
Тимашев С.Ф., Федянин В.К. // ДАН СССР. 1970. Т. 191. № 6. С. 1333.
Тимашев С.Ф., Федянин В.К. // ФTТ. 1971. Т. 13. С. 1196.
Федянин В.К. // ЖФХ. 1971. Т. 45. С. 2876
Темкин М.И. // ЖФХ. 1950. Т. 23. С. 169.
Пригожин И., Дефэй Р. Химическая термодинамика. Новосибирск: Наука, 1966. 510 с.
Энтелис C.Г., Тигер Р.П. Кинетика реакций в жидкой фазе. Количественный учет влияния среды. М.: Химия, 1973. 415 с.
Мелвин-Хьюз Е.А. Равновесие и кинетика реакций в растворах. М.: Химия, 1975. 470 с.
Товбин Ю.К., Вотяков Е.В. // ЖФХ. 1997. Т. 71. № 1. С. 271.
Товбин Ю.К., Титов С.В. // Сверхкритические флюиды: теория и практика. 2011. Т. 6. № 2. С. 35.
Товбин Ю.К. // ЖФХ. 1981 Т. 55. № 2. С. 284.
Товбин Ю.К. // Кинетика и катализ. 1982. Т. 23. № 4. С. 813.
Товбин Ю.К. // Кинетика и катализ. 1982. Т. 23. № 4. С. 821.
Товбин Ю.К. // Кинетика и катализ. 1982. Т. 23. № 5. С. 1231.
Товбин Ю.К. // Кинетика и катализ. 1983 Т. 24. № 2. С. 308.
Товбин Ю.К. // Кинетика и катализ. 1983 Т. 24. № 2. С. 317.
Товбин Ю.К. // ЖФХ. 2018. Т. 92. № 6. С. 929.
Зайцева Е.С., Товбин Ю.К. // ЖФХ. 2019. Т. 93. № 4. С. 508.
Зайцева Е.С., Товбин Ю.К. // Кинетика и катализ. 2019. Т. 60. № 4. С. 440.
Товбин Ю.К. // Теорет. и эксперим. химия. 1982. Т. 18. № 4. С. 417.
Товбин Ю.K. // Кинетика и катализ. 1978. Т. 20. № 5. С. 1226.
Onsager L. // Phys. Rev. 1931. V. 37. P. 405.
Onsager L. // Phys. Rev. 1931. V. 38. P. 2265.
Pauli W. Problems der Modernen Physik. Leipzig: Springer-Verlag, 1928. S. 30.
Полак Л.С. Неравновесная химическая кинетика и ее применение. М.: Наука, 1979. 248 с.
Товбин Ю.K. // ДАН СССР. 1982. Т. 267. № 6. С. 1415.
Товбин Ю.K. // Кинетика и катализ. 1984. Т. 25. № 1. С. 26.
Glauber J. // J. Math. Phys. 1963. V. 46. P. 541.
Товбин Ю.K. Малые системы и основы термодинамики. М.: Физматлит, 2018. 404 с.
Киселев В.Ф., Крылов О.В. Электронные явления в адсорбции и катализе на полупроводниках и диэлектриках. М.: Химия, 1979. 235 с.
Крылов О.В., Киселев В.Ф. Адсорбция и катализ на переходных металлах и их оксидах. М.: Химия, 1981. 287 с.
Товбин Ю.K. // Кинетика и катализ. 1978. Т. 20. № 6. С. 1453.
Товбин Ю.K. // Кинетика и катализ. 1980. Т. 22. № 5. С. 1165.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ