

Кинетика и катализ, 2019, T. 60, № 5, стр. 612-617
Моделирование окисления CO в присутствии циклических тиолатных комплексов золота: влияние лиганда
Н. А. Никитина a, *, Д. А. Пичугина a, Н. Е. Кузьменко a
a Московский государственный университет им. М.В. Ломоносова, химический факультет
119991 Москва, Ленинские горы, 1/3, Россия
* E-mail: nnikitina1719@gmail.com
Поступила в редакцию 26.10.2018
После доработки 25.03.2019
Принята к публикации 17.04.2019
Аннотация
Методом теории функционала плотности с использованием функционала PBE изучено взаимодействие СО и О2, а также дальнейшее окисление СО в присутствии циклических тиолатных и дитиолатных комплексов Au(I), представляющих модельные фрагменты кластеров золота, стабилизированных тиолатными лигандами. На основе рассчитанных значений показано, что О2 и СО слабо связываются с циклическим тиолатным комплексом Au(I). В присутствии дитиолатного комплекса происходит активация О2 и СО и дальнейшее окисление СО с низкими энергиями активации. Полученные результаты демонстрируют важную роль лиганда в каталитическом процессе.
Наночастицы золота являются известными катализаторами многих промышленно важных реакций окисления и гидрирования [1, 2], среди которых низкотемпературное каталитическое окисление СО служит модельной реакцией и часто применяется при тестировании новой серии катализаторов. Принципиально новыми каталитическими системами являются кластеры золота, стабилизированные лигандами различной природы (фосфин- и фенилацетиленсодержащие, тиолатные) [3–5]. Относительно недавно удалось получить тиолатный кластер состава Au25(SR)18, теоретически предсказать его структуру методом функционала плотности [6] и позднее подтвердить строение рентгеноструктурным анализом [7]. Этот кластер содержит ядро и оболочку. Ядро состоит из атомов золота, а в состав оболочки входит не только лиганд, но и атомы золота, образующие с лигандами скрепочные или циклические фрагменты различной протяженности –SR–(Au–SR–Au)x–.
Синтезированные серии кластеров золота, стабилизированных тиолатными лигандами [8–12], начали активно пробовать применять в различных областях, включая катализ. В частности, была показана их каталитическая активность в реакциях окисления СО и углеводородов, кросс-сочетания и гидрирования [13–15]. Несомненным преимуществом таких каталитических систем является формирование на поверхности гетерогенного катализатора кластеров металла определенного состава, что позволяет соотнести конкретное свойство (активность, селективность, стабильность) с определенным размером и структурой частицы. Подобные исследования были проведены в работе [16], в которой сравнили активность кластеров Aun(SR)m, нанесенных на гидроксоаппатит, в окислении стирола.
Многообещающее применение кластеров золота, стабилизированных тиолатными лигандами, в катализе сдерживается трудностью их получения в растворе ввиду того, что селективность синтеза крайне чувствительна к условиям проведения реакции. Кроме того, кластеры в растворе не устойчивы. Так, известно, что характерный пик в УФ-спектре определенного кластера в растворе исчезает через 1–2 сут после его получения [17]. При этом в спектре появляются сигналы, соответствующие скрепочным фрагментам и полимерным комплексам (Au(I)–SR)x [17, 18], что свидетельствует о полном или частичном разрушении кластера. Этот факт позволяет предположить образование комплексов (Au(I)–SR)x при использовании Aun(SR)m в качестве катализаторов как в гомогенной среде, так и при их нанесении на поверхность подложки. В настоящее время роль скрепочных фрагментов в каталитических процессах с участием кластеров золота, стабилизированных тиолатными лигандами, остается невыясненной, как и свойства кластера Aun(SR)m [19–22].
В данной статье приведены результаты квантово-химического моделирования взаимодействия циклических тиолатных и дитиолатных кластеров Au(I) с О2 и СО, а также дальнейшего окисления СО с целью установления каталитических свойств полимерных комплексов (Au(I)–SR)x.
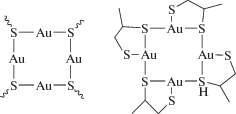
Формулы циклических тиолатных и дитиолатных комплексов золота
Дитиолатные комплексы наряду с тиолатными используются для стабилизации кластеров Aun(SR)m. Рассмотренные в работе циклические комплексы (AuSR)4 являются аналогами ранее синтезированных соединений Au4[SC(Si(CH3)3)3]4 [23].
МЕТОДИКА РАСЧЕТОВ
Квантово-химические расчеты проводили с помощью теории функционала плотности с использованием функционала РВЕ (Perdew−Burke−Ernzerhof) [24], базисного набора и псевдопотенциала SBKJC [25]. Данный метод уже применялся нами ранее для определения строения Au18(SR)15 [26], а также при моделировании каталитических и адсорбционных процессов с участием комплексов и кластеров золота [27–30].
Для исследуемых циклических комплексов, продуктов их взаимодействия с О2 и СО, а также возможных промежуточных комплексов окисления СО была рассчитана равновесная геометрия и полная энергия. В качестве органического фрагмента R в тиолатном лиганде SR рассматривали метильную группу. Вклад энергий нулевых колебаний определяли на основе частот колебаний, рассчитанных в гармоническом приближении. Для всех стадий реакции получены термодинамические характеристики, включая изменение полной энергии (∆Е) и стандартной энергии Гиббса (∆G0) при 298 К. Для наиболее важных стадий окисления СО проводили локализацию структуры переходного состояния (ПС) с помощью алгоритма оптимизации Берни [31] и рассчитывали энергии активации (Еа). ПС определяли с помощью исследования энергии реакционной системы в зависимости от координаты реакции методом IRC [32].
Все расчеты проводили в программе PRIRODA [33] при использовании суперкомпьютера МГУ “Ломоносов” [34].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
На первом этапе проведено исследование строения циклического тиолатного комплекса Au(I) и его реакционной способности по отношению к О2 и СО. Оптимизированная структура (AuSCH3)4 приведена на рис. 1. В комплексе фрагмент (AuS–)4 практически плоский. Анализ вида высших занятых молекулярных орбиталей (ВЗМО) показал, что связь Au–S имеет ковалентный характер. Ввиду значительной прочности связи Au–S в (AuSCH3)4 не происходит присоединения СО, и стабильный карбонильный комплекс (AuSCH3)4CO не образуется.
Рис. 1.
Оптимизированные структуры комплексов (AuSCH3)4 (а), (AuSCH3)4О2 (в) и визуализация ВЗМО (AuSCH3)4 (б). Ключевые межатомные расстояния указаны в ангстремах.
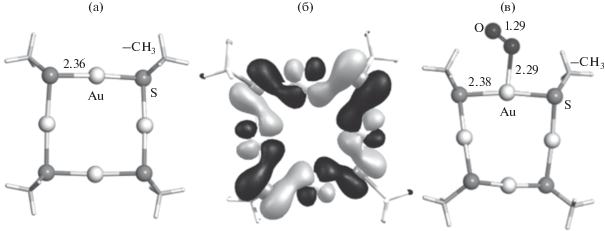
В качестве возможного продукта взаимодействия циклического комплекса с кислородом получено соединение (AuSCH3)4О2, находящееся в синглетном электронном состоянии. Его образование из триплетного кислорода не выгодно. Изменения полной энергии и энергии Гиббса при образовании (AuSCH3)4О2 из синглетного кислорода, который может получаться в присутствии тиолатных кластеров золота при фотоактивации [35, 36], незначительны (табл. 1). В (AuSCH3)4О2 кислород слабо активирован, на что указывает расстояние О–О, которое мало изменилось по сравнению с изолированной молекулой О2 (1.22 Å), а также частота колебания О–О (1225.3 см–1 по сравнению с 1542.7 см–1 в О2).
Таблица 1.
Термодинамические параметры взаимодействия циклических комплексов (AuSCH3)4 и (AuS CH2CHSCH3)4 с СО и О2
| Стадия | ∆E | ∆G298 |
|---|---|---|
| ккал/моль | ||
| (AuSCH3)4 + 1O2 → (AuSCH3)4O2 | –12.7 | –1.8 |
| (AuSCH2CHSCH3)4 + 3O2 → (AuSCH2CHSCH3)4O2 | –9.1 | –1.3 |
| (AuSCH2CHSCH3)4 + CO → (AuSCH2CHSCH3)4CO | –9.4 | –2.0 |
| (AuSCH2CHSCH3)4O2 + CO → (AuSCH2CHSCH3)4O2CO | –10.3 | –3.6 |
Таким образом, по данным расчета, (AuSCH3)4 не реагирует с СО, а слабая активация О2, требующая дополнительных условий, по-видимому, будет недостаточна для любых дальнейших процессов с участием кислорода.
Далее исследовали строение циклического дитиолатного комплекса Au(I) и его реакционной способности по отношению к О2 и СО. Оптимизированная структура (AuSCH2CHSCH3)4 приведена на рис. 2. Рассчитанные изменения полной энергии и стандартной энергии Гиббса представлены в табл. 1.
Рис. 2.
Оптимизированные структуры комплексов (AuSCH2CHSCH3)4 (а), (AuSCH2CHSCH3)4O2 (в), (AuSCH2CHSCH3)4CO (г), (AuSCH2CHSCH3)4O2CO (д) и визуализация ВЗМО (AuSCH2CHSCH3)4 (б). Ключевые межатомные расстояния указаны в ангстремах.
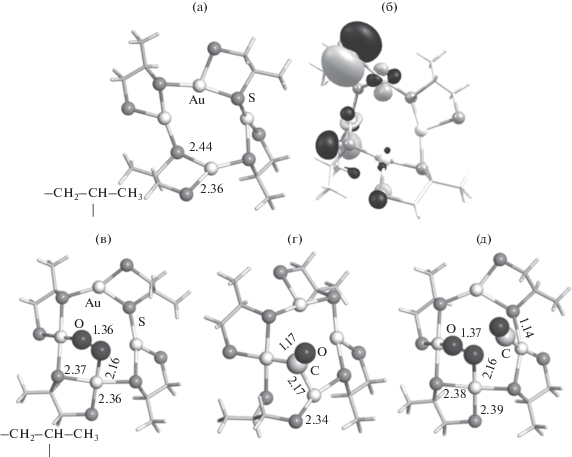
Замена тиолатного лиганда на дитиолатный существенно изменила строение (AuSR)4. Комплекс уже не является плоским, а расстояние Au–S увеличилось на 0.08 Å. Отличается и характер распределения электронной плотности вдоль связи Au–S (рис. 2б). ВЗМО локализована не по связи Au–S, как это происходит в (AuSCH3)4, а на атомах Au и S. Этот факт косвенно указывает на увеличение реакционной способности (AuS CH2CHSCH3)4. Действительно, была найдена структура стабильного карбонильного комплекса (AuSCH2CHSCH3)4CO, в котором фрагмент СО координирован по двум атомам Au. Изменение энергии при его образовании составляет ‒9.4 ккал/моль.
Устойчивый комплекс пероксо-типа образуется при взаимодействии дитиолатного комплекса с О2 (рис. 2б). В нем кислород одновременно связан с двумя атомами Au. Увеличенное межатомное расстояние О–О в (AuSCH2CHSCH3)4O2 на 0.07 Å и частота колебания О–О, равная 921.5 см–1, указывают на значительную активацию кислорода. Этот пероксо-комплекс может образовываться из триплетного кислорода с выигрышем по энергии –9.1 ккал/моль. Последующее присоединение СО к кислородному комплексу более выгодно, чем присоединение СО к (AuSCH2CHSCH3)4. В данном случае наблюдается эффект соадсорбции, заключающейся в положительном влиянии адсорбции одного реагента на последующую адсорбцию другого. Эффект соадсорбции О2 и CO хорошо известен для окисления СО на наночастицах золота [37, 38].
Найденный комплекс (AuSCH2CHSCH3)4O2CO (ПК0), судя по его строению, может выступать интермедиатом (промежуточным комплексом, ПК) дальнейшего окисления СО. При моделировании образования СО2 принимали во внимание один из механизмов, известный для наночастиц золота (Au) [39, 40]:
Для всех стадий этого процесса проведена оптимизация структуры промежуточных комплексов (ПК) и переходных состояний (ПС) (рис. 3) и рассчитаны соответствующие энергии. Диаграммы изменения энергии в стадиях окисления СО показаны на рис. 4.
Рис. 3.
Оптимизированные структуры промежуточных комплексов (ПК) и переходных состояний (ПС) стадий окисления СО из (AuSCH2CHSCH3)4O2CO. Ключевые межатомные расстояния указаны в ангстремах.
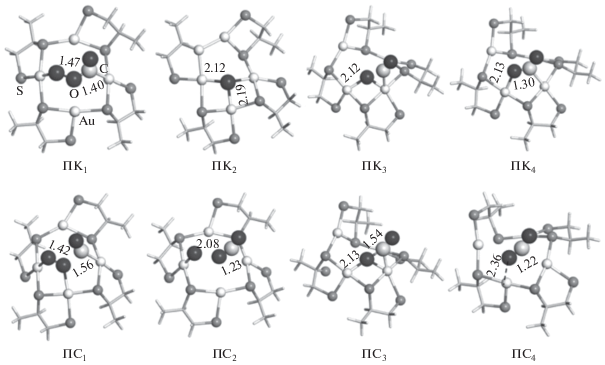
Рис. 4.
Энергетические диаграммы первых двух стадий окисления СО из исходного комплекса (AuSCH2CHSCH3)4O2CO (ПК0) (а) и двух стадий окисления второй молекулы СО из (AuSCH2CHSCH3)4OCO (ПК3) (б).
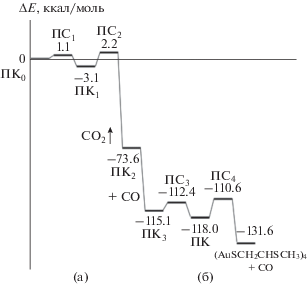
Образование первой молекулы СО2 происходит в две стадии за счет низких энергий активации (табл. 2) через интермедиат ПК1. В образовавшемся на заключительной стадии промежуточном комплексе ПК2 оставшийся атом кислорода координирован по трем атомам золота. Присоединение второй молекулы СО осуществляется по атому Au, связанному с O, с образованием интермедиата ПК3. Его превращение в СО2 и исходный кластер происходит в две стадии через промежуточный комплекс ПК4 с низкими энергиями активации.
Таблица 2.
Рассчитанные значения энергии активации Гиббса (Δ≠G298) и изменения энергии Гиббса (∆G) для стадий окисления СО в присутствии (AuS CH2CHSCH3)4
| Стадия | Δ≠G298 | ∆G298 |
|---|---|---|
| ккал/моль | ||
| ПК0 → ПК1 | 0.9 | –0.5 |
| ПК1 → ПК2 | 5.2 | –81.7 |
| ПК3 → ПК4 | 2.3 | –0.8 |
| ПК4 → (AuSCH2CHSCH3)4 + CO2 | 6.5 | –28.2 |
Таким образом, по данным расчета, комплекс (AuSCH2CHSCH3)4 по сравнению с (AuSCH3)4 будет более активен в активации O2 и CO и в дальнейшем окислении СО. На стадии активации реагентов и их превращений разрыва связей со стабилизирующими лигандами не происходит, что важно при сохранении размера и структуры комплекса. Можно предположить, что кластеры золота, стабилизированные аналогичными дитиолатными лигандами, будут более перспективными каталитическими системами окисления СО, чем кластеры, стабилизированные тиолатными группами. Синтез кластеров золота, стабилизированных дитиолатными лигандами, возможен путем лигандного обмена, например, между Au25(2-PET)18 и 1,1'-бинафил-2,2'-дитиол (BINAS) [41].
ВЫВОДЫ
Методом теории функционала плотности впервые исследовано строение и реакционная способность циклических тиолатных и дитиолатных комплексов Au(I), образованных скрепочным фрагментом –SR–Au–SR–Au–, типичным для всех Aun(SR)m. По данным расчета, тиолатный комплекс (AuSCH3)4 вследствие прочной связи Au–S является инертным по отношению к О2 и СО. При замене тиолатного лиганда на дитиолатный реакционная способность комплекса по отношению к тестовым молекулам существенно увеличивается. Показано, что (AuSCH2CHSCH3)4 или кластеры золота, содержащие аналогичные фрагменты, могут выступать катализаторами окисления СО. Полученные результаты указывают на важную роль лиганда не только в стабилизации размера кластера или сохранении состава комплекса, но и в изменении свойств комплекса.
Список литературы
Bond G.C., Louis C., Thompson D.T. Catalysis by Gold. London: Imperial College Press, 2007. 366 p.
Stratakis M., GarciaH. // Chem. Rev. 2012. V. 112. № 8. P. 4469.
Пичугина Д.А., Кузьменко Н.Е., Шестаков А.Ф. // Успехи химии. 2015. Т. 84. № 11. С. 1114.
Li G., Jin R. // Acc. Chem. Res. 2014. V. 47. № 3. P. 816.
Zhang G., Wang R., Li G. // Chin. Chem. Lett. 2018. V. 29. № 5. P. 687.
Akola J., Walter M., Whetten R.L., Häkkinen H., Grönbeck H. // J. Am. Chem. Soc. 2008. V. 130. № 12. P. 3756.
Heaven M.W., Dass A., White P.S., Holt K.M., Murray R.W. // J. Am. Chem. Soc. 2008. V. 130. № 12. P. 3754.
Qian H., Eckenhoff W.T., Zhu Y., Pintauer T., Jin R. // J. Am. Chem. Soc. 2010. V. 132. № 24. P. 8280.
Crasto D., Malola S., Brosofsky G., Dass A., Häkkinen H. // J. Am. Chem. Soc. 2014. V. 136. № 13. P. 5000.
Das A., Li T., Li G., Nobusada K., Zeng C., Rosi N.L., Jin R. // Nanoscale. 2014. V. 6. P. 6458.
Zeng C., Liu C., Chen Y., Rosi N.L., Jin R. // J. Am. Chem. Soc. 2014. V. 136. № 34. P. 11922.
Wang P., Xiong L., Sun X., Ma Z., Pei Y. // Nanoscale. 2018. V. 10. № 8. P. 3918.
Yoskamtorn T., Yamazoe S., Takahata R., Nishigaki J., Thivasasith A., Limtrakul J., Tsukuda T. // ACS Catal. 2014. V. 4. № 10. P. 3696.
Li G., Jin R. // Nanotechnol. Rev. 2013. V. 2. № 5. P. 529.
Li G., Jin R. // J. Am. Chem. Soc. 2014. V. 136. № 32. P. 11347.
Liu Y., Tsunoyama H., Akita T., Xie S., Tsukuda T. // ACS Catal. 2011. V. 1. № 1. P. 2.
Kurashige W., Yamaguchi M., Nobusada K., Negishi Y. // J. Phys. Chem. Lett. 2012. V. 3. № 18. P. 2649.
Harkness K.M., Cliffel D.E., McLean J.A. // Analyst. 2010. V. 135. № 5. P. 868.
Nie X., Qian H., Ge Q., Xu H., Jin R. // ACS Nano. 2012. V. 6. P. 6014.
Wu Z., Jiang D.E., Mann A.K., Mullins D.R., Qiao Z.A., Allard L.F., Zeng C., Jin R., Overbury S.H. // J. Am. Chem. Soc. 2014. V. 136. № 16. P. 6111.
Nie X., Zeng C., Ma X., Qian H., Ge Q., Xu H., Jin R. // Nanoscale. 2013. V. 5. P. 5912.
Li Y., Chen Y., House S.D., Zhao S., Wahab Z., Yang J.C., Jin R. // ACS Appl. Mater. Interfaces. 2018. V. 10. № 35. P. 29425.
Bonasia P.J., Gindelberger D.E., Arnold J. // Inorg. Chem. 1993. V. 32. № 23. P. 5126.
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. № 18. P. 3865.
Stevens W.J., Krauss M., Basch H., Jasien P.G. // Can. J. Chem. 1992. V. 70. № 2. P. 612.
Никитина Н.А., Пичугина Д.А., Кузьменко Н.Е. // Журн. физ. химии. 2017. Т. 91. № 8. С. 1364.
Пичугина Д.А., Шестаков А.Ф., Кузьменко Н.Е. // Кинетика и катализ. 2007. Т. 48. № 2. С. 321.
Askerka M., Pichugina D., Kuz’menko N., Shestakov A. // Phys. Chem. A. 2012. V. 116. № 29. P. 7686.
Пичугина Д.А., Николаев С.А., Мухамедзянова Д.Ф., Кузьменко Н.Е. // Журн. физ. химии. 2014. Т. 88. № 6. С. 991.
Polynskaya Y.G., Pichugina D.A., Kuz’menko N.E. // Comput. Theor. Chem. 2015. V. 1055. P. 61.
Schlegel H.B. // J. Comput. Chem. 1982. V. 3. № 2. P. 214.
Gonzalez C., Schlegel H.B. // J. Chem. Phys. 1989. V. 90. № 4. P. 2154.
Лайков Д.Н., Устынюк Ю.А. // Изв. АН. Сер. хим. 2005. № 3. С. 804.
Sadovnichy V., Tikhonravov A., Voevodin Vl., Opanasenko V. Contemporary High Performance Computing: From Petascale toward Exascale (Ser. Chapman & Hall/CRC Computational Science) Boca Raton: CRC Press, 2013. P. 283.
Kawasaki H., Kumar S., Li G., Zeng C., Kauffman D.R., Yoshimoto J., Iwasaki Y., Jin R. // Chem. Mater. 2014. V. 26. № 9. P. 2777.
Li Z., Liu C., Abroshan H., Kauffman D.R., Li G. // ACS Catal. 2017. V. 7. № 5. P. 3368.
Li L., Gao Y., Li H., Zhao Y., Pei Y., Chen Z., Zeng X.C. // J. Am. Chem. Soc. 2013. V. 135. № 51. P. 19 336.
Zeng W., Tang J., Wanga P., Pei Y. // RSC Adv. 2016. V. 6. P. 55 867.
Haruta M. // Gold Bull. 2004. V. 37. P. 27.
Bond G.C., Thompson D.T. // Gold Bull. 2000. V. 33. P. 41.
Knoppew S., Bürgi T. // Phys. Chem. Chem. Phys. 2013. V. 15. P. 15 816.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ